

Short abstract
The current speed of sequencing already exceeds the capability of annotation. A new method for functional annotation is proposed using the conservation patterns of gene clusters and has been applied to the genome of Escherichia coli and established functional relationships among 176 gene clusters, comprising 738 genes.
Abstract
Background
The current speed of sequencing already exceeds the capability of annotation, creating a potential bottleneck. A large proportion of the genes in microbial genomes remains uncharacterized. Here we propose a new method for functional annotation using the conservation patterns of gene clusters. If several gene clusters show the same coevolution pattern across different genomes it is reasonable to infer they are functionally related. The gene cluster phylogenetic profile integrates chromosomal proximity information and phylogenetic profile information and allows us to infer functional dependences between the gene clusters even at great distance on the chromosome.
Results
As a proof of concept, we applied our method to the genome of Escherichia coli K12 strain. Our method establishes functional relationships among 176 gene clusters, comprising 738 E. coli genes. The accuracy of pair phylogenetic profiles was compared with the single-gene phylogenetic profile and was shown to be higher. As a result, we are able to suggest functional roles for several previously unknown genes or unknown genomic regions in E. coli. We also examined the robustness of coevolution signals across a larger set of genomes and suggest a possible upper limit of accuracy for the phylogenetic profile methods.
Conclusions
The higher-order phylogenetic profiles, such as the gene-pair phylogenetic profiles, can detect functional dependences that are missed by using conventional single-gene phylogenetic profile or the chromosomal proximity method only. We show that the gene-pair phylogenetic profile is more accurate than the single-gene phylogenetic profiles.
Background
In the past 10 years we have witnessed an almost exponential growth of genomic sequence data [1,2]. This dramatic increase creates unique opportunities for comparative analysis leading to new insights into the behavior of living microorganisms. One of the burning questions of modern genomics research is the need to assign annotations to new genes whose biological function is yet to be understood. Computational tools based on sequence homology have proved to be most broadly applicable for effective and accurate functional annotations of genes in newly sequenced genomes. Among them, BLAST and PSI-BLAST [3] are widely used to assign functions to newly sequenced open reading frames (ORFs) in genome sequence. However, one of the most surprising outcomes of genome research is that roughly 20-40% of genes in newly sequenced genomes do not have statistically significant matches to functionally annotated sequences and are annotated as 'hypothetical proteins' [4].
Accordingly, several non-homology-based computational methods have been introduced recently in an attempt to provide putative functional assignments for those 'hypothetical proteins'. For example, among the most reliable methods, the Rosetta stone technique [5,6] detects functional associations based on protein-domain fusion events. Other methods include the chromosomal proximity method and the phylogenetic profile method.
The chromosomal proximity method of Overbeek et al. [7] is a popular technique that utilizes chromosomal proximity information to discover putative functional linkages between genes close to each other on the chromosome. When two genes appear as a neighboring gene pair in the genomes of several distantly related organisms (that is, they form a conserved gene cluster) it suggests the possibility that the genes might be functionally related [7]. In fact, the analysis of current data suggests that a cluster of two or more genes that appears in four or more distantly related microorganisms has a more than 90% probability of being involved in the same broad functional category (Y.Z., unpublished data).
Another seminal approach for establishing functional links between genes based on their coevolution patterns in different organisms was proposed and popularized by Pellegrini et al. [8]. Similar proposals have been made by Gaasterland et al. [9] and other groups. This method constructs a genetic phylogenetic profile for each gene. A phylogenetic profile of a gene indicates the presence or the absence of this gene in each organism by an entry of 1 or 0 in a long vector. In other words, each gene is assigned a binary vector of length N, where N is the number of organisms used to construct the phylogenetic profiles. The ith bit of the vector is set to 1 if a homologous gene exists in the ith genome; otherwise it is set to 0. Several variants of phylogenetic profiles have been described in the literature [10,11]. The functional linkage is established when two genes have similar phylogenetic profiles, that is, they show a correlated pattern of inheritance across the genomes examined.
Here we propose a new simple method for inferring functional linkages based on the phylogenetic profiles of gene clusters. This method simultaneously takes advantage of chromosomal proximity information and phylogenetic coevolution information. We demonstrate an enhanced ability to annotate a number of previously uncharacterized genes that are not yet functionally annotated and appear to resist the application of other computational techniques.
