

Abstract
The closely related mitogen-activated protein kinase isoforms extracellular signal-regulated kinase 1 (ERK1) and ERK2 have been implicated in the control of cell proliferation, differentiation and survival. However, the specific in vivo functions of the two ERK isoforms remain to be analysed. Here, we show that disruption of the Erk2 locus leads to embryonic lethality early in mouse development after the implantation stage. Erk2 mutant embryos fail to form the ectoplacental cone and extra-embryonic ectoderm, which give rise to mature trophoblast derivatives in the fetus. Analysis of chimeric embryos showed that Erk2 functions in a cell-autonomous manner during the development of extra-embryonic cell lineages. We also found that both Erk2 and Erk1 are widely expressed throughout early-stage embryos. The inability of Erk1 to compensate for Erk2 function suggests a specific function for Erk2 in normal trophoblast development in the mouse, probably in regulating the proliferation of polar trophectoderm cells.
Introduction
Mitogen-activated protein kinase (MAPK) pathways are evolutionarily conserved signalling modules through which cells transduce extracellular signals into intracellular responses (Lewis et al., 1998; Widmann et al., 1999; Pearson et al., 2001). The prototypical MAPK pathway is the extracellular signal-regulated kinase (ERK)1/2 pathway. Two related MAPK isoforms, known as ERK1 (p44mapk) and ERK2 (p42mapk), which share 84% amino-acid identity, have been identified in mammals (Boulton et al., 1990, 1991). The two proteins are ubiquitously expressed in cell lines and tissues, although their relative abundance is variable. ERK1 and ERK2 are activated by almost all mitogenic factors, differentiation stimuli, cytokines, and by other cellular perturbations. In fibroblasts, the two isozymes are coordinately regulated in response to serum (Meloche, 1995).
Although much still remains to be learned about the physiological functions of ERK1 and ERK2, several lines of evidence have implicated these enzymes in the control of cell proliferation and differentiation (Lewis et al., 1998; Whitmarsh & Davis, 2000). The ERK1/2 MAPK pathway has an important function in the regulation of cell-cycle re-entry and G1-phase progression (Pagès et al., 1993; Lavoie et al., 1996; Roovers & Assoian, 2000). Moreover, evidence suggests that this signalling pathway influences cell growth by modulating the rate of protein and pyrimidine-nucleotide biosynthesis (Servant et al., 1996; Graves et al., 2000). Activation of the ERK1/2 pathway also provides protection against apoptosis in several cell types when they are challenged by cellular stresses or chemotherapeutic drugs (Ballif & Blenis, 2001). However, many questions remain unanswered about the in vivo functions of ERK1 and ERK2 in normal development and growth. In particular, the functional specificity and redundancy of the two MAPK isoforms remain to be analysed by genetic approaches. Pagès et al. generated mice with a null mutation in the Erk1 gene (Pagès et al., 1999). Erk1−/− mice are viable, fertile and of normal size. However, the proliferation and maturation of thymocytes are affected in these mice, despite the expression and activation of Erk2.
We have used a gene-targeting approach to investigate the specific function of Erk2 in development. We found that mouse embryos lacking Erk2 die early during embryogenesis due to a defect in trophoblast development. Mutant embryos fail to form the ectoplacental cone and extra-embryonic ectoderm, which derive from the polar trophectoderm. Importantly, we show that Erk1 is widely expressed in normal and mutant embryos. The lack of compensation of Erk2 deficiency by Erk1 suggests that Erk2 has a specific function in the developing embryo.
