

Abstract
The serine protease plasmin can efficiently degrade amyloid peptide in vitro, and is found at low levels in the hippocampus of patients with Alzheimer's disease (AD). The cause of such paucity remains unknown. We show here that the levels of total brain plasminogen and plasminogen-binding molecules are normal in these brain samples, yet plasminogen membrane binding is greatly reduced. Biochemical analysis reveals that the membranes of these brains have a mild, still significant, cholesterol reduction compared to age-matched controls, and anomalous raft microdomains. This was reflected by the loss of raft-enriched proteins, including plasminogen-binding and -activating molecules. Using hippocampal neurons in culture, we demonstrate that removal of a similar amount of membrane cholesterol is sufficient to induce raft disorganization, leading to reduced plasminogen membrane binding and low plasmin activity. These results suggest that brain raft alterations may contribute to AD by rendering the plasminogen system inefficient.
Introduction
The plasminogen system comprises an inactive proenzyme, plasminogen, which is converted to the active enzyme plasmin on proteolytic cleavage (Plow et al., 1995). In the brain, the plasminogen system is involved in long-term potentiation (Qian et al., 1993) and learning (Calabresi et al., 2000), and mediates excitotoxicity after stroke and ischaemia (Chen & Strickland, 1997). Recently, plasmin has been related to Alzheimer's disease (AD) because it efficiently degrades amyloid peptide in vitro (Ledesma et al., 2000; Tucker et al., 2000) and its levels in the hippocampi and cortex of AD patients are significantly low (Ledesma et al., 2000).
Plasmin activity is regulated by plasminogen activators, plasminogen activator inhibitors and plasmin inhibitors (Collen, 1999). In addition, plasmin generation is dependent on the binding of plasminogen to the plasma membrane (Plow et al., 1995). This occurs via the association to molecules such as α-enolase (Miles et al., 1991), annexin II (Hajjar & Acharya, 2000), amphoterin (Parkkinen & Rauvala, 1991) or the ganglioside GM1 (Miles et al., 1989). Experimental data point to glycolipid-cholesterol detergent-resistant microdomains (DRMs or rafts; Simons & Toomre, 2000) as the sites where binding and activation take place. Thus, the urokinase-type plasminogen activator receptor (uPAR) is enriched in caveolae in a melanoma cell line, and interference with caveolae structure affects its function (Stahl & Mueller, 1995). Moreover, plasminogen-binding molecules are enriched in rafts of neurons (Ledesma et al., 2003), and plasmin is almost exclusively present in these neuronal microdomains (Ledesma et al., 2000). Given all this evidence, we reasoned that raft membrane domain disorganization might cause the low plasmin levels found in cases of AD. This hypothesis is validated in this work using human brain hippocampal tissue and rat hippocampal neurons in primary culture.
Results
Low levels of plasmin in AD ApoE4 brains
To determine the reason for the plasmin deficit observed in AD hippocampi (Ledesma et al., 2000), we repeated the plasmin level measurement in a larger sample population: 26 age-matched cases, 16 with AD and 10 nonaffected (see supplementary material and Table 1 online). Human hippocampi were analysed using an antiplasminogen antibody that also recognizes plasmin (Fig. 1A). This study revealed a statistically significant 67% average decrease in plasmin levels in ten of the AD samples, while the remaining six AD samples presented plasmin values similar to the ten control samples.
Figure 1.

Plasmin levels and activity, and plasminogen bound to the membrane are diminished in AD brains bearing the ApoE4 allele. Western blots with an equal protein amount from total and membrane extracts of control (C), AD non-ApoE4 (AD) and AD ApoE4 (AD4) human hippocampi. Left panels show representative examples of each group corresponding to (A) total plasmin, (B) total plasminogen and (C) membrane-bound plasminogen. Sample loading controls using antibodies against tubulin (for total extracts) and γ-adaptin (for membrane extracts) are also shown. Numbers on the left indicate molecular weights in kilodaltons. Right-side graphics show the mean values and standard deviations obtained for ten control, six AD non-ApoE4 and ten AD ApoE4 samples indicated in arbitrary units (a.u.). Statistical analysis revealed significant differences (*P<0.005) between AD ApoE4 samples versus control and AD non-ApoE4 only for plasmin and membrane-bound plasminogen. Graphs in (D) show plasmin activity in a.u. The scale range is equivalent in the two graphs. Data correspond to the absorbance at 405 nm produced on cleavage of a plasmin-specific chromogenic substrate after the addition of exogenous plasminogen to equal amounts of membrane extracts from control (a) and AD ApoE4 (b) hippocampi with similar post-mortem delays (5, 8, 10 and 15 h as indicated).