Our new method constructs gene cluster phylogenetic profiles by recording the conservation pattern of a gene cluster that contains two or more neighboring genes in a set of reference genomes. In this paper, we will focus on gene clusters of size two, that is, gene pairs. For a given gene pair AB (A and B are separate genes and are encoded continuously on the chromosome) in the target genome, the presence of AB in a reference genome is recorded when we detect the presence of either an A'B' or a B'A' gene cluster, where gene A' is a homolog of gene A and gene B' is a homolog of gene B. There are many established methods for detecting homology or orthology, for example, membership in the same COG (Clusters of Orthologous Genes) [12]. In this paper, homologs are detected by BLASTP with an E-value lower bound of 1e-10 to filter out statistically insignificant matches.
The implementation of the chromosomal proximity method does not strictly require successive ORFs in the genome. An important discovery in comparative genomics is that local gene rearrangements happen quite often during evolution, disrupting gene order in gene clusters [13]. To account for possible gene insertion and rearrangement events during evolution, a natural extension is to consider gene clusters with ORF gaps. That is, we extend the detection of A'B' clusters to include A'xB' and A'xyB' clusters in the reference genomes, where x and y are inserted genes (a maximum of two) and A' and B' are homologs of genes A and B. Similarly, to be symmetric, we allow the gapped gene pairs in the target genome, that is, AxB or AxyB pairs where x and y are genes between A and B in the target genome. The implementation of the gapped version of the gene cluster phylogenetic profile method increases the number of putative functional linkages between genes and thus improves the sensitivity of the method. Here we report results from the gapped version of the method. From now on, we will refer to a single gene phylogenetic profile as SGPP and to a gene pair phylogenetic profile as GPPP.
Results and discussion
Examples of functional dependences revealed by GPPP
We carried out an exhaustive grouping of the Escherichia coli gene pairs based on sharing the same GPPP (Hamming distance equal to zero). In E. coli, our non-gapped GPPP method detects 57 gene-pair clusters. These gene-pair clusters include 351 genes. Low-quality profiles, which refer to profiles with a norm of less than 4, are excluded. The norm of the profile is calculated by summing the 1s and 0s in the profile vector (see Figure 1 legend for the definition of profile norm). By using the gapped GPPP method, we were able to detect 176 functionally related gene clusters containing 738 genes. A two-dimensional representation of these clusters and their relationships is shown in Figure 1. As a result, by using the GPPP method, we could establish functional linkages among about 17% of the E. coli genome.
Figure 1.
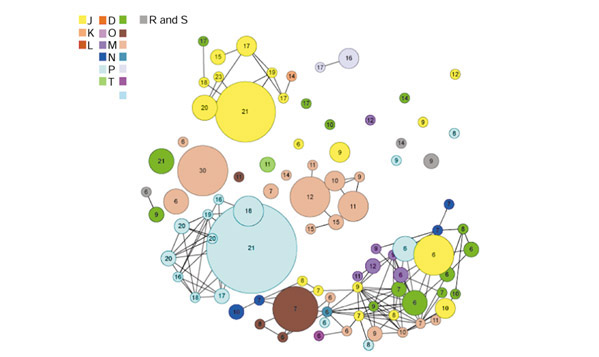
Visualization of gene clusters sharing the same profiles (GPPP) in a two-dimensional space. Each circular node represents a gene cluster grouped by the same pair phylogenetic profile. The radius of the circle is proportional to the size of the cluster. The number shown at the center of each circle is the norm of the profile vector for this cluster. For a profile vector (x1,x2,x3,...xN), the norm of this profile is calculated by 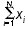 . All clusters are color-coded by COG's broad-function classification [25]. Links between clusters are present when the Hamming distance between two profiles is less than 5. We can see that for profiles with norms exceeding 10, similar profiles (Hamming distance less than 5), shown as clusters of connected nodes, tend to fall into the same functional category. When the profile norm is less than 10, broad functional categories for similar profiles begin to diverge. Several broad functions, for example, P (inorganic ion transport and metabolism), J (translation, ribosomal structure and biogenesis), and E (amino-acid transport and metabolism) can be well recognized by the phylogenetic method, while some are either absent or tend to mix with other broad functions. This shows the relative effectiveness of analyzing inheritance patterns of gene clusters for different broad functional categories. This figure is generated using the software package Pajek.s