Results and Discussion
To explore the function of Erk2 in development, we have inactivated the Erk2 gene by homologous recombination in embryonic stem (ES) cells (Fig. 1A). Targeted disruption of the Erk2 gene was confirmed by Southern analysis of genomic DNA (Fig. 1B) and immunoblot analysis of total protein (Fig. 1C) isolated from whole embryos. A significant percentage of embryos, probably corresponding to heterozygotes, expressed half the amount of Erk2 protein (Fig. 1C). No fragment of Erk2 was detected by immunoblot analysis using an amino-terminus-specific antibody, confirming that the targeting strategy resulted in a null allele. When Erk2 heterozygotes from a hybrid 129/Sv × C57Bl/6 background were intercrossed, no homozygous mutants were recovered from a progeny of 128 (Table 1), indicating that absence of Erk2 leads to embryonic lethality. Notably, Erk2+/− pups represented 50% of the total offspring, deviating from the expected normal mendelian frequency (p < 0.001). This indicates that some heterozygous animals also died during embryonic development. By contrast, in a CD1 background, the same cross (n = 394) produced 61.7% heterozygotes, which is close to the normal mendelian frequency. The cause of death of Erk2+/− animals in the 129/Sv × C57Bl/6 background remains to be investigated. In contrast to the heterozygotes, the phenotype of homozygous embryos was identical in the two genetic backgrounds.
Figure 1.
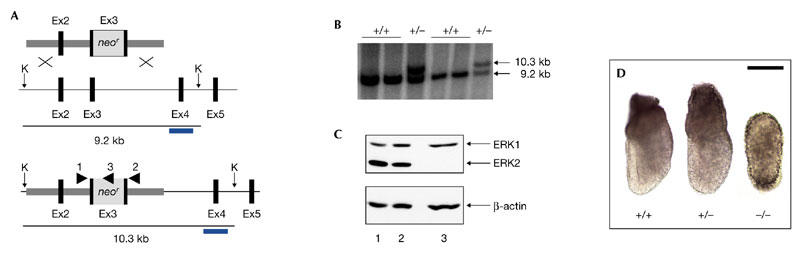
Targeted disruption of the mouse Erk2 gene. (A) The targeting vector (top), the wild-type Erk2 locus (middle) and the mutant Erk2 locus (bottom) are shown. Black boxes represent Erk2 exons (Ex2–Ex5) and the grey box represents the neor (neomycin resistance gene)–poly(A) cassette. The arrowheads indicate the positions and orientations of PCR primers that were used for genotyping analysis. The 3′ external probe used for Southern analysis is shown as blue bars. KpnI restriction sites (K) are indicated. (B) Southern blot analysis of wild-type and heterozygous embryonic stem cell DNA. DNA samples were digested with KpnI and hybridized with the 3′ external probe. The positions and sizes of wild-type and mutant fragments are indicated. (C) Immunoblot analysis of whole extracts from embryos obtained from Erk2 intercrosses. Top panel, blotting using the α1cp44 antibody, which recognizes both Erk1 and Erk2 isoforms. Lower panel, control blot using anti-β-actin. (D) Gross morphology of wild-type, heterozygous and homozygous Erk2 mutant embryos at embryonic day 6.5. Scale bar, 100 μm. Erk2, extracellular signal-regulated kinase 2.
Table 1.
Genotype of progeny from Erk2 intercrosses
| Stage | +/+ | +/− | −/− | Resorbed embryos recovered |
|---|---|---|---|---|
| Weaning | 63 (49.2%) | 65 (50.8%) | 0 (0%) | – |
| E9.5 | 16 (28.6%) | 27 (48.2%) | 0 (0%) | 13 (23.2%) |
| E8.5 | 27 (26.5%) | 56 (54.9%) | 9 (8.8%) | 10 (9.7%) |
| E7.5 | 10 (21.3%) | 26 (55.3%) | 11 (23.4%) | – |
| E6.5 | 19 (28.8%) | 31 (47.0%) | 16 (24.2%) | – |
| Expected | 25% | 50% | 25% | – |
E, embryonic day.