The existence of two groups of AD patients with different plasmin levels suggests that disease-associated features such as inflammation or cell death are not responsible for plasmin paucity. This view is reinforced by the fact that the amount of amyloid plaques and tau filament aggregation are similar for all AD samples (see supplementary material and Table 1 online). Furthermore, variables such as age, sex, post-mortem brain sampling delay, hippocampal area analysed, degree of dementia or cause of death could also not be correlated to the alteration observed. On the other hand, we observed that all individuals with normal plasmin levels, AD and non-AD, carried either the E3/E3 or E3/E2 alleles of the apolipoprotein E (ApoE), whereas all individuals with low plasmin content carried at least one ApoE4 allele, a known risk factor for AD (Corder et al., 1993). Although these data point to the existence of a link between AD ApoE4 and low plasmin, it is beyond the scope of this work to determine if ApoE4 is a primary cause of this defect. Our goal remained to elucidate what cellular requirements might have failed in this group of patients.
Reduced plasminogen binding in AD hippocampi
To identify the possible cause of low plasmin in human AD brains, we measured the levels of the plasmin precursor plasminogen. Figure 1B shows that total plasminogen levels are similar in all human hippocampi, irrespective of plasmin levels. To analyse whether low plasmin is due to decreased plasminogen membrane binding, a requisite for its activation into plasmin, this event was next measured. Indeed, membranes from the low-plasmin group contain a significant 70% reduction in bound plasminogen (Fig. 1C). To demonstrate directly that the reduced levels of membrane-bound plasminogen reflect low binding and activation capacities, all human membranes were incubated in the presence of exogenous human plasminogen, followed by a chromogenic-based plasmin activity assay (see Methods). In membranes derived from biopsy samples obtained within the first 15 h of death, plasmin activity was observed. However, this was significantly lower in membranes from the low-plasmin AD group (Fig. 1D).
The above data show that low-plasmin AD hippocampi have a defect in plasminogen membrane binding and activation. We reasoned that this could be due to either downregulation of plasminogen-binding molecules or to defects in the membrane domains required for plasminogen binding.
Abnormal rafts in AD hippocampi
Molecules such as α-enolase, amphoterin, annexin II and the ganglioside GM1 are known to mediate binding of plasminogen to the surface of different cell types (see Introduction), including hippocampal neurons (Ledesma et al., 2003). Consequently, the levels of these proteins were measured in all human hippocampi. Figure 2 shows that all molecules are at similar levels in all samples.
Figure 2.
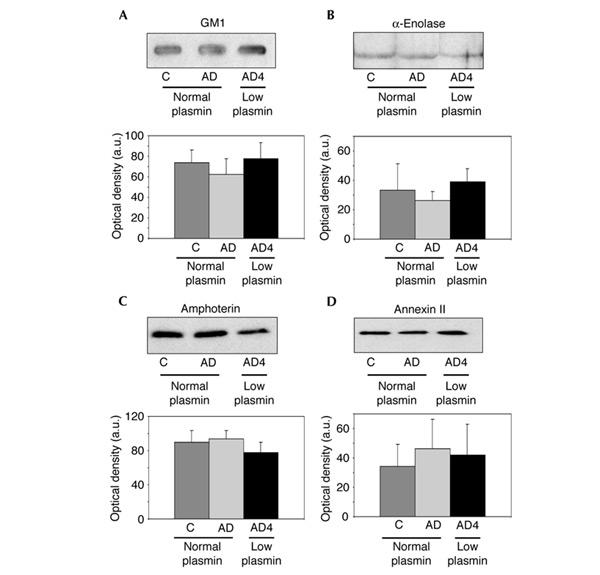
Levels of plasminogen-binding molecules show no significant differences. (A) GM1 levels were detected by slot-blot using cholera toxin linked to peroxidase. Western blots of hippocampal membranes using antibodies against α-enolase (B), amphoterin (C) and annexin II (D) from control (C) and AD non-ApoE4 patients (AD) showing normal levels of plasmin and AD ApoE4 patients (AD4) with low plasmin. Representative examples of each group are shown on the autoradiograms. Graphs represent mean values and standard deviations in arbitrary units (a.u.), obtained in the densitometric quantification of the blots from ten control, six AD non-ApoE4 and ten AD ApoE4 brains. The Student's t-test revealed no significant differences (P>0.005).