. All clusters are color-coded by COG's broad-function classification [25]. Links between clusters are present when the Hamming distance between two profiles is less than 5. We can see that for profiles with norms exceeding 10, similar profiles (Hamming distance less than 5), shown as clusters of connected nodes, tend to fall into the same functional category. When the profile norm is less than 10, broad functional categories for similar profiles begin to diverge. Several broad functions, for example, P (inorganic ion transport and metabolism), J (translation, ribosomal structure and biogenesis), and E (amino-acid transport and metabolism) can be well recognized by the phylogenetic method, while some are either absent or tend to mix with other broad functions. This shows the relative effectiveness of analyzing inheritance patterns of gene clusters for different broad functional categories. This figure is generated using the software package Pajek.s
In many cases the GPPP method is able to establish functional linkages that are missed by the application of the SGPP method or the chromosomal proximity method independently. There are numerous examples where gene pairs share a common GPPP and have a functional linkage, although the individual genes may not have similar SGPPs. Our method provides a new way to establish functional linkages between distant coevolved gene clusters on the chromosome, enhancing the ability to assign gene functions consistently in a broader genomic context. One such example consists of the E. coli gene pairs: b1129(gi|1787374)/b1130(gi|1787375) and b4398(gi|1790860)/b4399(gi|1790861). These two gene pairs share the same GPPP (Figure 2), whereas individual genes do not have the same SGPP (Figure 2). Hamming distances between the individual genes are shown in Table 1.
Figure 2.

Comparison of gene cluster profile and single-gene profile. Filled cells (black) represent the presence of a gene or a gene pair in the genome and empty cells represent its absence.
Table 1.
Hamming distances between the single gene profiles in two gene clusters that share the same pair profile
| b1129 | b1130 | b4398 | b4399 | |
| b1129 | 0 | 7 | 4 | 3 |
| b1130 | 7 | 0 | 3 | 10 |
| b4398 | 4 | 3 | 0 | 7 |
The gene pair b1129/b1130 in E. coli encodes a two-component regulatory system PhoP-PhoQ [14,15]. This two-component system is also present in several other Gram-negative bacteria and is associated with virulence, adaptation to Mg2+-limiting environments and other cellular activities [15]. The gene b4399 has been annotated as a 'catabolite repression sensor kinase for PhoB, an alternative sensor for PhoB', although it is far from phoB (b0399) on the chromosome. In fact, PhoB forms another two-component system with the product of its neighboring gene, PhoR (b0400), which is responsible for phosphate regulation [15]. The gene b4398 has been assigned a general function as a catabolic regulation response regulator. As these two gene pairs b4398/b4399 and b1129/b1130 share the same coevolution pattern, revealed by the gene-pair phylogenetic profiles, we suggest that the gene pair b4398/b4399 probably encodes another two-component system in E. coli. This two-component system may be functionally closer to the PhoP-PhoQ system than to the PhoB-PhoR system. Interestingly, as no individual gene has a similar SGPP (see Table 1), relying on SGPP would miss this highly coupled functional linkage.