We next analysed timed pregnancies of Erk2 heterozygous matings to determine the stage of embryonic development at which lethality occurred. Embryos collected from embryonic days E6.5–E7.5 were found to have a normal mendelian frequency (Table 1). However, at E8.5, severely resorbed mutant embryos were found, suggesting that the embryos die before this stage. Earlier, at E6.5 and E7.5, Erk2−/− embryos were significantly smaller than their wild-type or heterozygous littermates (Fig. 1D; and data not shown). Mutant embryos at E6.5 showed an abnormal morphology, being oval in shape, with no obvious proximodistal and anteroposterior polarities, which can be easily distinguished in wild-type embryos at this stage (Fig. 1D). Analysis of blastocyst outgrowth showed no difference between genotypes, suggesting that Erk2 is not required for pre-implantation development (data not shown).
Histological analysis was performed to examine defects at the tissue and cellular levels in early post-implantation embryos. At E5.5, wild-type embryos had developed into egg cylinders with organized embryonic and extra-embryonic structures and a proximal–distal polarity that was marked by the ectoplacental cone (Fig. 2A). Mutant embryos were oval in shape, with inner and outer epithelia, and lacked identifiable ectoplacental cones (Fig. 2B). At E6.5, the inner layers of mutant embryos looked disorganized (Fig. 2D), instead of appearing as single layers of pseudostratified epithelia as in wild-type embryos at the early-streak stage of gastrulation (Fig. 2C). The outer epithelial layer, known as the visceral endoderm, was formed in mutant embryos, but visceral endoderm cells accumulated in the proximal and distal regions of the E5.5 and E6.5 embryos, respectively (red arrows in Fig. 2B,D). Parietal endoderm cells were also seen along the uterine walls, suggesting that this differentiation step of the primitive endoderm had occurred in mutant embryos (black arrow in Fig. 2B). In addition, primary giant cells were identified (black arrow in Fig. 2D), indicating that further differentiation of trophectoderm cells takes place in the mutants.
Figure 2.
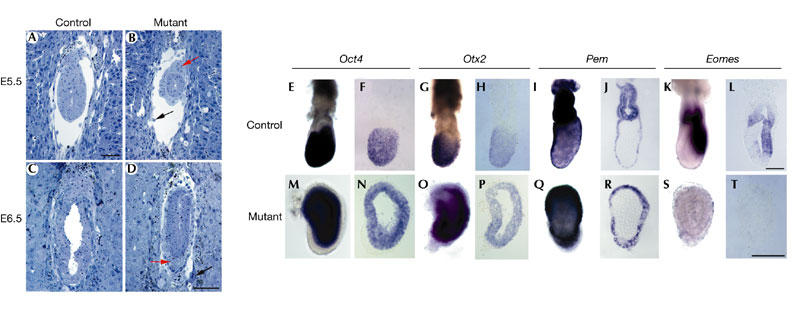
Impairment of extra-embryonic ectoderm development in Erk2−/− embryos. (A–D) Histological sections of control (A,C) and mutant embryos (B,D) at early post-implantation stages (embryonic days (E) 5.5 and 6.5) show the absence of ectoplacental cones and reduced size of the mutant embryo. (E–T) Marker gene expression in control (E–L) and mutant (M–T) embryos by whole-mount in situ hybridization followed by section analyses reveal that the extra-embryonic ectoderm domains of Pem (n = 2) (I,J,Q,R) and eomesodermin (Eomes; n = 2) (K,L,S,T) are not expressed in the mutant embryos, whereas the embryonic domains of Oct4 (n = 2) (E,F,M,N) and Otx2 (n = 2) (G,H,O,P) are still present. (A–D) are shown at the same magnification, as are (E–L) and (M–T). Scale bars, 100 μm. Erk2, extracellular signal-regulated kinase 2.