To determine whether the problem is at the level of the plasma membrane, the organization of rafts that have been implied in plasminogen activation was analysed by conventional detergent extraction-gradient procedures. Figure 3A shows that the membranes derived from the normal plasmin levels group, AD and non-AD, contain an equivalent protein content in the raft fraction: 13±5 and 15.3±6% of the total protein, respectively (Fig. 3Aa). Conversely, the membranes from the low-plasmin samples have a more than twofold, statistically significant decrease in raft protein content (6.2±2% of the total protein; P<0.005) (Fig. 3Aa). No significant differences in protein content were observed between the two groups in any of the other gradient fractions (Fig. 3Ab). Although the reduced amount of proteins in rafts of AD patients with low plasmin suggests that rafts are no longer efficient as a means of clustering proteins, we tested this directly by analysing the migration pattern of two raft markers: the glycolipid GM1 and the protein flotillin 1 (Fig. 3B). As predicted, GM1 and flotillin 1 appeared to be enriched in the raft fractions of normal-plasmin human samples. By contrast, GM1 and flotillin 1 appeared displaced to non-raft heavier fractions in the low-plasmin human samples. In addition, uPAR migrated with typical raft protein characteristics in normal-plasmin human membranes but not in low-plasmin samples (Fig. 3B). These results indicate that human AD brains with low plasmin have anomalous raft microdomains, resulting in the altered distribution of plasminogen-binding and -activating molecules.
Figure 3.

Raft alteration and reduced membrane cholesterol in low-plasmin AD brains. (A) Rafts are altered in the hippocampus of low-plasmin AD brains. Hippocampal samples with an equal amount of protein from control individuals (C) and AD non-ApoE4 patients (AD) with normal levels of plasmin, and AD ApoE4 patients (AD4) with low plasmin levels were detergent extracted on ice and floated in an Optiprep gradient. Equal volumes of each fraction (40%, 30%, interphase 30–5% (rafts) and 5% Optiprep) were analysed by SDS–PAGE and stained with Coomassie blue. (a) Representative gels for each group. Numbers on the left indicate molecular weights in kilodaltons. (b) Quantification with mean values and standard deviations of the samples with normal plasmin (n=16) or low plasmin (n=10). Average percentages of total protein in the different fractions of the Optiprep gradients and their standard deviations were 61.3±8 (40% fraction), 13.5±4.1 (30% fraction), 14.1±5.5 (raft fraction) and 11±4.2% (5% fraction) in the normal-plasmin group, and 64.6±6.8 (40% fraction), 17.2±4.3 (30% fraction), 6.2±2 (raft fraction) and 12±5% (5% fraction) in the low-plasmin group. The amount of protein is significantly diminished (*P<0.005) in the raft fraction of the low-plasmin brains only. (B) Changes in the flotation profile of the raft markers GM1 and flotillin 1 and the uPAR. Representative example of the sucrose gradient of normal- and low-plasmin human hippocampal samples after detergent extraction on ice. GM1 was detected by slot-blot and cholera toxin linked to peroxidase. Flotillin 1 and uPAR were detected by western blot with specific antibodies. (C) Brain membrane (Mbr. chol.) but not total cholesterol (Tot. chol.) levels are diminished in low-plasmin hippocampi. Total cholesterol in membrane (a) and total extracts (b) of normal-plasmin individuals' (n=16) and low-plasmin AD patients' (n=10) hippocampi (bars indicate standard deviations). A statistically significant (*P<0.005) 36% cholesterol reduction was found in the membranes of low-plasmin AD patients while levels of cholesterol in total extracts were similar.