Functional dependence between genes as a selective pressure sometimes favors gene clusters over random gene arrangement along the chromosome [16]. The chromosomal proximity method aims to detect local functional dependences ('intracluster' dependences) established by conserved proximity among distantly related genomes. However, functional dependencis between distant gene clusters ('intercluster' functional dependences) on the chromosome usually cannot be resolved by the chromosomal proximity method. Instead, GPPP can reveal even distant functional dependences between gene clusters that participate in closely coupled processes or pathways. An example is the E. coli murG cluster (b0089, b0090, b0092) and the lpxD cluster (b0177, b0179) which share the same GPPP; their current annotations are shown in Table 2. They are both present in Chlamydophila pneumoniae, Chlamydia trachomatis, Xylella fastidiosa, Haemophilus influenzae, Neisseria meningitidis and Pseudomonas aeruginosa and are absent in all the other genomes included. MurG, the last enzyme in the intracellular phase of peptidoglycan synthesis, is essential for the production of the layers of peptidoglycan that protect cells from rupturing under high internal osmotic pressure [17]. LpxD is a key enzyme in lipid A biosynthesis [18]. Lipid A is a glucosamine-based phospholipid that makes up the monolayer of the outer membrane outside the peptidoglycan layers. None of the genes in these clusters is homologous to any of the others by BLASTP. The conventional non-homology-based chromosomal proximity method can imply functional dependences inside each cluster separately. For example, as b0089, b0090, b0092 are conserved among the six organisms above, we can infer functional linkage and interpret the collective function as cell-membrane maintenance from their current annotations (Table 2). However, with additional information from GPPP, we can establish a distant intercluster functional dependence in addition to the intracluster dependences found by the chromosomal proximity method. As both clusters are associated with the outer membrane of the cell and are essential for survival of the bacteria [19,20], this dependence may suggest an inherent functional linkage between them. Noticing there is a hypothetical protein (b0177) in the lpxD gene cluster, we then carried out sequence analysis on this gene and its encoded protein, expecting that it might be associated with the outer membrane of the cell. A simple BLASTP search tells us it has significant homology (E-value 0.0) to the outer-membrane antigen present in many other bacteria. Given that the lipid A layer provides anchoring sites for bacterial surface antigens such as lipopolysaccharide (LPS), this discovery again supports the reliability of our prediction.
Table 2.
Description of the murG and lpxD gene clusters
| Gene name | Synonym (Genbank id) | Current annotation |
| murG cluster | ||
| ftsW | b0089 (1786277) | Cell division; membrane protein involved in shape determination |
| murG | b0090 (1786278) | UDP-N-acetylglucosamine:N-acetylmuramyl-(pentapeptide) pyrophosphoryl-undecaprenol N-acetylglucosamine transferase |
| lpxD cluster | ||
| ddlB | b0092 (1786280) | D-alanine-D-alanine ligase B, affects cell division |
| yaeT | b0177 (1786374) | ORF, hypothetical protein |
| lpxD | b0179 (1786376) | UDP-3-O-(3-hydroxymyristoyl)-glucosamine N-acyltransferase; third step of endotoxin (lipidA) synthesis |
Previously uncharacterized genes in E. coli
By applying the GPPP method, a number of previously uncharacterized genes in E. coli with hypothetical or unknown annotation can now be functionally linked to characterized gene pairs. Some of these genes have not been assigned a function because of the lack of sufficient data at the time of annotation. We then carried out additional sequence analyses of these genes using BLAST, Pfam [21] and COG to confirm our prediction. A number of previously unknown genes that can be annotated by our method and can be confirmed by additional analysis are listed in Table 3. It can be seen in Table 3 that many of the predictions made by GPPP agree with more detailed sequence analysis.
Table 3.
List of previously unknown genes that can be annotated using GPPP
| E. coli gene name (Genbank id) | Previous annotation | Predicted function by GPPP | Additional evidences from BLAST/Pfam/COG |
| b1681 (1787971) | Hypothetical protein | Transport system, membrane protein | COG0719: predicted membrane components of ABC-transporter SufB |
| b1683 (1787973) | Hypothetical protein | Transport system, membrane protein | |
| b2608 (1788960) | Hypothetical protein | RNA processing | Pfam domain: RimM. RimM is essential for processing of 16S rRNA. |
| b2766 (1789125) | Hypothetical protein | Flavorprotein, electron transport | COG0644:dehydrogenase/flavoprotein |
| b0407 (1786608) | Hypothetical protein | Protein secretion | Pfam domain: DUF219, (uncharacterized secreted protein) |