The phenotype of Erk2 mutant embryos was characterized further by examining the expression of appropriate marker genes for specific tissues in embryos at E6.5. Expression of Oct4 and Otx2 is usually restricted to all distal ectoderm or epiblast cells (Ang et al., 1994; Scholer et al., 1990), and is not detectable in the proximal extra-embryonic ectoderm of wild-type embryos at this stage (Fig. 2E,F and Fig. 2G,H, respectively). However, all cells of the inner layer expressed these genes in Erk2-deficient embryos (Fig. 2M,N and Fig. 2O,P, respectively), suggesting that the epiblast, but not the extra-embryonic ectoderm, was formed in these mutants. This result was confirmed by the absence of expression of two markers of the extra-embryonic ectoderm, Pem (Lin et al., 1994) and eomesodermin (Eomes; Russ et al., 2000), in mutant embryos (Fig. 2Q,R and Fig. 2S,T, respectively). Pem and Otx2 were expressed in the outer layer of wild-type as well as mutant embryos (Fig. 2R and Fig. 2P, respectively), indicating that the visceral endoderm is specified to some extent in these mutants. The histological and molecular data indicate that inactivation of Erk2 leads to a specific loss of the extra-embryonic ectoderm and ectoplacental cone, whereas other tissues are formed in the mutant embryos. As the extra-embryonic ectoderm and ectoplacental cone are usually derived from the polar trophectoderm, which corresponds to the subset of trophectoderm cells that are in contact with the inner cell mass (ICM) in the blastocyst, our results strongly suggest that the morphological defects in Erk2 mutants may result from a primary defect at the level of the polar trophectoderm.
Recent molecular and genetic findings indicate that fibroblast growth factor 4 (Fgf4) is the main signal derived from the ICM that is required for the proliferation of polar trophectoderm cells. Direct evidence for a role of Fgf4 in promoting trophoblast proliferation comes from the treatment of wild-type and Oct4 mutant blastocysts with Fgf4, which increases trophectoderm cell numbers (Nichols et al., 1998), and the ability to generate trophoblast stem (TS) cell lines from blastocysts and extra-embryonic ectoderm in the presence of Fgf4 and fibroblast-conditioned medium (Tanaka et al., 1998). Fgf receptor 2 (Fgfr2), which is strongly expressed in the proliferating trophoblast, is a likely candidate receptor for receiving the Fgf4 signal. This model is consistent with genetic studies that show that Fgf4- and Fgfr2-deficient embryos show similar phenotypes at the peri-implantation stage. As ERK1/2 MAPKs are important downstream effectors of FGF receptors (Ornitz & Itoh, 2001), it is tempting to speculate that Erk2 functions downstream of Fgfr2 and is required cell autonomously in polar trophectoderm cells to transduce the proliferative signal of Fgf4.
To distinguish specific functions of Erk2 in embryonic versus extra-embryonic tissues, we generated Erk2−/− <-> +/+ chimeric embryos with wild-type extra-embryonic tissues and >95% Erk2−/− epiblast cells by injecting Erk2−/− ES cells into Rosa26 lacZ-expressing heterozygous embryos (Beddington & Robertson, 1989). The Erk2−/− ES cells were isolated from a heterozygous intercross (see supplementary information online). The resulting chimaeras (n = 5) developed ectoplacental cones and extra-embryonic ectoderm abutting the embryonic ectoderm (Fig. 3A,B; and data not shown), indicating that the failure of these tissues to develop correctly in Erk2−/− mutant embryos is not because of a function of Erk2 in the epiblast. This result suggests a specific function for Erk2 in extra-embryonic tissues in the development of the extra-embryonic ectoderm.
Figure 3.