To determine the cause of raft disorganization in the AD low-plasmin samples, cholesterol levels were quantified in the membrane and total cell extracts from all human samples. Membrane cholesterol was 0.7±0.2 nmol μg−1 protein in the low-plasmin group and 1.1±0.3 nmol μg−1 protein in the normal-plasmin group. This represents a statistically significant 36% reduction in membrane cholesterol in the low-plasmin group (Fig. 4Ca). By contrast, total cholesterol levels were very similar in both groups (0.49±0.05 and 0.48±0.1 nmol μg−1 protein in the normal and deficient raft groups, respectively; Fig. 4Cb).
Figure 4.
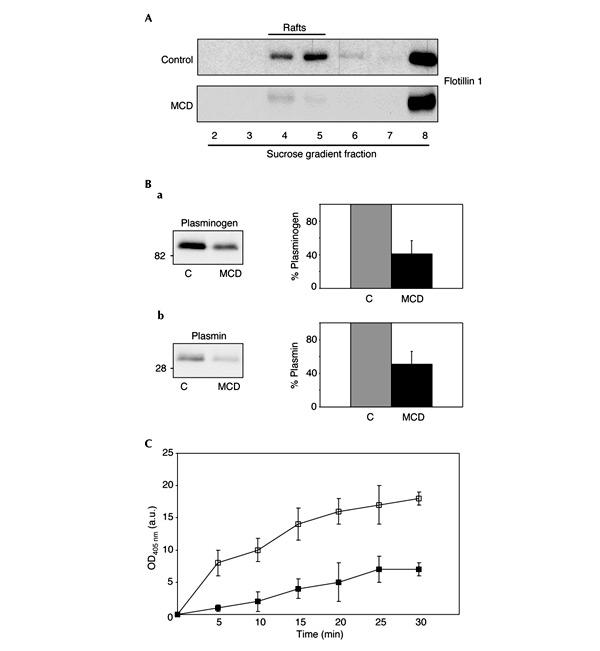
Mild cholesterol extraction leads to raft disruption and reduced plasmin production and activity in cultured hippocampal neurons. (A) Mild cholesterol extraction leads to raft disruption in cultured hippocampal neurons. Raft disruption was confirmed by detergent extraction on ice and sucrose gradients of an equivalent number of control and methyl-β-cyclodextrin (MCD)-treated neurons. While in control neurons the raft marker flotillin 1 floats to light density fractions (4 and 5) corresponding to rafts; the protein is displaced to heavy fractions in MCD-treated cells. (B) Mild cholesterol extraction inhibits plasminogen binding and plasmin production. Rat hippocampal neurons treated (MCD) or not (C) with MCD were incubated with human plasminogen. We measured the amount of bound plasminogen by western blot of membrane extracts using an antibody against human plasminogen (a) that also recognizes the 30 kDa plasmin (b) but not the rodent homologue. Numbers on the left indicate molecular weights in kilodaltons. Graphics show mean values with standard deviations of four independent experiments. Not only plasminogen binding was reduced (in average 59%) but also its activation to plasmin (in average 49%) in MCD-treated neurons. (C) Mild cholesterol extraction reduces plasmin enzymatic activity. Plasmin activity after the addition of exogenous plasminogen, indicated in arbitrary units after measuring absorbance at 405 nm on the same number of hippocampal neurons treated (filled squares) or not (open squares) with MCD. Cholesterol-depleted neurons show lower plasmin activity at all the times analysed (5–30 min).
Raft disruption reduces plasmin activity in primary neurons
The studies in human tissue reveal that low membrane cholesterol, raft disorganization, reduced plasminogen binding and poor plasmin activity coexist in the same samples. To test directly whether membrane cholesterol reduction triggers all the other events, hippocampal neurons were exposed to low concentrations of mevilonin and methyl-β-cyclodextrin (MCD) for 5 days, until 30–35% of total membrane cholesterol was extracted. In the treated neurons, membrane rafts appeared to be disrupted by cholesterol removal as shown by the displacement, from the raft fraction to heavier fractions, of the marker protein flotillin 1 (Fig. 4A) as well as GM1 (not shown). To test that raft disorganization was due to membrane cholesterol reduction rather than secondary to cell death, apoptotic rates were measured and were similar: 16±3 and 17±2% of total cells in normal and low cholesterol-containing neurons, respectively. This was also confirmed by the lack of disturbance of the neurons' overall architecture and molecular polarization (see supplementary figure B online).