| b1395 (1787661) | Putative enzyme | Part of fad operon | BLAST: 3-hydroxyacyl-CoA dehydrogenase |
| b2341 (1788682) | Putative enzyme | Part of fad operon | BLAST: 3-hydroxyacyl-CoA dehydrogenase; enoyl-CoA isomerase/hydrotase |
| b0284 (1786478) | Hypothetical protein | Neighboring with xanthine dehydrogenase gene (b0286) | BLAST: Putative oxidoreductase |
| b2866 (1789230) | Hypothetical protein | Neighboring with xanthine dehydrogenase (b2868) | BLAST: Probable aldehyde oxidase and xanthine dehydrogenase family protein |
| b1674 (1787963) | Hypothetical protein | Oxidoreductase | BLAST: Aldehyde ferredoxin oxidoreductase |
| b2371 (1788714) | Hypothetical protein | Enzyme in carnitine metabolism | Pfam domain: CAIB-BAIF, domain involved in carnite metabolism |
| b2374 (1788717) | Hypothetical protein | Enzyme in carnitine metabolism | Pfam domain: CAIB-BAIF, domain involved in carnite metabolism |
| b2125 (2367130) | Hypothetical protein | Response regulator in 2-component system | Pfam domains: response-reg, REC probable cheY receiver domain |
| b0936 (1787167) | Hypothetical protein | Transport system | Pfam domain: PBPb, transport system related |
| b2075 (1788390) | Hypothetical protein | Integral transmembrane protein | Pfam: ACR_tran, SecD_SecF, Patched, transmembrane domain related |
| b2076 (1788391) | Hypothetical protein | Integral transmembrane protein | Pfam: ACR_tran |
| b1086 (1787327) | Hypothetical protein | RNA processing | BLAST: RNA pseudouridylate synthase |
Accuracy of the GPPP method
We have compared the accuracy of GPPP with that of the SGPP method using COG's broad-function classification system [12]. Genes in each cluster grouped by the same profile are labeled using COG's 18 broad functional categories excluding category R (general function) and category S (function unknown). A good method for establishing functional linkages will tend to cluster genes within the same broad functional category. To this end, we devised two separate procedures to compare the effectiveness of GPPP and SGPP, with the results summarized below. Because it is hard to calculate the number of false negatives, which are functionally dependent genes or gene clusters that do not show a common coevolution pattern, we did not compare the sensitivities of these methods.
The first accuracy measure is based on the proportion of 'pure' clusters among all the clusters. We defined satisfying pure clusters heuristically, considering the intrinsic vagueness of the concept 'broad category' and the fact that it is difficult to classify proteins' functional roles precisely using a one-dimensional classification schema [22]. If more than 80% of the members in a cluster stay within a certain COG broad functional category, which means that they might be involved in the same biological process, we consider this cluster as a pure cluster. The proportion of such pure clusters among the total clusters serves as a coarse measure of the specificity of the phylogenetic profile method.
We plotted this measure versus the norm of the profile for both GPPP and SGPP (Figure 3a). To account for the possible systematic bias of this measure toward sizes of the clusters, we also plotted the average cluster size versus the norm of the profile (Figure 3b). Figure 3b shows that except at the very ends of the norm axis (norm = 1, 2, 30) the average cluster sizes from both GPPP and SGPP are close to each other.
Figure 3.
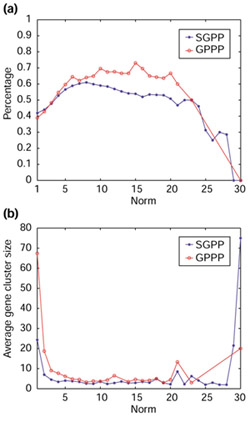
Comparison of the accuracy of GPPP and SGPP. (a) Comparison of the accuracy of GPPP and SGPP using the proportion of 'pure' clusters. The x-axis represents the norm of the profiles and the y-axis the percentage of 'pure' clusters among all clusters. (b) Average cluster size of GPPP and SGPP. In this and the following figures, the data points from GPPP are marked by circles and those from SGPP are marked by stars.
In the other experiment to compare accuracy, we simply examined all pairs of proteins that end up in the same cluster and calculated the frequency with which two such proteins are from the same functional category. This measure is essentially the same as the Jaccard coefficient (C) referred to in [10]. Given a gene cluster, let N be the number of all pairs of genes chosen from this cluster and S be the number of all pairs of genes that are chosen from this cluster and are from the same COG category. Then the Jaccard coefficient is calculated by C = S/N. C varies from zero to one and is less dependent on cluster size, unlike the previous measure. C is plotted versus the norm of the profile in Figure 4. In both experiments (Figures 3a,4), we see that GPPP achieves a higher accuracy (an increase of 10% on average) than the conventional SGPP method, especially in the norm range 5 to 20.
Figure 4.
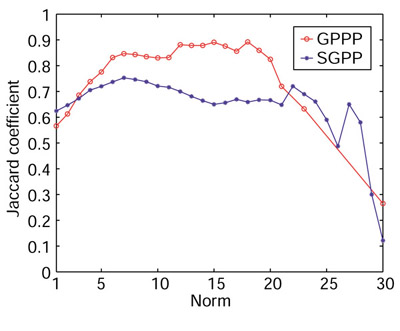
Comparison of the accuracy of GPPP and SGPP using C (Jaccard coefficient).