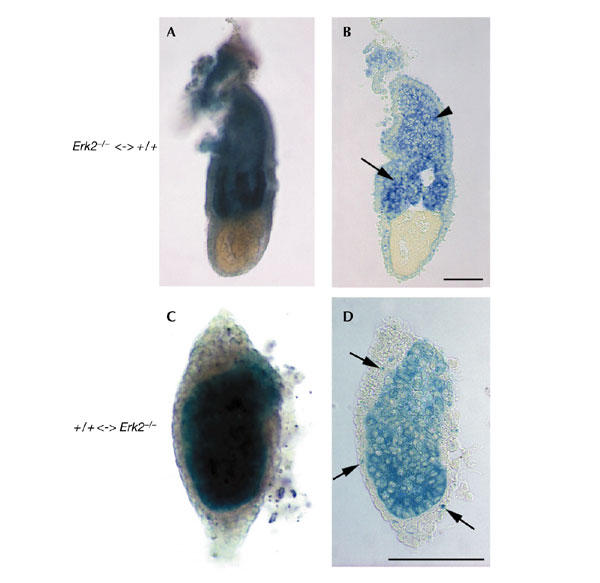
Erk2 is required in extra-embryonic tissues for the development of the extra-embryonic ectoderm and the ectoplacental cone. Chimeric embryos at embryonic day (E) 6.5 stained for β-galactosidase, and the corresponding sagittal sections, are shown. (A,B) Chimeric embryo with wild-type extra-embryonic tissues (blue) and a predominantly Erk2−/− epiblast (white), showing rescue of extra-embryonic ectoderm (arrow in (B)) and ectoplacental cone (arrowhead in (B)) development. (C,D) By contrast, a chimeric embryo with Erk2−/− extra-embryonic tissues (white) and a predominanly wild-type (blue) epiblast phenocopies Erk2−/− embryos. In the latter chimaera, a significant number of wild-type cells also contributed to the visceral endoderm (arrows in (D)). Magnifications are the same in (A,B) and in (C,D). Scale bars, 100 μm. Erk2, extracellular signal-regulated kinase 2.
We also generated reciprocal +/+ <-> Erk2−/− chimeric embryos, with Erk2−/− extra-embryonic tissues and predominantly wild-type epiblast cells by injecting LacZ-positive wild-type ES cells into embryos collected from Erk2+/− intercrosses. These chimaeras (n = 3), with a contribution of 50–90% from wild-type ES-derived cells, morphologically phenocopied the Erk2−/− mutant embryos. Histological sections of these chimaeras revealed the absence of extra-embryonic ectoderm and ectoplacental cones (Fig. 3C,D; and data not shown). In addition, cells accumulated in the distal and/or proximal regions of the visceral endoderm in the chimaeras, as seen in Erk2−/− embryos, and the epiblast cells failed to develop into a columnar epithelium. The presence of a high contribution of normal ES-derived cells in embryonic tissues of Erk2−/− <-> +/+ chimeric embryos did not rescue the development of the extra-embryonic tissues. Altogether, the data obtained from the chimeric studies show that Erk2 functions autonomously in extra-embryonic tissues, probably in the polar trophectoderm, for the development of the ectoplacental cone and the extra-embryonic ectoderm. The visceral endoderm and epiblast defects observed in Erk2−/− mutant embryos and +/+ <-> Erk2−/− chimeric embryos may be indirect and will require further investigation.
Disruption of the Erk1 gene did not result in any developmental phenotype (Pagès et al., 1999), indicating that Erk1 is dispensable during embryonic development or, alternatively, that Erk2 can compensate for the loss of Erk1. A possible explanation for the embryonic lethality seen on inactivation of the Erk2 gene is that Erk1 is not expressed in early-stage embryos, and thus cannot functionally rescue the loss of Erk2. To determine whether Erk1 and Erk2 differ in the timing of their expression during early embryonic development, we performed radioactive in situ hybridization on sagittal sections of wild-type and mutant embryos. Both Erk2 and Erk1 were found to be widely expressed throughout the whole embryo (Fig. 4A,F and Fig. 4C,H, respectively). Immunohistochemical analysis of E5.5 embryos using a phosphospecific antibody indicated that Erk1 and Erk2 are activated in the extra-embryonic ectoderm, ectoplacental cone and trophectoderm giant cells (see supplementary information online). We also found that Erk1 and Erk2 proteins are expressed and phosphorylated in blastocyst-derived TS cells (data not shown). As Erk1 is still expressed in Erk2 mutants (Fig. 4E,J; n = 2), the lack of compensation for the loss of Erk2 by Erk1 strongly suggests that Erk2 has a specific function in the developing embryo.