The method for cholesterol reduction in cultured neurons reproduces the situation found in human samples regarding cholesterol loss and raft disruption without affecting cell viability. Once this had been confirmed, treated and untreated rat hippocampal neurons were incubated with human plasminogen, and the conversion of plasminogen to plasmin was analysed by western blotting and proteolytic activity. Neurons with intact rafts showed a 59±16% higher plasminogen binding (Fig. 4Ba), which correlated with a 49±15% higher plasmin production compared to neurons with disrupted rafts (Fig. 4Bb). Moreover, plasmin enzymatic activity was significantly lower in the membrane cholesterol-reduced neurons than in the normal-cholesterol cells (Fig. 4Bc). Together, these data indicate that the low cholesterol, anomalous rafts, low plasminogen binding and low plasmin activity present in a group of human AD hippocampi are directly linked.
Discussion
The question of the cellular requirements for brain plasmin generation in normal and pathological conditions was addressed here by performing biochemical analysis in human samples with normal and low levels of plasmin, and in rodent hippocampal neurons in culture. The results presented indicate that neuronal membrane rafts are essential for plasmin generation, by concentrating plasminogen-binding and -activating molecules. Thus, human AD brain samples with low plasmin activity show a low amount of membrane-bound plasminogen and raft anomalies. The fact that in the same human membranes cholesterol was significantly, although not dramatically, reduced indicates that cholesterol loss may be a primary cause for raft disruption. This is supported by the fact that rodent neurons in culture, with a 30–35% reduction of membrane cholesterol, equivalent to that observed in the human samples with low plasmin, presented disorganized rafts, reduced membrane plasminogen binding and low plasmin activity. Since the disorganized rafts were observed exclusively in brain samples from AD patients, we reason that raft disorganization could lead to AD because of a failure in the activation of a plasmin-mediated amyloid-clearance pathway. Such a postulate is in apparent contradiction to that in which raft disorganization may prevent AD by reducing amyloid peptide production (Simons et al., 1998; Ehehalt et al., 2003). The amount of cholesterol removal, and the type of cell used, may explain the different outcomes of raft disruption. Thus, we observed loss of plasmin activity when cholesterol reduction was not higher than 36% (human samples) or 35% (rat hippocampal neurons). Differently, inhibition of amyloid-β production was in one case (Simons et al., 1998) observed after the acute removal of more than 60% of membrane cholesterol, and in the other (Ehehalt et al., 2003) found in undifferentiated N2A cells. In any event, it appears clear that the lack of proper raft organization reported here in a group of AD patients would also produce deficits in raft-mediated functions other than plasmin activation. It is an important task for the future to determine which functions are affected and how they relate to AD.
We have also shown that plasmin paucity and raft defects in the human samples correlate with the presence of the E4 allele of ApoE, an essential protein in brain cholesterol homeostasis (Herz & Beffert, 2000). Although it is not possible to draw any definitive conclusion until a larger population is studied, the possibility that this correlation holds true will open new perspectives for the implication of this known risk factor. In that scenario, the ApoE4 allele, in predisposed individuals, could contribute to raft alterations through deficient cholesterol homeostasis. In support of this, transgenic mice carrying the human ApoE4 allele have low brain cholesterol (Hamanaka et al., 2000), and their astrocytes, a cellular system responsible for neuronal cholesterol, release less cholesterol than astrocytes from ApoE3 mice (Gong et al., 2002).
Irrespective of the origin of raft defects, the demonstration that rafts are altered in AD cases with low plasmin highlights the importance of proper membrane organization in the maintenance of amyloid clearance. These findings stress that prevention of progressive membrane disorganization and/or plasmin inactivation could become important therapeutic targets for AD.
Methods
Cell culture
Primary cultures of hippocampal neurons from rat embryos were maintained in serum-free medium (N2) as in Goslin & Banker (1991).