From the information theory perspective, we know that the predictive quality of a profile is reflected by its mutual information:
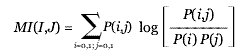
where P(i) is the probability of seeing i (i = 0,1) in the profile vector and P(i,j) is the probability of seeing (i,j) jointly in two aligned profiles I and J. In theory, the predictive value is maximized when half the entries in a profile are 1s and the others are 0s (high mutual information (MI) regions). The bell-like accuracy curve for the profile methods (Figures 3a,4) can be explained by considering the information content of profiles, which is low when the norm of a profile is close to 0 (a vector with all entries 0) or N (a vector with all entries 1) (low-MI regions). Intuitively, the fact that certain gene clusters appear in every organism or appear in only one organism does not necessarily indicate functional relationship. We can see that when the profiles are near these low-MI regions, the size of the clusters tends to increase dramatically, suggesting the corruption of coevolution signals (Figures 3b, 5b). It is important to realize which regions our functional linkages are established from when we use these phylogenetic profile methods.
Figure 5.
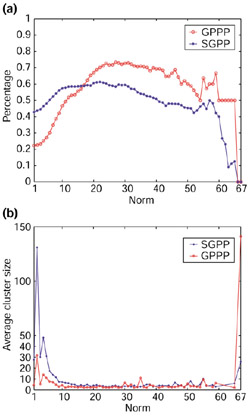
Comparison of accuracy and average cluster size using a larger set of organisms. (a) Comparison of accuracy of GPPP and SGPP using 68 organisms. (b) Average cluster size of GPPP and SGPP using 68 organisms.
As more and more fully sequenced genomes of microorganisms have become available, it is natural to ask whether the accumulation of new genomes will help us improve the accuracy of the phylogenetic profile methods. With more than 77 sequenced genomes now available, we were able to expand the phylogenetic profile analysis to a larger set of organisms. Using total 68 microorganisms, in Figure 5a we plot the accuracy versus the norm and in Figure 5b we plot the average size distribution for both GPPP and SGPP. We can see that the accuracy of the GPPP method is improved about 5% on average when more genomes are included and the cluster sizes tend to become smaller. However, we did not see dramatic improvements when using the larger set of genomes, which made us think there may be an upper limit to the accuracy of the phylogenetic profile method. When more genomes are included, both the coevolution signal and the noise signal are 'amplified', so we would not expect the accuracy of the phylogenetic method to improve dramatically when a larger, randomly selected genome set is sampled unless a clever sampling strategy is used. All accuracy curves (Figures 3a,4,5a) show that the GPPP method outperforms the SGPP method. The improved accuracy makes GPPP a possible complementary annotation tool to aid conventional homology-based sequence comparison.
To measure the robustness of the GPPP method for a larger sample of genomes, we also examined whether the functional linkages previously established by 30-dimensional profiles can still be recovered by 67-dimensional profiles. We find that the previously reported functional linkages can be completely reconfirmed (data not shown), which suggests that the true coevolution patterns of certain gene clusters are robust when a wider range of genomes are sampled and appear to resist the noise due to evolutionary diversity that could be introduced when more genomes are used. Additionally, the GPPP method using 68 genomes generates additional putative functional associations (see [23] for a complete list). As some microorganisms are closely related to each other (for example, different strains of the same organism), it is important to realize that the number of 'informative' genomes is less than the number of genomes included. The discriminative power of phylogenetic profiles will be improved when a proper strategy for sampling organisms in different taxa is developed.
In summary, gene cluster phylogenetic profiles combine and improve on the chromosomal proximity method and the single-gene phylogenetic profile method. A gene cluster phylogenetic profile with a large norm simply states the fact that this gene cluster is highly conserved across different organisms, which is equivalent to the chromosomal proximity method. By clustering gene clusters with the same phylogenetic profiles, we are able to detect functional linkages between distant genomic regions on the chromosome based on their pattern of coevolution. A phylogenetic profile of a single gene could be corrupted by many genomic events during evolution, such as gene duplication or the possible loss of gene functions after speciation [24], which introduces noise into the coevolution patterns. As the requirement for the presence of a gene cluster is stricter than for the presence of a single gene, the pair profiles help to obtain an improvement in the accuracy of functional linkage detection.
Genes in microorganisms are known to form operons, two-component systems, paralogous gene clusters, and other functionally related genomic clusters. As described in here, the implementation of GPPPs gives us a tool for establishing functional linkages between these genomic elements even when they are not physically close on the chromosome. In some cases, these functional associations can help us understand the dependencies between gene clusters in biological processes, such as the murG and lpxD clusters described in this paper.
In addition to GPPPs, we could naturally develop software for detecting higher-order profiles of bigger gene clusters; however, we would expect to see a smaller coverage with a possibly higher accuracy. In fact, we observed that some gene pairs with the same phylogenetic profile reside in a close proximity on the chromosome, which suggests a longer conserved gene cluster (for example, ribosomal gene clusters). Ultimately, all these techniques are based on the identification of gene clusters that show similar inheritance patterns across genomes.
Homology-based annotation tools aim to detect sequence similarity between new genes and known genes by following a one-by-one gene annotation methodology. The GPPP, however, detects functional relationship between clusters of genes on the basis of their coevolution patterns across genomes, and is able to assign gene functions in groups by considering a wider genomic context. With the accumulation of fully sequenced genomes, the information content in gene cluster phylogenetic profiles is expected to increase, as does the accuracy of the proposed methodology. The GPPPs, and possibly higher-order gene cluster phylogenetic profiles, together with other non-homology methods, are likely to substantially increase our ability to assign function to a large number of putative genes.
Materials and methods
We initially chose 31 fully sequenced microbial genomes, including 8 archaeal genomes and 23 bacterial genomes. While this work was in progress the number of fully sequenced microbial genomes grew to more than 70. We then expanded our study to a total of 68 organisms to estimate the robustness of the phylogenetic profile method and present the results on accuracy evaluation. All protein sequences were retrieved from the National Center for Biotechnology Information (NCBI) genome repository. We chose E. coli K12 as the target genome for functional linkage detection and the other genomes as reference genomes for constructing the gene cluster phylogenetic profiles. We performed pairwise one-against-all BLAST searches to identify all homologous E. coli genes in other organisms.
By determining the presence or absence of all possible neighboring E. coli gene-pair clusters in 30 other genomes, we were able to get a set of 30 × 1 binary profile vectors that are similar in spirit to the ones obtained by the SGPP method. The profile of a gene cluster is simply a binary vector that has a 1 in coordinate K if the gene cluster occurs in the Kth genome; otherwise it has 0 in that coordinate. To measure the similarity between two phylogenetic profiles, we use the Hamming distance, simply expressed by the number of vector entries that need to be changed to obtain one profile from the other profile. Other natural techniques can include mutual information (MI) or correlation coefficients (CC) that measure the statistical dependence of two discrete distributions of coevolution patterns.
The list of possible functionally linked gene clusters reported by both the gapped and non-gapped versions of GPPP can be accessed at [23].
Acknowledgments
Acknowledgements
Y.Z. would like to thank Megon Walker, Mike Schaffer and Jie Wu for proofreading the manuscript and inspiring discussions. This work has been partly supported by grant NSF-KDI0196227. Scripts related to this study are available upon request.