Figure 4.
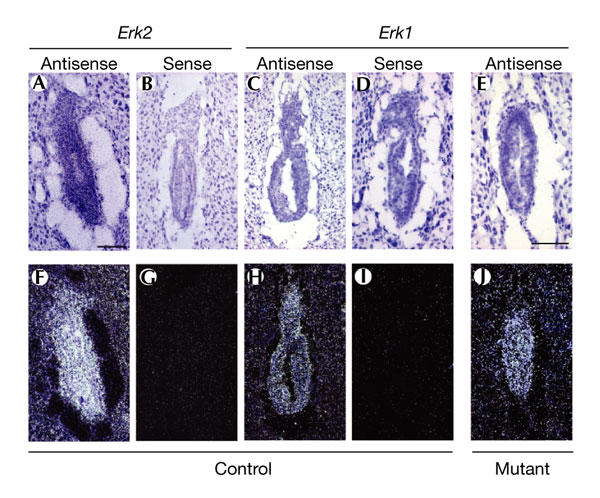
Gene expression patterns for Erk1 and Erk2 at E6.5. Brightfield (A–E) and corresponding darkfield images (F–J) are shown. Erk2 (A,B,F,G) and Erk1 (C–E,H–J) are ubiquitously expressed throughout the whole embryo in control embryos ((A,F) and (C,H), respectively) at E6.5, as assessed by 35S radioactive in situ hybridization. Erk1 is expressed in mutant embryos (E,J). 3′ UTR sense and antisense probes were used. Magnifications are the same in (A–D,F–I) and (E,J). Scale bars, 100 μm. Erk2, extracellular signal-regulated kinase
Our results show that Erk2 has an essential function in embryonic development in the generation of extra-embryonic ectoderm, which gives rise to the fetal part of the placenta and the secondary trophoblast giant cells. Hence, Erk2 is required for mouse trophoblast development, probably in signalling that is required for the proliferation of TS cells in the polar trophectoderm. This hypothesis is supported by the observation that TS cell lines cannot be generated in the presence of specific inhibitors of the upstream kinases MAPK/ERK kinase 1 (MEK1) and MEK2 (Rossant, 2001). Interestingly, the activation of ERK1/2 was recently suggested to be dispensable for the proliferation of ES cells, but might instead have a more important function in their differentiation (Burdon et al., 1999). In support of this idea, we found no significant change in the proliferative rate of Erk2−/− epiblast cells in mutant embryos when compared to wild-type littermates at E6.5 (see supplementary information online). All these findings indicate that the early cell lineages in the conceptus respond differently to ERK1/2 signalling. TS cells and ES cells might therefore be good in vitro model systems for examining how specific cellular responses to the ERK1/2 MAPK pathway are generated.
Methods
Gene disruption.
The targeting vector was constructed by inserting a PGK–neor cassette into exon 3 of Erk2, which contains protein kinase subdomains V and VI. The linearized vector was electroporated into the 129-derived ES cell line D3. The presence of the targeted allele of Erk2 was determined by restriction-fragment-length-polymorphism analysis of genomic DNA extracted from ES cells using an external 3′ probe corresponding to a 1.5-kb XbaI fragment containing exon 4. The presence of a single neor cassette was confirmed by Southern blot analysis using a neor gene fragment as a probe (data not shown). Cells heterozygous at the Erk2 locus (Erk2+/−) were used to generate chimeric mice by injection into blastocyst-stage embryos. Chimeric males were bred with C57Bl/6 females to produce F1 heterozygotes. Germline transmission was confirmed by Southern blot analysis. F1 heterozygous males and females were mated to produce homozygous mutant animals. DNA was extracted from tail biopsies obtained three weeks after birth. Genotypes were determined by Southern blot hybridization using an external 3′ probe or by PCR (see supplementary information online).
Generation of chimeric embryos.