Total and membrane hippocampal extracts We homogenized hippocampal tissue in phosphate-buffered saline containing 9% sucrose and protease inhibitors (Clap: pepstatin, antipain, chymostatin, leupeptin, each at 25 μg ml−1) using a dounce homogenizer and through ten passages in a 22-gauge syringe. The samples were centrifuged for 10 min at 400g and the postnuclear supernatants were considered as total extracts. A further centrifugation was performed at 100,000g for 1 h at 4°C to pellet the membrane fraction. Protein concentration was quantified by the BCA method (Biorad).
Quantification of western blots and statistical analysis Specific primary and peroxidase-linked secondary antibodies and the ECL method (Amersham) were used. We measured GM1 by slot-blot using cholera toxin subunit B linked to peroxidase (Sigma). Quantification was carried out by densitometry of the autoradiograms using the NIH-image software. Data were analysed using the Student's t-test. P-values less than 0.005 were considered to be statistically significant.
Raft isolation on Optiprep and sucrose gradients We incubated human hippocampal total extracts (80 μg of total protein in each case) at 4°C for 45 min in 1% Triton X-100, 10 mM MES (2-[N-morpholino]ethanesulphonic acid) pH 7.00, 2 mM EDTA, 1 mM DTT and Clap. We brought extracts to 40% Optiprep™ (Nycomed), overlaid a step gradient of 30 and 5% Optiprep™, and centrifuged for 5 h at 100,000g and 4°C. Equal volumes of the 40%, 30%, 30–5% interphase (rafts) and 5% fractions were collected. For sucrose gradients, extracts containing 100 μg of total protein were detergent extracted as above and brought to 60% sucrose. A discontinuous gradient was overlaid with 35 and 5% sucrose. After centrifugation at 100,000g for 18 h at 4°C, 11 fractions were collected.
Membrane cholesterol reduction Hippocampal neurons were treated at day 5 in vitro with 0.4 μM mevilonin and 0.5 mM MCD. At day 10, a membrane pellet (100,000g) was obtained from cellular postnuclear supernatants. Protein concentration was quantified by the BCA method (Biorad).
Plasminogen binding Neurons were incubated with 0.2 μM human plasminogen in Hank's balanced saline solution (HBSS) and 0.1% ovalbumin for 5 min at 37°C. Cells were washed twice in HBSS for 5 min and processed for western blot using an antibody against human plasminogen that also recognizes the plasmin fragment but not the rat proteins. Although the neurons are cultured in serum-free medium, thus avoiding an excessive contribution of endogenous bound plasminogen, experiments were also performed including a pretreatment with the lysine analogue N-acetyl-L-lysine (Sigma) at 40 mM for 12 h at 37°C. The neuronal medium was then removed and the cells were washed twice with HBSS prior to the addition of human plasminogen. The results obtained with or without lysine pretreatment were very similar.
Cholesterol determination We measured total cholesterol in extracts containing an equal amount of protein using Ecoline 25 (Merck), which relies on the enzymatic oxidation of cholesterol and its esters. We used pure cholesterol (Sigma) solutions as standards.
Plasmin activity measurements Plasmin enzymatic activity was assayed using the chromogenic substrate S-2251 (Chromogenix) specific for this protease in both human samples and cultured neurons. In the former, 200 μg of freshly prepared membrane extracts from frozen human hippocampi was obtained as described above and resuspended in HBSS containing 1 mM CaCl2, 1 mM MgCl2 and 0.1% ovalbumin (Sigma). They were placed in a 96-well plate in the presence of 2 μM human plasminogen and 2 mM chromogenic peptide. In the case of neuronal cultures, 25,000 cells per point were plated in 96-well plates in N2 medium and treated or not with cholesterol-reducing drugs (see above). At day 10 in vitro, neurons were washed twice with HBSS, and 2 μM human plasminogen and 2 mM chromogenic peptide were added as for the human membranes. Absorbance was measured in all samples at 37°C and 405 nm in an ultra-microplate reader.
Supplementary data is available at EMBO reports online (http://www.nature.com/embor/journal/vaop/ncurrent/extref/4-7400021-s1.pdf).
Supplementary Material
supplementary material
Acknowledgments
We are grateful to E. Cassin and B. Hellias for the preparation of hippocampal neurons. We thank M. Medina and J. Santos Da Silva for discussions. This work was supported by EU grant DIADEM (QLK-3-CT-2001-02362) to C.G.D. and M.D.L.