References
- The Institute for Genomic Research: comprehensive microbial resource http://www.tigr.org/tigr-scripts/CMR2/CMRGenomes.spl
- National Center for Biotechnology Information: Entrez Genome http://www.ncbi.nlm.nih.gov/PMGifs/Genomes/ micr.html
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenstein E, Gilliland G, Herzberg O, Moult J, Orban J, Poljak RJ, Banerjei L, Richardson D, Howard A. Biological function made crystal clear - annotation of hypothetical proteins via structural genomics. Curr Opin Biotechnol. 2000;11:25–30. doi: 10.1016/s0958-1669(99)00063-4. [DOI] [PubMed] [Google Scholar]
- Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D. Detecting protein function and protein-protein interaction from genome sequences. Science. 1999;285:751–753. doi: 10.1126/science.285.5428.751. [DOI] [PubMed] [Google Scholar]
- Enright AJ, Iliopoulos I, Kyrpides NC, Ouzounis CA. Protein interaction maps for complete genomes based on gene fusion events. Nature. 1999;402:86–90. doi: 10.1038/47056. [DOI] [PubMed] [Google Scholar]
- Overbeek R, Fonstein M, D'Souza M, Pusch GD, Maltsev N. The use of gene clusters to infer functional coupling. Proc Natl Acad Sci USA. 1999;96:2896–2901. doi: 10.1073/pnas.96.6.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc Natl Acad Sci USA. 1999;96:4285–4288. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaasterland T, Ragan MA. Microbial genescapes: phyletic and functional patterns of ORF distribution among prokaryotes. Microb Comp Genomics. 1998;3:199–217. doi: 10.1089/omi.1.1998.3.199. [DOI] [PubMed] [Google Scholar]
- Marcotte EM, Xenarios I, van der Bliek AM, Eisenberg D. Localizing proteins in the cell from their phylogenetic profiles. Proc Natl Acad Sci USA. 2000;97:12115–12120. doi: 10.1073/pnas.220399497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlidis P, Grundy WN. Combining microarray expression data and phylogenetic profiles to learn gene functional categories using support vector machines. Columbia University Computer Science Department Technical Report CUCS-011-00. April 2000.
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- Dandekar T, Snel B, Huynen M, Bork P. Conservation of gene order: a fingerprint of proteins that physically interact. Trends Biochem Sci. 1998;23:324–328. doi: 10.1016/s0968-0004(98)01274-2. [DOI] [PubMed] [Google Scholar]
- Kato A, Tanabe H, Utsumi R. Molecular characterization of the PhoP-PhoQ two-component system in Escherichia coli K-12: identification of extracellular Mg2+-responsive promoters. J Bacteriol. 1999;181:5516–5520. doi: 10.1128/jb.181.17.5516-5520.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groisman EA. The pleiotropic two-component regulatory system PhoP-PhoQ. J Bacteriol. 2001;183:1835–1842. doi: 10.1128/JB.183.6.1835-1842.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Szustakowski JD, Fortnow L, Roberts RJ, Kasif S. Computational identification of operons in microbial genomes. Genome Res. 2002;12:1221–1230. doi: 10.1101/gr.200602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Men H, Ha S, Ye XY, Brunner L, Hu Y, Walker S. Intrinsic lipid preferences and kinetic mechanism of Escherichia coli MurG. Biochemistry. 2002;41:6824–6833. doi: 10.1021/bi0256678. [DOI] [PubMed] [Google Scholar]
- Vaara M, Nurminen M. Outer membrane permeability barrier in Escherichia coli mutants that are defective in the late acyltransferases of Lipid A biosynthesis. Antimicrob Agents Chemother. 1999;43:1459–1462. doi: 10.1128/aac.43.6.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengin-Lecreulx D, Texier L, Rousseau M, van Heijenoort J. The murG gene of Escherichia coli codes for the UDP-N-acetylglucosamine: N-acetylmuramyl-(pentapeptide) pyrophosphoryl-undecaprenol N-acetylglucosamine transferase involved in the membrane steps of peptidoglycan synthesis. J Bacteriol. 1991;173:4625–4636. doi: 10.1128/jb.173.15.4625-4636.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuorio R, Vaara M. Comparison of the phenotypes of the lpxA and lpxD mutants of Escherichia coli. FEMS Microbiol Lett. 1995;134:227–232. doi: 10.1111/j.1574-6968.1995.tb07942.x. [DOI] [PubMed] [Google Scholar]
- Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, Griffiths-Jones S, Howe KL, Marshall M, Sonnhammer EL. The Pfam protein families database. Nucleic Acids Res. 2002;30:276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley M, Serres MH. Interim report on genomics of Escherichia coli. Annu Rev Microbiol. 2000;54:341–411. doi: 10.1146/annurev.micro.54.1.341. [DOI] [PubMed] [Google Scholar]
- Pair phylogenetic profile http://genomics4.bu.edu/profiles
- McGuire AM, Church GM. Predicting regulons and their cis-regulatory motifs by comparative genomics. Nucleic Acids Res. 2000;28:4523–4530. doi: 10.1093/nar/28.22.4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COGs functional annotation http://www.ncbi.nlm.nih.gov/cgi-bin/COG/palox?fun=all