The Erk2−/− ES cell line B1 was generated by the in vitro culture of blastocysts (Hogan et al., 1994) isolated from Erk2 heterozygous intercross matings. Cytogenetic analysis of the B1 line was performed by the Banque de Cellules Leucémique du Québec and showed a normal karyotype. The morulae-stage (E2.5) embryos used to generate chimaeras were obtained from intercrosses of ROSA26/+ and CD1 mice. These embryos were injected with approximately ten Erk2−/− ES cells from the B1 line and ten LacZ-positive ES cells from the Rosa26.1 line (Varlet et al., 1997), respectively, and were subsequently reimplanted into pseudopregnant females. Chimeric embryos were harvested at E6.5 and E7.5 and processed for β-galactosidase staining as described previously (Hogan et al., 1994).
Immunoblot analysis.
Protein extracts were prepared by homogenization of embryos or cells in lysis buffer. Equal amounts of protein were resolved by SDS–polyacrylamide gel electrophoresis and transferred to nylon membrane. The membrane was blocked in TBS, 0.1% Tween 20, 4% non-fat dried milk, and probed with antibody α1cp44 (1:5000 dilution), which recognizes Erk1 and Erk2 isoforms (Meloche, 1995). To control for protein loading, the blot was stripped and reprobed with anti-β-actin (1:10,000 dilution). We also used an Erk2 N-terminal-specific polyclonal antibody (AB3055; Chemicon) to confirm the absence of expression of N-terminal fragments of Erk2.
Supplementary information is available at EMBO reports online (http://www.nature.com/embor/journal/vaop/ncurrent/extref/4-embor939-s1.pdf).
Supplementary Material
Supplementary information
Acknowledgments
We thank A. Dierich for the culture of ES cells, H. Scholer and A. Russ for providing Oct4 and eomesodermin probes, E. Robertson for providing Rosa26.1 ES cells, and J. Rossant for critical reading of the manuscript and for the phospho-ERK1/2 immunostaining protocol. We acknowledge C. Benoist and D. Mathis for their contribution during the early phase of this work. This work was supported by grants from the Cancer Research Society and Canadian Institutes of Health Research (CIHR) to S.M., by grants from the European Community Biotech Programme and Association pour la Recherche Contre le Cancer, and by funds from the INSERM, the Centre National de la Recherche Scientifique and the Hôpital Universitaire de Strasbourg to S.-L.A. S.M. is an Investigator of the CIHR.
References
- Ang S.L, Conlon R.A., Jin O. & Rossant J. ( 1994) Positive and negative signals from mesoderm regulate the expression of mouse Otx2 in ectoderm explants. Development, 120, 2979–2989. [DOI] [PubMed] [Google Scholar]
- Ballif B.A. & Blenis J. ( 2001) Molecular mechanisms mediating mammalian mitogen-activated protein kinase (MAPK) kinase (MEK)–MAPK cell survival signals. Cell Growth Differ., 12, 397–408. [PubMed] [Google Scholar]
- Beddington R.S. & Robertson E.J. ( 1989) An assessment of the developmental potential of embryonic stem cells in the midgestation mouse embryo. Development, 105, 733–737. [DOI] [PubMed] [Google Scholar]
- Boulton T.G. et al. ( 1990) An insulin-stimulated protein kinase similar to yeast kinases involved in cell cycle control. Science, 249, 64–67. [DOI] [PubMed] [Google Scholar]
- Boulton T.G. et al. ( 1991) ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell, 65, 663–675. [DOI] [PubMed] [Google Scholar]
- Burdon T., Stracey C., Chambers I., Nichols J. & Smith A. ( 1999) Suppression of SHP-2 and ERK signalling promotes self-renewal of mouse embryonic stem cells. Dev. Biol., 210, 30–43. [DOI] [PubMed] [Google Scholar]
- Graves L.M. et al. ( 2000) Regulation of carbamoyl phosphate synthetase by MAP kinase. Nature, 403, 328–332. [DOI] [PubMed] [Google Scholar]
- Hogan B., Beddington R., Constantini F. & Lacy E. ( 1994) Manipulating the Mouse Embryo: A Laboratory Manual, 2nd edn. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, USA. [Google Scholar]
- Lavoie J.N., Rivard N., L'Allemain G. & Pouyssegur J. ( 1996) A temporal and biochemical link between growth factor-activated MAP kinases, cyclin D1 induction and cell cycle entry. Prog. Cell Cycle Res., 2, 49–58. [DOI] [PubMed] [Google Scholar]
- Lewis T.S., Shapiro P.S. & Ahn N.G. ( 1998) Signal transduction through MAP kinase cascades. Adv. Cancer Res., 74, 49–139. [DOI] [PubMed] [Google Scholar]
- Lin T.P. et al. ( 1994) The Pem homeobox gene is X-linked and exclusively expressed in extraembryonic tissues during early murine development. Dev. Biol., 166, 170–179. [DOI] [PubMed] [Google Scholar]
- Meloche S. ( 1995) Cell cycle reentry of mammalian fibroblasts is accompanied by the sustained activation of p44mapk and p42mapk isoforms in the G1 phase and their inactivation at the G1/S transition. J. Cell Physiol., 163, 577–588. [DOI] [PubMed] [Google Scholar]
- Nichols J. et al. ( 1998) Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell, 95, 379–391. [DOI] [PubMed] [Google Scholar]
- Ornitz D.M. & Itoh N. ( 2001) Fibroblast growth factors. Genome Biol., 2, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagès G., Lenormand P., L'Allemain G., Chambard J.C., Meloche S. & Pouyssegur J. ( 1993) Mitogen-activated protein kinases p42mapk and p44mapk are required for fibroblast proliferation. Proc. Natl Acad. Sci. USA, 90, 8319–8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagès G. et al. ( 1999) Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science, 286, 1374–1377. [DOI] [PubMed] [Google Scholar]
- Pearson G. et al. ( 2001) Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr. Rev., 22, 153–183. [DOI] [PubMed] [Google Scholar]
- Roovers K. & Assoian R.K. ( 2000) Integrating the MAP kinase signal into the G1 phase cell cycle machinery. Bioessays, 22, 818–826. [DOI] [PubMed] [Google Scholar]
- Rossant J. ( 2001) Stem cells from the mammalian blastocyst. Stem Cells, 19, 477–482. [DOI] [PubMed] [Google Scholar]
- Russ A.P. et al. ( 2000) Eomesodermin is required for mouse trophoblast development and mesoderm formation. Nature, 404, 95–99. [DOI] [PubMed] [Google Scholar]
- Scholer H.R., Dressler G.R., Balling R., Rohdewohld H. & Gruss P. ( 1990) Oct-4: a germline-specific transcription factor mapping to the mouse t-complex. EMBO J., 9, 2185–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant M.J., Giasson E. & Meloche S. ( 1996) Inhibition of growth factor-induced protein synthesis by a selective MEK inhibitor in aortic smooth muscle cells. J. Biol. Chem., 271, 16047–16052. [DOI] [PubMed] [Google Scholar]
- Tanaka S., Kunath T., Hadjantonakis A.K., Nagy A. & Rossant J. ( 1998) Promotion of trophoblast stem cell proliferation by FGF4. Science, 282, 2072–2075. [DOI] [PubMed] [Google Scholar]
- Varlet I., Collignon J. & Robertson E.J. ( 1997) nodal expression in the primitive endoderm is required for specification of the anterior axis during mouse gastrulation. Development, 124, 1033–1044. [DOI] [PubMed] [Google Scholar]
- Whitmarsh A.J. & Davis R.J. ( 2000) A central control for cell growth. Nature, 403, 255–256. [DOI] [PubMed] [Google Scholar]
- Widmann C., Gibson S., Jarpe M.B. & Johnson G.L. ( 1999) Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol. Rev., 79, 143–180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information