References
- Calabresi P. et al. ( 2000) Tissue plasminogen activator controls multiple forms of synaptic plasticity and memory. Eur. J. Neurosci., 12, 1002–1012. [DOI] [PubMed] [Google Scholar]
- Chen Z.L. & Strickland S. ( 1997) Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell, 91, 917–925. [DOI] [PubMed] [Google Scholar]
- Collen D. ( 1999) The plasminogen (fibrinolytic) system. Thromb. Haemost., 82, 259–270. [PubMed] [Google Scholar]
- Corder E.H. et al. ( 1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science, 261, 921–923. [DOI] [PubMed] [Google Scholar]
- Ehehalt R. et al. ( 2003) Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. J. Cell Biol., 160, 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J.S. et al. ( 2002) Apolipoprotein E (ApoE) isoform-dependent lipid release from astrocytes prepared from human ApoE3 and ApoE4 knock-in mice. J. Biol. Chem., 277, 29919–29926. [DOI] [PubMed] [Google Scholar]
- Goslin K. & Banker G. ( 1991) in Culturing Nerve Cells (eds Goslin, K. & Banker, G.) 251–281. MIT Press, Cambridge, Massachusetts, USA. [Google Scholar]
- Hajjar K.A. & Acharya S.S. ( 2000) Annexin II and regulation of cell surface fibrinolysis. Ann. NY Acad. Sci., 902, 265–271. [DOI] [PubMed] [Google Scholar]
- Hamanaka H. et al. ( 2000) Altered cholesterol metabolism in human apolipoprotein E4 knock-in mice. Hum. Mol. Genet., 9, 353–361. [DOI] [PubMed] [Google Scholar]
- Herz J. & Beffert U. ( 2000) Apolipoprotein E receptors: linking brain development and Alzheimers' disease. Nature Rev. Neurosci., 1, 51–58. [DOI] [PubMed] [Google Scholar]
- Ledesma M.D. et al. ( 2000) Brain plasmin enhances APP α-cleavage and Aβ degradation and is reduced in Alzheimer's disease brains. EMBO Rep., 1, 530–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledesma M.D. et al. ( 2003) Proteomic characterization of neuronal sphingolipid-cholesterol microdomains: role in plasminogen activation. Brain Res. 987, 107–116. [DOI] [PubMed] [Google Scholar]
- Miles L.A. et al. ( 1989) Gangliosides interact directly with plasminogen and urokinase and may mediate binding of these fibrinolytic components to cells. Biochemistry, 28, 9337–9343. [DOI] [PubMed] [Google Scholar]
- Miles L.A. et al. ( 1991) Role of cell-surface lysines in plasminogen binding to cells: identification of α-enolase as a candidate plasminogen receptor. Biochemistry, 30, 1682–1691. [DOI] [PubMed] [Google Scholar]
- Parkkinen J. & Rauvala H. ( 1991) Interactions of plasminogen and tissue plasminogen activator (t-PA) with amphoterin. Enhancement of t-PA-catalyzed plasminogen activation by amphoterin. J. Biol. Chem., 266, 16730–16735. [PubMed] [Google Scholar]
- Plow E.F. et al. ( 1995) The cell biology of the plasminogen system. FASEB J., 9, 939–945. [DOI] [PubMed] [Google Scholar]
- Qian Z. et al. ( 1993) Tissue-plasminogen activator is induced as an immediate-early gene during seizure, kindling and long-term potentiation. Nature, 361, 453–457. [DOI] [PubMed] [Google Scholar]
- Simons M. et al. ( 1998) Cholesterol depletion inhibits the generation of β-amyloid in hippocampal neurons. Proc. Natl Acad. Sci. USA, 95, 6460–6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K. & Toomre D. ( 2000) Lipid rafts and signal transduction. Nature Rev. Mol. Cell Biol., 1, 31–39. [DOI] [PubMed] [Google Scholar]
- Stahl A. & Mueller B.M. ( 1995) The urokinase-type plasminogen activator receptor, a GPI-linked protein, is localized in caveolae. J. Cell Biol., 129, 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker H.M. et al. ( 2000) The plasmin system is induced by and degrades amyloid-β aggregates. J. Neurosci., 20, 3937–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary material