

Abstract
MAP kinases (MAPKs) form a complex with MAPK kinases (MAPKKs), MAPK-specific phosphatases (MKPs) and various targets including MAPKAPKs. These docking interactions contribute to regulation of the specificity and efficiency of the enzymatic reactions. We have previously identified a docking site on MAPKs, termed the CD (common docking) domain, which is utilized commonly for docking interactions with MAPKKs, MKPs and MAPKAPKs. However, the CD domain alone does not determine the docking specificity. Here we have identified a novel site on p38 and ERK2 MAPKs that regulates the docking specificity towards MAPKAPKs. Remarkably, exchange of two amino acids in this site of ERK2 for corresponding residues of p38 converted the docking specificity for MAPKAPK-3/3pk, which is a dominant target of p38, from the ERK2 type to the p38 type, and vice versa. Furthermore, our detailed analyses with a number of MAPKAPKs and MKPs suggest that a groove in the steric structure of MAPKs, which comprises the CD domain and the site identified here, serves as a common docking region for various MAPK-interacting molecules.
Keywords: docking interaction/MAP kinase/phosphorylation/specificity
Introduction
Regulation of cellular functions and responses utilizes a number of signal transduction pathways. Each pathway should transduce signals with high efficiency and fidelity in order to avoid unnecessary cross-talk. A recent advance in the understanding of determinants for this efficiency and fidelity is the determination of the domains for protein– protein interactions, such as SH2, SH3, PDZ, etc. (Pawson, 1995; Hunter, 2000; Pawson and Nash, 2000). Each protein recognizes and interacts with specific partners through these domains with various efficiencies. These domains often form modular structures, and the amino acid composition and sequence of the domains determine the specificity of the interactions.
Members of the MAP kinase (MAPK) family are known to be activated by a wide variety of extracellular signals. There are three major subgroups of the MAPK family: ERK, p38 and JNK/SAPK. ERK is activated mainly by growth factors and phorbol esters, and is associated with cellular proliferation and differentiation. JNK/SAPK and p38 are activated by extracellular stresses, such as UV irradiation and osmotic stress, and by inflammatory cytokines. Activation of these protein kinases leads to variable responses, such as gene expression, cell proliferation, differentiation, cell cycle arrest, apoptosis, early development, etc., depending on the cell type (Sturgill and Wu, 1991; Ahn et al., 1992; Nishida and Gotoh, 1993; Marshall, 1995; Kyriakis and Avuruch, 1996; Treisman, 1996; Robinson and Cobb, 1997; Ip and Davis, 1998; Schaeffer and Weber, 1999). The MAPK pathways are characterized by a cascade of multiple kinases: MAPK kinase kinase (MAPKKK), MAPK kinase (MAPKK), MAPK and MAPK-activated protein kinase (MAPKAPK). Each MAPK has specific activators (MAPKKs) and substrates (MAPKAPKs). For example, MEK1 is a specific activator for ERK, and RSKs are specific MAPKAPKs for ERK. The signal is transduced in the form of the phosphorylation event from an upstream kinase to a downstream one. The phosphorylation event, the kinase reaction, must be efficient and specific in order to avoid unnecessary cross-talk among the MAPK cascades or with other signaling pathways. The molecular basis for regulating the efficiency and fidelity of these reactions, however, is poorly understood. It has been reported recently that MAPKs form a stable complex with cognate MAPKKs, MAPKAPKs and specific phosphatases, and that there is a good correlation between the binding specificity and the enzymatic specificity; for example, ERK forms a complex with RSKs, but p38 and JNK/SAPK do not (Bardwell and Thorner, 1996; Fukuda et al., 1997; Muda et al., 1998; Pulido et al., 1998; Xia and Karin, 1998; Gavin and Nebreda, 1999; Holland and Cooper, 1999; Jacobs et al., 1999; Smith et al., 1999; Zuniga et al., 1999; Tanoue et al., 2000). This is surprising because MAPKs form a globular structure without a distinct motif beyond the kinase domain (Zhang et al., 1994; Wilson et al., 1996; Wang et al., 1997). We have shown that a C-terminal portion of MAPKs is utilized commonly for binding to MAPKKs, MAPKAPKs and phosphatases (Tanoue et al., 2000). We named this site the CD (common docking) domain. The CD domain is characterized by negatively charged amino acids [Asp316 and Asp319 for ERK2 (rat), and Asp313, Asp315 and Asp316 for p38α (human)], and is located on the opposite side to the active center in the steric structure of the molecules (see Figures 1B and 4A). The corresponding MAPK-binding sites of MAPKKs, MAPKAPKs and phosphatases are shown to exist outside their catalytic domain in the primary sequence. Therefore, such binding is not achieved by a transient enzyme– substrate interaction through the active center. Thus, this interaction is called the docking interaction, which was first described by Kallunki et al. (1996). Conceptually, the recognition between MAPKs and the interacting molecules involves both the docking interaction and the transient enzyme–substrate interaction through the active center, and the docking interaction may help to regulate the efficiency and specificity of the enzymatic reactions. Previous studies have demonstrated that the efficiency of these enzymatic reactions is regulated by the docking interactions (Muda et al., 1998; Xia and Karin, 1998; Gavin et al., 1999; Smith et al., 1999; Tanoue et al., 2000).
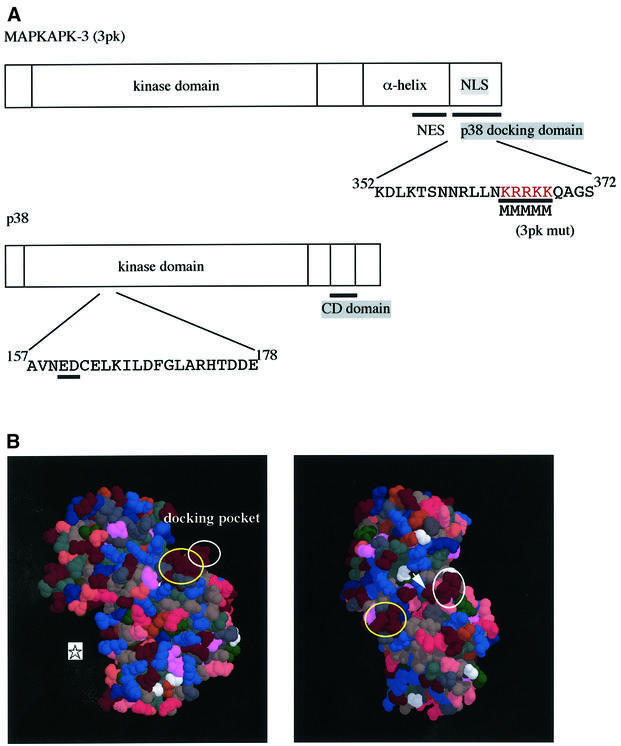
Fig. 1. Structures of p38 and MAPKAPK-3/3pk. (A) Schematic representation of the primary structures of 3pk and p38. (B) Three-dimensional structure of p38α. Yellow circles indicate acidic amino acids of the CD domain (Asp313, Asp315 and Asp316 for human p38α), and white circles the ED site (Glu160 and Asp161 for human p38α). An asterisk indicates the position of the active center. An arrowhead indicates Glu163. See text for details. This figure was made using RasMol, based on the crystallographic data (Wilson et al., 1996; Wang et al., 1997).
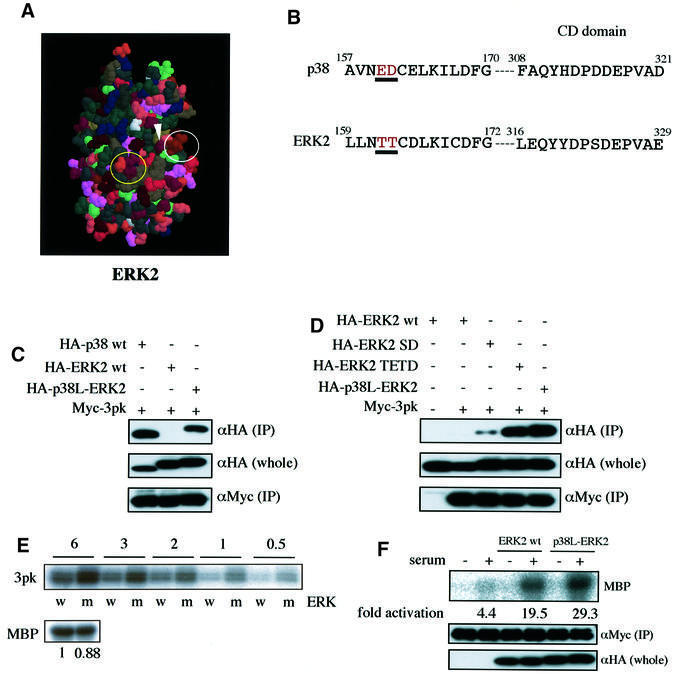
Fig. 4. Glu160 and Asp161 in p38 and Thr157 and Thr158 in ERK2 are the determinants for the docking specificity towards 3pk. (A) Three-dimensional structure of ERK2. A yellow circle indicates the CD domain (Asp316 and Asp319 for rat ERK2, and Asp321 and Asp324 for Xenopus ERK2). A white circle indicates the site corresponding to the ED site of p38 (Thr157 and Thr158 for rat ERK2, and Thr162 and Thr163 for Xenopus ERK2). This figure was made using RasMol, based on the crystallographic data (Zhang et al., 1994). (B) The primary sequences of the ED site (or the TT site) and the CD domain of p38 and ERK2. The numbers shown are the sequences of Xenopus ERK2 and human p38. (C) The binding of p38-like ERK2 (p38L-ERK2) to 3pk. Lysates of NIH 3T3 cells co-transfected with the indicated combinations of constructs were immunoprecipitated with anti-Myc antibody. Co-immunoprecipitated wild-type (wt) p38, wild-type (wt) ERK2 or p38-like ERK2 (p38L-ERK2) were detected [upper panel, αHA (IP)]. The expression levels of these HA-tagged constructs were similar [middle panel, αHA (whole)]. Comparable amounts of 3pk were immunoprecipitated in each lane [lower panel, αMyc (IP)]. Similar results were obtained in three different experiments. (D) The binding of the mutant forms of ERK2 to 3pk. Lysates of NIH 3T3 cells co-transfected with the indicated combinations of constructs were immunoprecipitated with anti-Myc antibody. Co-immunoprecipitated wild-type or the mutant forms of ERK2 were detected [upper panel, αHA (IP)]. The expression levels of these HA-tagged constructs were similar [middle panel, αHA (whole)]. Comparable amounts of 3pk were immunoprecipitated in each lane [lower panel, αMyc (IP)]. Similar results were obtained in three different experiments. (E) The ability of wild-type ERK2 (w) or p38-like ERK2 (m) to phosphorylate 3pk in vitro (upper panel). GST–3pk was produced in bacteria. The GST portion was cut off by precision protease treatment, and the remaining 3pk was used as substrate. The amount of 3pk used was 6, 3, 2, 1 or 0.5 µg in 20 µl of reaction buffer, as indicated. The HA-tagged wild-type ERK2 or the HA-tagged p38-like ERK2 was expressed in ΔB-Raf:ER cells (Pritchard et al., 1995) and activated by estrogen treatment. The activated ERK2 constructs were then immunoprecipitated and used. Comparable amounts were immunoprecipitated (data not shown), and they showed comparable kinase activity towards MBP (lower panel; left, wild-type ERK2; right, p38-like ERK2). Similar results were obtained in three different experiments. (F) Activation of 3pk in cells by wild-type ERK2 or p38-like ERK2. NIH 3T3 cells co-transfected with SRα-Myc 3pk and SRα-HA-tagged wild-type (wt) ERK2 or HA-tagged p38-like ERK2 (p38L-ERK2) were serum deprived and then stimulated by 10% FCS for 15 min. Myc-3pk was then immunoprecipitated and assayed for kinase activity using MBP as substrate (upper panel, MBP). To remove co-precipitated ERK2, the immunoprecipitated 3pk was washed extensively with a high-salt buffer before kinase assay. Comparable amounts of 3pk were immunoprecipitated in each lane [middle panel, αMyc (IP)]. Comparable amounts of ERK2 were expressed in each lane [lower panel, αHA (whole)].
The next open question is how the specificity of the docking interactions of MAPKs is determined. Because the CD domain alone did not fully explain the difference in the docking specificity among different MAPKs (Tanoue et al., 2000), we hypothesized the existence of another region that might regulate the specificity. In this study, we have identified a novel site on MAPKs, near the CD domain in the steric structure, which determines the docking specificity towards MAPKAPKs. Surprisingly, exchange of only two amino acids in this site of p38 and ERK (Glu160 and Asp161 in p38, and Thr157 and Thr158 in ERK2) converts the docking specificity. We named this site the ED site. The ED site is also located on the opposite side from the active center and utilized differently for docking interactions with each interacting molecule. Furthermore, we show here that amino acid substitutions in the CD domain also lead to changes in the docking affinity of p38 and ERK2 for various MAPKAPKs. We then propose a model in which a groove in the steric structure of MAPKs, which comprises the CD domain and the ED site, serves as a common docking groove. Every MAPK-interacting molecule may bind to this docking groove. However, each amino acid residue in the docking groove is involved differently in each docking interaction. Thus, this study defines the molecular basis for determining the specificity and strength of docking interactions of MAPKs.
Results
Identification of a novel docking site on p38α for MAPKAPK-3/3pk
We have shown previously that the CD domain on p38 MAPK, which is characterized by three aspartic acids (Asp313, Asp315 and Asp316; see Table I), is utilized commonly for docking to MKK6 (p38 activator), MKP-5 (a member of the MKPs) and MNK1 (a member of the MAPKAPKs) (Tanoue et al., 2000). To examine whether the CD domain is also utilized for docking to MAPKAPK-3/3pk (see Figure 1A), the other member of the MAPKAPKs that is specific for p38, we performed a co-immunoprecipitation binding assay. Wild-type p38 co-immunoprecipitated well with wild-type 3pk (Figure 2A, lane 2). When a mutant p38 (p38 CDm), in which the three aspartic acids in the CD domain were replaced by asparagines (see Table I), was assayed, the docking ability was reduced compared with wild-type p38, but the p38 CDm still bound to 3pk significantly (Figure 2A, lane 5). This result suggested the possibility that a site different from the CD domain might also participate in the docking interaction. As the C-terminal portion shows the greatest diversity in primary sequence among members of the MAPK family, we first assumed that a cluster of negatively charged amino acids [Asp354, Glu356, Glu357 and Glu359 (for human p38)], which is located in the most C-terminal portion of the molecule, might be involved in regulation of the docking interaction. However, even when all these four negatively charged amino acids of this region, in addition to the three aspartic acids of the CD domain, were replaced by asparagine or glutamine, the docking ability of this mutant p38 to 3pk was almost the same as that of p38 CDm (data not shown). Thus, this most C-terminal region may not be involved in the docking interaction. We then noticed another cluster of negatively charged amino acids [Glu160 and Asp161 (for human p38)] near the CD domain in the steric structure of p38 (see Figure 1B). We tentatively named this site the ED site. To test the possible involvement of the ED site in the docking interaction, we constructed a mutant form of p38 in which Glu160 and Asp161 were replaced by threonines (p38 ETDT). p38 ETDT also showed a decreased ability to bind to 3pk compared with wild-type p38 (Figure 2A, lane 4). Furthermore, when both the CD domain and the ED site were mutated (p38 all), the binding was almost completely lost (Figure 2A, lane 6). Glutathione S-transferase (GST) pull-down assays using bacterially expressed GST–3pk protein gave essentially the same results (Figure 2B, upper panels). In addition, the bacterially expressed wild-type GST–p38 also bound directly to the bacterially expressed His-3pk in vitro and the GST–p38 all showed much weaker affinity for His-3pk (Figure 2B, lower panels). Thus, both the CD domain and the ED site are important for the docking interaction. Next, to show that a putative p38 docking site of 3pk is indeed a docking site, we constructed a mutant 3pk in which five consecutive basic amino acids (KRRKK) in the putative docking site were replaced by methionines (3pk mut, see Figure 1A), and examined its docking ability. Whereas wild-type 3pk co-immunoprecipitated with wild-type p38, the mutant 3pk did not (Figure 2A). This result was also confirmed by the GST pull-down assay using bacterially expressed GST–p38 protein (Figure 2C). We then performed the GST pull-down assay using a fusion between GST and a C-terminal portion of 3pk (residues 326–382). While wild-type p38 co-precipitated well with this fragment of 3pk, p38 ETDT, p38 CDm and the double mutant (p38 all) did not (Figure 2D). These results suggest that the C-terminal, basic region of 3pk serves as a direct docking site for p38. However, we cannot exclude the possibility that the other site of 3pk might also participate in the docking interaction between 3pk and p38, because the C-terminal portion of 3pk bound to p38 more weakly than full-length 3pk (see Figure 2B and D).
Table I. p38 mutants and ERK2 mutants used in this study.
| Name of mutant | Mutations introduced | |||
|---|---|---|---|---|
| p38 | p38 CDm | 308FAQYHDPDDEPVA320 | → | FAQYHNPNNEPVA |
| p38 ETDT | 154SNLAVNEDCELKIL167 | → | SNLAVNTTCELKIL | |
| p38 all | 154SNLAVNEDCELKIL167 | → | SNLAVNTTCELKIL | |
| 308FAQYHDPDDEPVA320 | → | FAQYHNPNNEPVA | ||
| ERKL-p38 | 154SNLAVNEDCELKIL167 | → | SNLAVNTTCELKIL | |
| 308FAQYHDPDDEPVA320 | → | FAQYHDPSDEPVA | ||
| ERK2 | ERK2 CDm | 316LEQYYDPSDEPVA328 | → | LEQYYNPSNEPVA |
| ERK2 SD | 316LEQYYDPSDEPVA328 | → | LEQYYDPDDEPVA | |
| ERK2 TETD | 156SNLLLNTTCDLKIC169 | → | SNLLLNEDCDLKIC | |
| p38L-ERK2 | 156SNLLLNTTCDLKIC169 | → | SNLLLNEDCDLKIC | |
| 316LEQYYDPSDEPVA328 | → | LEQYYDPDDEPVA |
The numbers shown are the sequences of Xenopus ERK2 and human p38α.
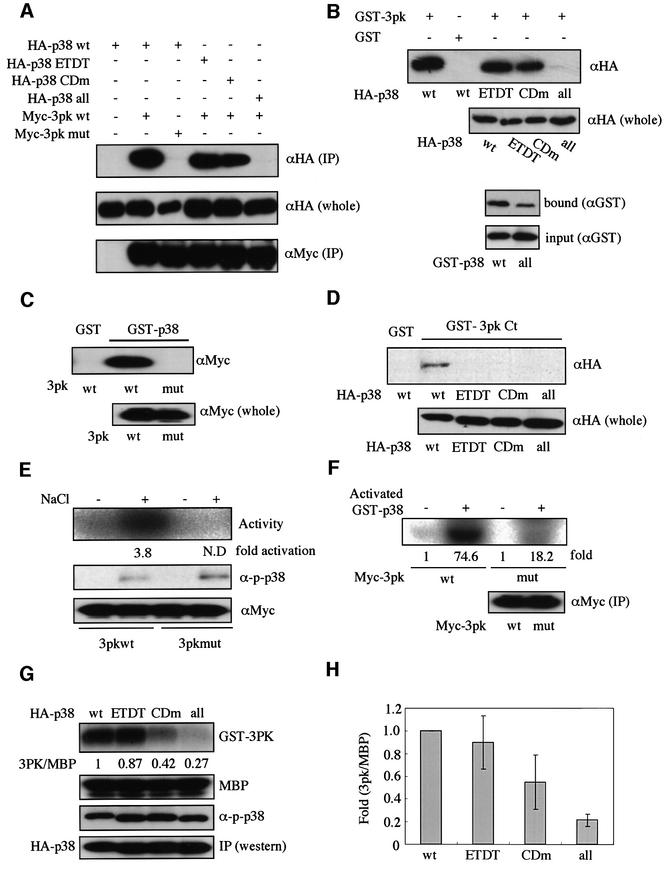
Fig. 2. Glu160 and Asp161 of p38α are necessary for the docking interaction with 3pk. (A) Lysates of NIH 3T3 cells co-transfected with the indicated combinations of constructs were immunoprecipitated with anti-Myc antibody. Co-immunoprecipitated wild-type (wt) and mutant forms of p38 were detected [upper panel, αHA (IP)]. The expression levels of p38 in each sample were similar [middle panel, αHA (whole)]. Comparable amounts of 3pk were immunoprecipitated in each lane [lower panel, αMyc (IP)]. Similar results were obtained in three different experiments. (B) An equal amount (7 µg) of GST or GST–3pk was incubated with each lysate from NIH 3T3 cells expressing HA-wild-type (wt) or mutant forms of p38 (upper two panels). The proteins were precipitated with glutathione–Sepharose beads and analyzed by immunoblotting with anti-HA antibody (αHA). Comparable amounts of p38 were expressed [αHA (whole)]. Similar results were obtained in three different experiments. The direct binding between p38 and 3pk in vitro was examined using GST–p38 and His-3pk (lower two panels). GST fusions of p38 and His-3pk were expressed in bacteria and purified by the method described previously (Tanoue et al., 2000). Purified His-3pk (1 µg) was mixed with purified GST–p38 wt (∼1 µg) or GST–p38 all (∼1 µg) in a solution containing 50 mM HEPES pH 7.4 and 150 mM NaCl, and incubated for 1 h at 4°C. His-3pk was then precipitated with Probond resin (Invitrogen) and co-precipitated GST–p38 was detected by immunoblotting with anti-GST antibody (bound). (C) The binding between wild-type p38 and wild-type or mutant 3pk was examined by GST pull-down assay. An equal amount (10 µg) of GST or GST–p38 was incubated with each lysate from NIH 3T3 cells expressing Myc-wild-type (wt) 3pk or a mutant (mut) 3pk. Co-precipitated 3pk was detected (upper panel, αMyc). Comparable amounts of 3pk were expressed [lower panel, αMyc (whole)]. Similar results were obtained in three different experiments. (D) The binding between the mutant forms of p38 and the C-terminal portion of 3pk (residues 326–382) (GST–3pk Ct) was examined by GST pull-down assay. An equal amount (30 µg) of GST or GST–3pk Ct was incubated with each lysate from NIH 3T3 cells expressing HA-wild-type (wt) or mutant forms of p38. Co-precipitated p38 was detected (upper panel, αHA). Comparable amounts of p38 were expressed [lower panel, αHA (whole)]. Similar results were obtained in three different experiments. (E) NIH 3T3 cells transfected with either SRα-Myc-wild-type 3pk (wt) or SRα-Myc-mutant 3pk (mut) were exposed to 0.5 M NaCl for 15 min. Immunoprecipitated 3pk activity was measured using MBP as substrate (Activity). Comparable amounts of endogenous p38 were activated in both cell types after the stimulation (α-p-p38). Comparable amounts of 3pk were immunoprecipitated (αMyc). Similar results were obtained in three different experiments. (F) The ability of p38 to phosphorylate the docking-deficient 3pk (3pk mut) in vitro. Myc-3pk (wt) or Myc-3pk (mut) was prepared by immunoprecipitation from cells transfected with each construct. Activated p38 was prepared by incubating with His-tagged MKK6. The fold phosphorylation rates are shown (fold). Similar results were obtained in three different experiments. (G) The ability of the mutant forms of p38 to phosphorylate GST–3pk in vitro. Various forms of HA-tagged p38 were prepared by immunoprecipitation from COS7 cells transfected with each construct. The expression levels of the mutant forms of p38 are shown (IP). The p38 in the immunoprecipitates was then activated in vitro by incubating with His-tagged MKK6 (for 1 h at 37°C). Comparable phosphorylation levels of various forms of p38 were comfirmed by immunoblotting with anti-phospho-p38 (α-p-p38). The phosphorylation of MBP by these phosphorylated (thus activated) forms of p38 is shown (MBP). The rate of GST–3pk phosphorylation relative to that of MBP was quantified and the value relative to the wild-type p38 is shown (3pk/MBP). (H) Quantification of the data from four independent experiments of (G).
Both the CD domain and the ED site of p38 are essential for the optimal, efficient activation of 3pk
We showed previously that the docking interactions through the CD domain were indispensable for the efficient enzymatic reactions between MAPKs and various MAPK-interacting molecules (Tanoue et al., 2000). Thus, we examined whether both the CD domain and the ED site of p38 are required for the efficient phosphorylation of 3pk. As shown in Figure 2G and H, the ability of both mutants p38 ETDT and p38 CDm to phosphorylate 3pk was markedly decreased compared with that of wild-type p38, and the double mutant showed the lowest ability, whereas all the mutant forms of p38 exhibited almost the same ability as wild-type p38 to phosphorylate myelin basic protein (MBP), which appears not to have a docking site. The decrease in the ability to phosphorylate 3pk correlated roughly with the decrease in the docking affinity (see Figure 2A, B, G and H). These results thus suggest that both the CD domain and the ED site are required for the efficient phosphorylation of 3pk by p38. We then tested a docking-deficient mutant of 3pk. The mutant 3pk was not significantly activated in response to osmotic stress, although we could not measure the fold activation of the docking-deficient 3pk after osmotic stress accurately because the activity was too low to quantitate (Figure 2E). The phosphorylation rate by p38 in vitro was also decreased greatly in the docking-deficient 3pk (Figure 2F). These results, taken together, suggest the importance of the docking interaction in the efficient phosphorylation and activation of 3pk by p38.
The docking interaction regulates the subcellular localization of 3pk
It has been reported that MAPKAPK-2 has a nuclear export signal (NES) sequence and a putative nuclear localization signal (NLS) in tandem in the C-terminal portion. Under unstimulated conditions, MAPKAPK-2 is present in the nucleus, and upon stimulation it becomes phosphorylated by p38 and is consequently exported from the nucleus probably due to unmasking of its NES (Ben-Levy et al., 1998; Engel et al., 1998). The primary sequence of 3pk is similar to that of MAPKAPK-2, and both the NES and NLS-like sequences are conserved in 3pk (Engel et al., 1998; see Figure 1A). To test whether the putative NES and NLS of 3pk are functional, we constructed a fusion [green fluorescent protein (GFP)–3pk Ct] between GFP and a C-terminal portion of 3pk (residues 326–382) that contains both the NES and NLS-like sequences. When expressed in NIH 3T3 cells, GFP–3pk Ct localized in the cytoplasm (Figure 3A, left), suggesting that the activity of the NES, if exposed, is stronger than that of the NLS. When the cells were treated with leptomycin B (a specific inhibitor of NES-mediated active nuclear export), GFP–3pk Ct translocated to the nucleus and accumulated there (Figure 3A, right). Thus, 3pk has both the NES and NLS in the C-terminal portion, like MAPKAPK-2. Next, we examined the subcellular localization of full-length 3pk before and after osmotic stress. GFP-tagged wild-type 3pk was present in the nucleus before stimulation and translocated to the cytoplasm after osmotic stress (Figure 3B, 3pk wt). Since this behavior is identical to that of MAPKAPK-2 (Engel et al., 1998), it is likely that the stimulus-dependent change in the subcellular localization of 3pk is also caused by the phosphorylation by p38. When the docking-deficient mutant of 3pk (3pk mut) was examined for its subcellular distribution, it localized in both the nucleus and the cytoplasm under unstimulated conditions, and this distribution did not change after stress stimulation (Figure 3B, 3pk mut). The pan-cellular localization of 3pk mut before stimulation may be due to the partial disruption of NLS by docking-disrupting mutagenesis, as the five basic amino acids (residues 364–368; see Figure 1A) are essential for both the docking and the NLS. The inability to change its localization upon stimulation may be due to the inability of 3pk mut to be phosphorylated by p38, suggesting that the docking interaction is essential for the phosphorylation of 3pk by p38 in cells.
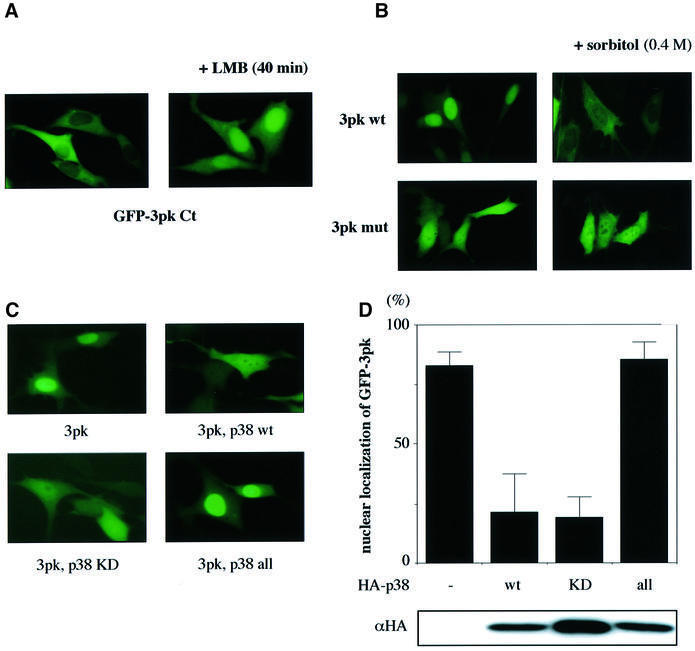
Fig. 3. The possible role of the docking interaction in the subcellular localization of 3pk. (A) Subcellular localization of a GFP-tagged C-terminal portion of 3pk (residues 326–382) (GFP–3pk Ct) in cultured cells. NIH 3T3 cells were transfected with GFP–3pk Ct. The cells were treated with leptomycin B (LMB, 8 ng/ml) for 40 min (right panel). (B) The subcellular localization of GFP-tagged wild-type 3pk (3pk wt) or a GFP-tagged docking-deficient 3pk (3pk mut) in cultured cells. The cells were stimulated with osmotic stress (0.4 M sorbitol) for 20 min (right panels). (C) Subcellular localization of GFP-tagged wild-type 3pk in the absence or presence of overexpressed wild-type p38 (p38 wt), kinase-negative p38 (p38 KD) or p38 all (p38 all). Lys53 was replaced by methionine in the kinase-negative form of p38. p38 KD, p38 all and p38 wt showed the same pan-cellular localization (data not shown). (D) Quantification of the data from three independent experiments in (C). The three experiments gave essentially the same results. The cells in which the concentration of GFP–3pk in the nucleus was much higher than that in the cytoplasm (see C, 3pk alone) were regarded as the cells showing ‘nuclear localization of GFP–3pk’, and are shown as percentages of total cells examined. In each case, 300 cells were examined in total. Comparable amounts of HA-p38 were expressed (lower panel, representative of three experiments).
As the p38 docking site of 3pk overlaps the NLS (see Figure 1A), when co-expressed with p38, wild-type 3pk must have the same localization pattern as the docking-deficient 3pk because the NLS sequence might be masked by the docking of p38; this was found to be the case. As shown in Figure 3C and D, GFP–3pk became localized in both the nucleus and the cytoplasm when co-expressed with wild-type (p38 wt) or kinase-negative p38 (p38 KD). In contrast, co-expression of the docking-deficient p38 (p38 all) did not induce the pan-cellular localization of 3pk; 3pk remained in the nucleus. All these results are consistent with our argument that the docking interaction between p38 and 3pk is achieved through the direct interaction of the C-terminal portion of 3pk (the NLS-like sequence) with the CD domain and the ED site on p38.
The ED site regulates the docking specificity
The amino acid residues in ERK2 that correspond to the ED site (Glu160 and Asp161) of p38 are Thr157 and Thr158 (for rat ERK2) (Figure 4B). They are also expected to be surface exposed and to be localized in a position almost identical to that of the ED site on p38 in the steric structure (see Figures 4A and 1B, right). Glutamic acid and aspartic acid are negatively charged, whereas threonine is not. As we have shown previously that the electrostatic interaction is important for the docking interactions of MAPKs (Tanoue et al., 2000), we then postulated that these two amino acids might regulate the docking specificity of ERK2 and p38. We first constructed a mutant ERK2 (ERK2 TETD), in which the two threonines were replaced by glutamic acid and aspartic acid, respectively. While wild-type ERK2 did not bind to 3pk in the co-immunoprecipitation assay, ERK2 TETD bound significantly (Figure 4D, lane 4), indicating the importance of the ED site in docking to 3pk. In the CD domain, p38 has three acidic amino acids (Asp313, Asp315 and Asp316) that are surface exposed, while the residue corresponding to Asp315 is serine in ERK2 (Figure 4B and see Tanoue et al., 2000). We then constructed a mutant ERK2 (ERK2 SD), in which this serine was replaced by aspartic acid. ERK2 SD bound to 3pk, although its binding ability was much weaker than that of ERK2 TETD (Figure 4D, lanes 3 and 4). When both mutations (TETD and SD) were introduced, the resulting ERK2 (termed p38-like ERK2) bound to 3pk more strongly than ERK2 TETD (Figure 4D, lane 5). The binding ability of p38-like ERK2 (p38L-ERK2) was comparable to that of p38 (Figure 4C). Thus, both the ED site and the CD domain play a role in regulating the docking interaction, and the ED site contributes more to regulation of the docking specificity towards 3pk. In agreement with this, the mutant p38 (p38 ETDT), in which the ED site sequence of p38 was changed into the ERK2 sequence, bound to 3pk much more weakly than wild-type p38, although the binding was not completely lost (Figure 2A and B).
We next examined the efficiency of the enzymatic reaction. Activated wild-type ERK2 and activated p38-like ERK2 were prepared and assayed for the ability to phosphorylate 3pk in vitro. As shown in Figure 4E, p38-like ERK2 (m) had stronger enzymatic activity than wild-type ERK2 (w), whereas both had comparable kinase activity towards MBP, which appears not to have a MAPK-docking site. When co-expressed in cells, p38-like ERK2 activated 3pk more strongly than wild-type ERK2 (Figure 4F). However, the difference was modest, probably because endogenous p38 and ERKs might affect the result; for example, the activity of endogenous molecules might be inhibited to different extents by the two exogenously expressed molecules.
A docking groove on p38 and ERK MAPKs
We extended our analyses to other p38-specific MAPKAPKs, MSK2 and PRAK (Deak et al., 1998; New et al., 1998; Pierrat et al., 1998). In the co-immunoprecipitation assay, MSK2 bound to wild-type p38, but not to wild-type ERK2 (Figure 5B). The CD domain of p38 was essential for the docking (Figure 5A). p38 ETDT showed almost the same ability to bind to MSK2 as wild-type p38 (Figure 5A), and ERK-like p38 showed a marked decrease in this ability (Figure 5B). Although ERK2 SD did not bind to MSK2, like wild-type ERK2, ERK2 TETD bound MSK2 slightly. Moreover, p38-like ERK2 bound to MSK2 more strongly than ERK2 TETD (Figure 5C). Thus, both the CD domain and the ED site regulate the docking specificity of p38 and ERK2 towards MSK2.
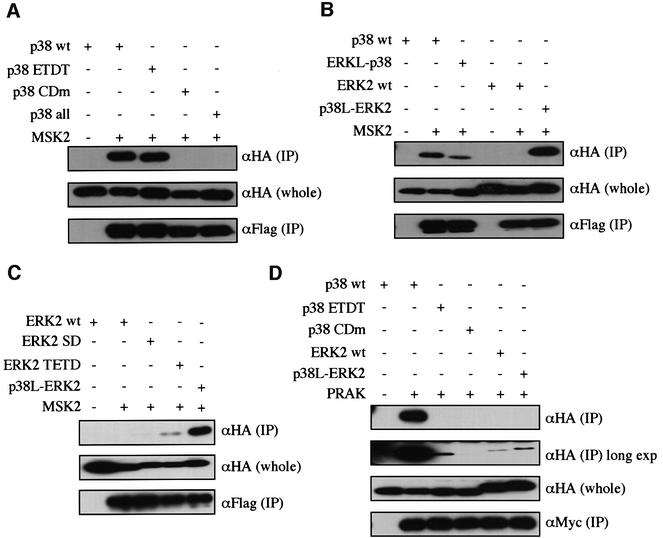
Fig. 5. Involvement of the ED site in the docking interaction of MAPKs with MSK2 and PRAK. (A) The binding of the mutant forms of p38 to MSK2. Lysates of NIH 3T3 cells co-transfected with the indicated combinations of constructs were immunoprecipitated with anti-Flag antibody for Flag-MSK2. Co-immunoprecipitated wild-type p38 (wt) and mutant forms of p38 (HA-tagged) were detected [upper panel, αHA (IP)]. The expression levels of p38 in each sample were similar [middle panel, αHA (whole)]. Comparable amounts of Flag-MSK2 were immunoprecipitated in each lane [lower panel, αFlag (IP)]. pMT-Flag-MSK2 was used. Similar results were obtained in three different experiments. (B) The binding of p38-like ERK2 or ERK2-like p38 to MSK2 was examined as in (A). Similar results were obtained in three different experiments. (C) The binding of the mutant forms of ERK2 to MSK2 was examined as in (A). SRα-HA-wild-type ERK2, HA-ERK2 SD, HA-ERK2 TETD and HA-p38-like ERK2 were used. Similar results were obtained in three different experiments. (D) The binding of the mutant forms of p38 and ERK2 to PRAK was examined as in (A). SRα-Myc-PRAK was used. A longer exposure of the most upper panel is shown in the second upper panel [αHA (IP) long exp]. Similar results were obtained in three different experiments.
PRAK bound to wild-type p38, but not to wild-type ERK2 (Figure 5D). The CD domain of p38 was essential for the docking. The mutation in the ED site of p38 induced a dramatic decrease in the docking ability (p38 ETDT), and p38-like ERK2 bound to PRAK slightly more strongly than wild-type ERK2 (Figure 5D). Thus, in the case of PRAK, both the CD domain and the ED site are important for determining the docking specificity, but amino acid residues other than those of ED (TT) and the CD domain may also participate in regulating the specificity.
We next examined RSK2, a MAPKAPK specific for ERK2. RSK2 bound to ERK2, but not to p38 (Figure 6A). The CD domain of ERK2 was essential for the docking (Figure 6B). p38-like ERK2 showed a decreased ability to bind to RSK2, whereas ERK-like p38 did not bind to RSK2. Thus, the amino acids in the ED site and the CD domain are not the only determinants of the docking specificity.
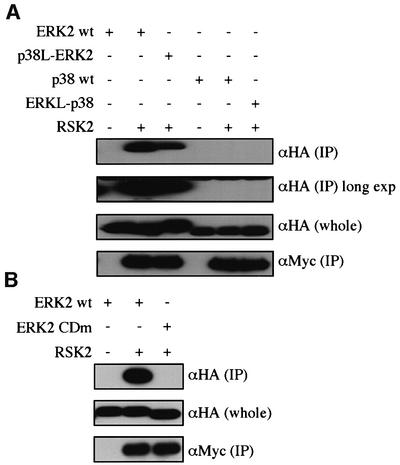
Fig. 6. Docking interactions of p38 and ERK2 with RSK2. (A) The binding of the mutant forms of p38 and ERK2 to RSK2. Lysates of NIH 3T3 cells co-transfected with the indicated combinations of constructs were immunoprecipitated with anti-Myc antibody for Myc-RSK2. Co-immunoprecipitated HA-ERK2 wt, HA-p38L-ERK2, HA-p38 wt and HA-ERK2L-p38 were detected [upper panel, αHA (IP)]. The same membrane was exposed for a longer time (long exp). The expression levels of these HA-tagged constructs were similar [middle panel, αHA (whole)]. Comparable amounts of Myc-RSK2 were immunoprecipitated in each lane [lower panel, αMyc (IP)]. Similar results were obtained in three different experiments. (B) The binding of ERK2 CDm to RSK2 was examined as in (A). Similar results were obtained in three different experiments.
We next examined MSK1 and MNK1, members of the MAPKAPK subfamily that are specific for both p38 and ERK2 (Fukunaga and Hunter, 1997; Waskiewicz et al., 1997; Deak et al., 1998; New et al., 1999). MSK1 bound to both wild-type p38 and wild-type ERK2 (Figure 7B). The CD domain was essential for the MSK1 docking to p38 and ERK2 (Figure 7A and C). p38 ETDT and ERK-like p38 showed an increased ability to bind to MSK1 compared with wild-type p38, while ERK2 SD, ERK2 TETD and p38-like ERK2 also showed an increased ability in docking to MSK1 compared with wild-type ERK2 (Figure 7D). MNK1 also bound to both wild-type p38 and wild-type ERK2, and the CD domain was essential (Tanoue et al., 2000). ERK-like p38 showed a decreased docking ability compared with wild-type p38, and p38-like ERK2 also showed a decreased docking ability compared with wild-type ERK2 (Figure 7E). Therefore, in the case of these two p38- and ERK-specific MAPKAPKs, the docking interactions require the CD domain, and changes in amino acid compositions or sequences in the ED site and the CD domain could affect the docking affinity.
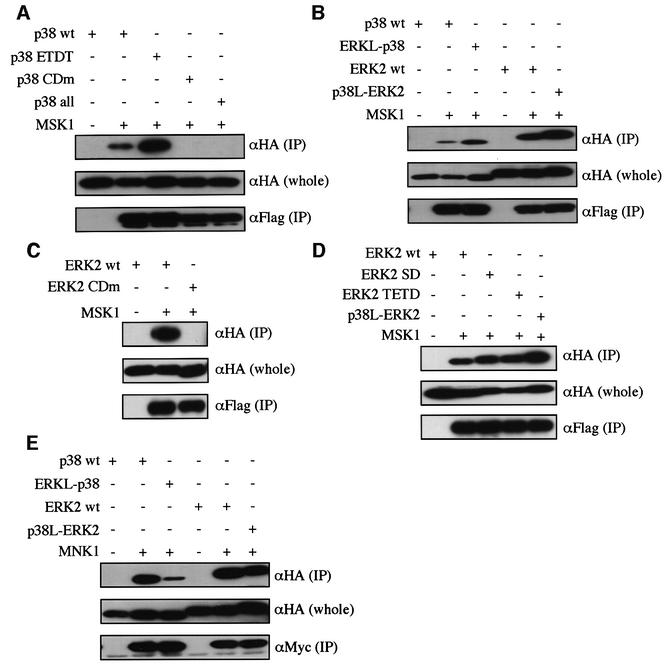
Fig. 7. Docking interactions of p38 and ERK2 with MSK1 or MNK1. (A) The binding of the mutant forms of p38 to MSK1. Lysates of NIH 3T3 cells co-transfected with the indicated combinations of constructs were immunoprecipitated with anti-Flag antibody for Flag-MSK1. Co-immunoprecipitated HA-p38 wt, HA-p38 ETDT, HA-p38 CDm and HA-p38 all were detected [upper panel, αHA (IP)]. The expression levels of these HA-tagged constructs were similar [middle panel, αHA (whole)]. Comparable amounts of Flag-MSK1 were immunoprecipitated in each lane [lower panel, αFlag (IP)]. pMT-Flag-MSK1 was used. Similar results were obtained in three different experiments. (B) The binding of p38-like ERK2 or ERK2-like p38 to MSK1 was examined as in (A). SRα-HA-ERKL-p38 and SRα-HA-p38L-ERK2 were used. Similar results were obtained in three different experiments. (C) The binding of ERK2 CDm to MSK1 was examined as in (A). Similar results were obtained in three different experiments. (D) The binding of the mutant forms of ERK2 to MSK1 was examined as in (A). SRα-HA-ERK2 wt, SRα-HA-ERK2 SD, SRα-HA-ERK2 TETD and SRα-HA-p38L-ERK2 were used. Similar results were obtained in three different experiments. (E) The binding of the mutant forms of p38 and ERK2 to MNK1. Myc-tagged MNK1 was expressed. Similar results were obtained in three different experiments.
The above results taken together suggest that a region comprising both the ED site and the CD domain on p38 and ERK MAPKs is a docking surface for MAPKAPKs. By looking at the steric structure of p38 and ERK2, we found that there is a groove comprising both the CD domain and the ED (TT) site (see Figures 1B and 4A). We propose that the overall structure of this groove and the location and number of charged amino acids in the groove are important for determining both the docking specificity and the affinity of docking interactions (see Figure 9). We call this groove a docking groove. It is likely that each amino acid residue in the docking groove is utilized differently in each docking interaction.
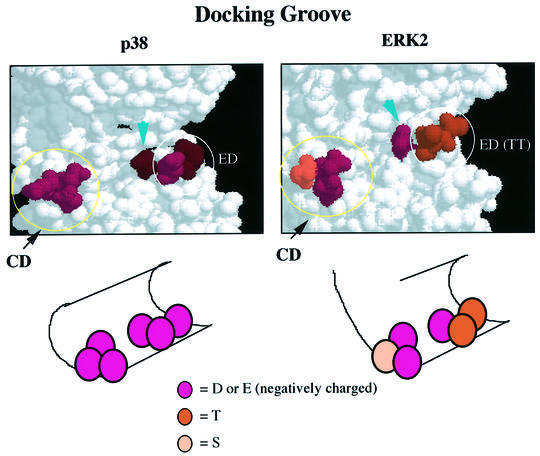
Fig. 9. Schematic representation of the docking groove. The three-dimensional structures of the docking pocket of p38 and ERK2 are shown (upper panels). The CD domain and the ED (TT) site are indicated. The arrows indicate Glu163 of p38 or Asp160 of ERK2. See text for details.
The docking groove is also utilized in the docking interaction between MAPKs and MKPs
We examined the docking between MAPKs and MKP-5 or MKP-3. MKP-5 is a dual-specificity phosphatase specific for p38 and JNK/SAPK (Tanoue et al., 1999). While wild-type p38 bound strongly to MKP-5, wild-type ERK2 bound only slightly to MKP-5. p38-like ERK2 bound to MKP-5 slightly more strongly than wild-type ERK2, and p38 ETDT showed almost the same ability to bind to MKP-5 as wild-type p38 (Figure 8A). MKP-3 is a dual-specificity phosphatase specific for ERK (Muda et al., 1998). MKP-3 bound to wild-type ERK2, but not to p38 (Figure 8B). p38-like ERK2 lost the ability to bind to MKP-3, although p38 ETDT did not bind to MKP-3 (Figure 8B). It was shown previously that the CD domain is indispensable for the docking interaction between MAPKs and MKP-5 or MKP-3 (Tanoue et al., 2000). Collectively, these results indicate that the docking groove of p38 and ERK MAPKs may also be utilized for the docking interactions with MKPs, and that each amino acid in both the ED (TT) site and the CD domain may affect the docking specificity in a distinct way.
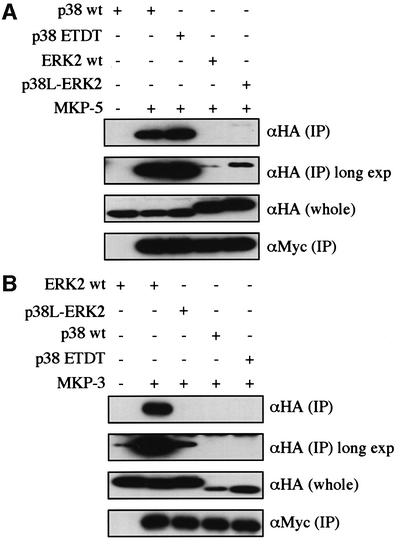
Fig. 8. Involvement of the ED site in the docking interaction of MAPKs with MKPs. (A) The binding of the mutant forms of p38 and ERK2 to MKP-5. Lysates of NIH 3T3 cells co-transfected with the indicated combinations of constructs were immunoprecipitated with anti-Myc antibody for Myc-MKP-5. Co-immunoprecipitated HA-p38 wt, HA-p38 ETDT, HA-ERK2 wt and HA-p38L-ERK2 were detected [upper panel, αHA (IP)]. The same membrane was exposed for a longer time (long exp). The expression levels of these HA-tagged constructs were similar [middle panel, αHA (whole)]. Comparable amounts of Myc-MKP-5 were immunoprecipitated in each lane [lower panel, αMyc (IP)]. SRα-Myc-MKP-5 was used. Similar results were obtained in three different experiments. (B) The binding of the mutant forms of p38 and ERK2 to MKP-3. SRα-Myc-MKP-3 was used. Similar results were obtained in three different experiments.
Discussion
In this study, we have identified a novel site (termed the ED site) on p38 (Glu160 and Asp161 of human p38) that is needed for the docking interactions with several MAPKAPKs. Furthermore, this site on p38 and the corresponding site on ERK2 (Thr156 and Thr157 of rat ERK2) are shown to regulate the docking specificity of p38 and ERK2 for MAPKAPKs. Thus, when the two amino acid residues of ERK2, Thr157 and Thr158, were replaced by glutamic acid and aspartic acid, respectively, ERK2 bound to 3pk and MSK2, indicating that the exchange of amino acids in the ED site converts the docking specificity of ERK2 to that of the p38 type. However, the reverse exchange in p38 did not convert the docking specificity of p38 toward RSK2 from the p38 type to the ERK2 type. This implies that the other amino acid residues also participate in the docking interaction. The amino acids comprising the CD domain and the ED site form a groove-like structure in the steric structure of p38 and ERK2 (see Figures 1B and 4A). Thus, we propose the concept of a docking groove (see Figure 9). The structural difference in the docking groove between p38 and ERK2 might affect both the affinity and specificity of docking. Each amino acid residue in the docking groove appears to contribute differently to each docking interaction. In agreement with this idea, although the CD domain is important in the same way for all the docking interactions between MAPKs and MAPKAPKs examined in this study, the ED site is involved differently in each interaction. For example, while the mutation of the ED site of p38 (Glu160 and Asp161 were replaced by threonines) weakened its docking interaction with PRAK, the same mutation strengthened the docking interaction with MSK1. However, not only the ED site but also the CD domain is important for regulating the docking specificity of p38 and ERK2. For example, ERK2 SD, in which the CD domain of ERK2 was converted to that of p38, was able to bind to 3pk, like p38, while wild-type ERK2 was unable to bind to 3pk. In addition, the conversion of both the CD domain and the ED (TT) site of ERK2 to those of p38 converted the docking specificity of ERK2 towards MSK2 to that of p38. Moreover, it is likely that other amino acids near the CD domain and the ED site within the docking groove are also involved in the docking interactions. The difference in the amino acid composition and sequence of the MAPK docking sites of the MAPK-interacting molecules (MAPKKs, MKPs and MAPKAPKs) might affect the specificity and affinity of their docking interactions with the docking groove of MAPKs.
Asp319 (in rat ERK2) in the CD domain is mutated to asparagine in the sevenmaker mutant of Drosophila ERK/Rolled (Brunner et al., 1994). It has been reported recently that a mutation of Asp160 (in rat ERK2) to asparagine leads to a phenotype similar to that of the sevenmaker mutant (Lim et al., 1999), suggesting involvement of Asp160 in the docking interaction. Asp160 (see Figure 4A, arrowhead) is located adjacent to Thr158 of the ED (TT) site in the steric structure of ERK2 (Figure 4B). The corresponding amino acid in p38 is Glu163 (see Figure 1B, right, arrowhead). It is clear that both Asp160 of ERK2 and Glu163 of p38 are located within the docking groove and are expected to be surface exposed (see Figure 9). Therefore, this recent observation is consistent with our idea of a docking groove.
Does the docking interaction alone determine the substrate specificity?
Previously, it was reported that the N-terminal portion (Cdc25-like domain) of dual-specificity phosphatases (MKP-3 and M3/6) determines both the docking specificity and the enzymatic specificity (Muda et al., 1998). This is a case where the docking interaction determines the enzymatic specificity. It was reported, on the other hand, that multiple regions of ERK2 and p38 are important for the determination of the enzymatic specificity of upstream kinases (MAPKKs) towards MAPKs (Brunet and Pouyssegur, 1996; Wilsbacher et al., 1999). Recently it has been shown that the substitution of amino acid residues within the T-loop of p38β2 greatly affects the activatability by MKK3 (Enslen et al., 2000), indicating the importance of the transient enzyme–substrate interaction in determining the substrate specificity. Our results here suggest that the docking interaction through the docking groove helps to regulate the specificity of the enzymatic reaction towards MAPKAPKs in most cases, but is not the sole determinant for the substrate specificity.
The docking interactions in the MAPK cascades
The docking groove is located on the opposite side of the active center in the steric structure. Thus, the docking interaction is different from the transient enzyme– substrate interaction achieved through the active center. It has been shown that several enzymes need a co-factor(s) or a specific interaction sequence of their own for an efficient and specific enzymatic reaction. For example, PP2A requires specific co-factors or subunits to recognize each substrate (Millward et al., 1999), and PDK1 has a hydrophobic docking pocket for the interaction with its substrates, PKA, PKCζ and PRK2 (Balendran et al., 2000; Ricardo et al., 2000). However, it is not known whether such interactions besides the transient enzyme–substrate interaction through the active center are always necessary for kinase–substrate recognition. Interestingly, other substrates of MAPKs, such as transcription factors PDE and SOS, have also been shown to have a docking site for MAPKs (Kallunki et al., 1996; Yang et al., 1998, 1999; Jacobs et al, 1999; T.Tanoue and E.Nishida, unpublished observations). Although it is not known at present whether the docking groove of MAPKs is also utilized in these docking interactions, they are in fact docking interactions different from the transient enzyme–substrate interaction, as their proposed docking sites are located far from the phosphorylation sites in the primary sequence.
What is the role of the docking interaction between MAPKs and their substrates? While it is often the case that the consensus sequence of the phosphorylation site in the substrate for protein kinases involves charged amino acids, such as the sequence RXRXXS/TP-Hyd (Hyd is a hydrophobic residue) for Akt/PKB (Alessi et al., 1996), the sequence S/TPXR/K for cyclin-dependent kinases, a group of acidic amino acids for casein kinase II, etc. (Pinna and Ruzzene, 1996), the consensus sequence for MAPKs, PXS/TP or S/TP (Clark-Lewis et al., 1991; Gonzalez et al., 1991), does not contain charged amino acids. Thus, it can be deduced that MAPKs utilize the docking interactions involving the charged amino acids in order to increase their efficiency and specificity for the substrates. However, this idea does not necessarily explain why the same docking groove is utilized for the docking interactions with MAPKKs and phosphatases. Therefore, the docking interactions in the MAPK cascades might have other roles. First, as the docking interactions of MAPKs with MAPKKs, phosphatases and MAPKAPKs are mutually exclusive, they may underlie the molecular basis for the sequential and specific activation and inactivation of MAPKs. Secondly, the docking interactions may function to determine the subcellular localization of MAPKs and MAPK-interacting molecules. For example, the N-terminal portion of MEK1 is the ERK docking domain, and MEK1 can function as a cytoplasmic anchor for ERK2 (Fukuda et al., 1997). In addition, we have shown here that the docking interaction with p38 regulates the subcellular localization of 3pk, a member of the MAPKAPKs.
Materials and methods
Plasmids
The expression vector used for 3pk, MNK1, RSK2, PRAK, MKP-5 (human) and MKP-3 (rat) (Fukunaga and Hunter, 1997; Muda et al., 1998; New et al., 1998; Tanoue et al., 1999) is pDL-SRα-3XMyc. For ERK2 (Xenopus) and p38α (human), pDL-SRα-HA was used. For MSK1 and -2 (human) (Deak et al., 1998; New et al., 1999), pMT-Flag was used.
Mutagenesis
The mutants used were constructed by PCR-based mutagenesis. PCR was performed using Pfu polymerase (Stratagene). A DpnI restriction enzyme (Stratagene)-treated PCR product was transformed into Escherichia coli. Positive clones were picked up and mutagenesis was verified by sequencing. The primers used for p38 CDm were 5′-ctttgctcagtaccacaatcctaataatgaaccagtggccg-3′ and 5′-cggccactggttcattattaggattgtggtactgagcaaag-3′; 5′-gtaatctagctgtgaatacaacatgtgagctgaagattctgg-3′ and 5′-ccagaatcttcagctcacatgttgtattcacagctagattac-3′ for p38 ETDT; 5′-ctttgctcagtaccacaatcctaataatgaaccagtggccg-3′, 5′-cggccactggttcattattaggattgtggtactgagcaaag-3′, 5′-gtaatctagctgtgaatacaacatgtgagctgaagattctgg-3′ and 5′-ccagaatcttcagctcacatgttgtattcacagctagattac-3′ for p38 all; 5′-gctcagtaccacgatcctagtgatgaaccagtggccg-3′, 5′-cggccactggttcatcactaggatcgtggtactgagc-3′, 5′-gtaatctagctgtgaatacaacatgtgagctgaagattctgg-3′ and 5′- ccagaatcttcagctcacatgttgtattcacagctagattac-3′ for ERKL-p38; 5′-ctggagcagtattataacccaagtaatgagcctgtagctg-3′ and 5′-cagctacaggctcattacttgggttataatactgctccag-3′ for ERK2 CDm; 5′-gcagtattatgacccagatgatgagcctgtagctgaagc-3′ and 5′-gcttcagctacaggctcatcatctgggtcataatactgc-3′ for ERK2 SD; 5′-caaatttgctgcttaacgaggactgtgatctcaagatctg-3′ and 5′-cagatcttgagatcacagtcctcgttaagcagcaaatttg-3′ for ERK2 TETD; and 5′-gcagtattatgacccagatgatgagcctgtagctgaagc-3′, 5′-gcttcagctacaggctcatcatctgggtcataatactgc-3′, 5′-caaatttgctgcttaacgaggactgtgatctcaagatctg-3′ and 5′-cagatcttgagatcacagtcctcgttaagcagcaaatttg-3′ for p38L-ERK2.
Cell cultures and transfection
NIH 3T3 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% calf serum. ΔB-Raf:ER cells (Pritchard et al., 1995) were cultured in DMEM containing 10% fetal bovine serum and 25 mM HEPES pH 7.4 without phenol red. These cells were maintained in 5% CO2 at 37°C. Cells were split onto 35 or 60 mm dishes at 2 × 105 or 5 × 105 cells per dish, respectively. After 19 h, cells were transfected using Lipofectamine Plus reagent (Gibco-BRL) according to the manufacturer’s protocol.
Co-immunoprecipitation
Cells were lysed in 50 mM HEPES pH 7.4, 10% glycerol, 2 mM EGTA, 2 mM MgCl2, 1% NP-40, 1 mM phenylmethylsulfonyl fluoride (PMSF) and 20 µg/ml aprotinin. Tagged proteins were immunoprecipitated from cell lysates (∼3 × 106 cells in each sample) by incubation with 5 µg of anti-c-Myc antibody (9E10) (Santa Cruz) or 5 µg of anti-Flag antibody (Kodak) and protein A–Sepharose beads (25 µl) (Pharmacia) for 2 h at 4°C. The precipitates were then washed twice with lysis buffer. The proteins were separated by SDS–PAGE and analyzed by immunoblotting.
GST pull-down assay
Cells were lysed in 50 mM HEPES pH 7.4, 2 mM EGTA, 2 mM MgCl2, 10% glycerol, 1% NP-40, 1 mM PMSF and 20 µg/ml aprotinin. The lysates were incubated with GST recombinant proteins and glutathione–Sepharose beads for 16 h. The precipitates were then washed twice with lysis buffer. The proteins were separated by SDS–PAGE and analyzed by immunoblotting.
Kinase assay
Cells were lysed in lysis buffer (20 mM Tris–HCl pH 7.5, 12 mM 2-glycerophosphate, 150 mM NaCl, 1.5 mM MgCl2, 2 mM EGTA, 10 mM NaF, 1% Triton X-100, 1 mM PMSF, 1 mM sodium vanadate and 20 µg/ml aprotinin). Tagged proteins were immunoprecipitated from cell lysates (∼1 × 106 cells in each sample) by incubation with 2 µg of appropriate antibody and protein A–Sepharose beads (15 µl) (Pharmacia) for 2 h at 4°C. Each precipitate was washed twice with TBS [20 mM Tris–HCl pH 7.5, 0.5 M NaCl, 1 mM PMSF, 2 mM dithiothreitol (DTT), 1 mM sodium vanadate and 20 µg/ml aprotinin], and then washed with Tris buffer (20 mM Tris–HCl pH 7.5). The washed beads were mixed with substrates in a kinase reaction buffer [20 mM Tris–HCl pH 7.5, 10 mM MgCl2 and 100 µM ATP (2 µCi of [γ-32P]ATP)], and incubated for 15 min at 37°C. The reaction was stopped by addition of Laemmli’s sample buffer. Substrate phosphorylation was detected by autoradiography and BAS 2500 (Fuji Film) after SDS–PAGE.
Acknowledgments
Acknowledgements
We are grateful to R.Fukunaga, S.Arkinstall, D.R.Alessi and M.McMahon for providing us with MNK1, MKP-3, MSK1, -2 and ΔB-Raf:ER cells, respectively; C.Bjørbæk, N.Masuyama and Y.Gotoh for RSK2; M.Yoshida for leptomycin B; and T.Moriguchi and members of our laboratory for helpful discussion. T.T. and M.A. are Research Fellows of the Japan Society for the Promotion of Science. This work was supported by grants from the Ministry of Education, Science and Culture of Japan (to E.N.).
References
- Ahn N.G., Seger,R. and Krebs,E.G. (1992) The mitogen-activated protein kinase activator. Curr. Opin. Cell Biol., 4, 992–999. [DOI] [PubMed] [Google Scholar]
- Alessi D.R., Caudwell,F.B., Andjelkovic,M., Hemmings,B.A. and Cohen,P. (1996) Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett., 399, 333–338. [DOI] [PubMed] [Google Scholar]
- Balendran A., Biondi,R.M., Cheung,P.C.F., Casamayor,A., Deak,M. and Alessi,D.R. (2000) A PDK1 docking site is required for the phosphorylation of PKCζ and PRK2 by PDK1. J. Biol. Chem., 275, 36324–36333. [DOI] [PubMed] [Google Scholar]
- Bardwell L. and Thorner,J. (1996) A conserved motif at the amino termini of MEKs might mediate high-affinity interaction with the cognate MAPKs. Trends Biochem. Sci., 21, 373–374. [PubMed] [Google Scholar]
- Ben-Levy R., Hooper,S., Wilson,R., Paterson,H.F. and Marshall,C.J. (1998) Nuclear export of the stress-activated protein kinase p38 mediated by its substrate MAPKAP kinase-2. Curr. Biol., 8, 1049–1057. [DOI] [PubMed] [Google Scholar]
- Brunet A. and Pouyssegur,J. (1996) Identification of MAP kinase domains by redirecting stress signals into growth factor responses. Science, 272, 1652–1655. [DOI] [PubMed] [Google Scholar]
- Brunner D., Oellers,N., Szabad,J., Biggs,W.H., Zipursky,S.L. and Hafen,E. (1994) A gain-of-function mutation in Drosophila MAP kinase activates multiple receptor tyrosine kinase signaling pathways. Cell, 76, 875–888. [DOI] [PubMed] [Google Scholar]
- Clark-Lewis I., Sanghera,J.S. and Pelech,S.L. (1991) Definition of a consensus sequence for peptide substrate recognition by p44mpk, the meiosis-activated myelin basic protein kinase. J. Biol. Chem., 266, 15180–15184. [PubMed] [Google Scholar]
- Deak M., Clifton,A.D., Lucocq,L.M. and Alessi,D.R. (1998) Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J., 17, 4426–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel K., Kotlyarov,A. and Gaestel,M. (1998) Leptomycin B-sensitive nuclear export of MAPKAP kinase 2 is regulated by phosphorylation. EMBO J., 17, 3363–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enslen H., Brancho,D.M. and Davis.R.J. (2000) Molecular determinants that mediate selective activation of p38 MAP kinase isoforms. EMBO J., 19, 1301–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M., Gotoh,Y. and Nishida,E. (1997) Interaction of MAP kinase with MAP kinase kinase: its possible role in the control of nucleocytoplasmic transport of MAP kinase. EMBO J., 16, 1901–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga R. and Hunter,T. (1997) MNK1, a new MAP kinase-activated protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J., 16, 1921–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin A.C. and Nebreda,A.R. (1999) A MAP kinase docking site is required for phosphorylation and activation of p90RSK/MAPKAPK-1. Curr. Biol., 9, 281–284. [DOI] [PubMed] [Google Scholar]
- Gonzalez F.A., Raden,D.L. and Davis.R.J. (1991) Identification of substrate recognition determinants for human ERK1 and ERK2 protein kinases. J. Biol. Chem., 266, 22159–22163. [PubMed] [Google Scholar]
- Holland P.M. and Cooper,J.A. (1999) Protein modification: docking sites for kinases. Curr. Biol., 9, 329–331. [DOI] [PubMed] [Google Scholar]
- Hunter T. (2000) Signaling—2000 and beyond. Cell, 100, 113–127. [DOI] [PubMed] [Google Scholar]
- Ip Y.T. and Davis,R.J. (1998) Signal transduction by the c-Jun N-terminal kinase (JNK)—from inflammation to development. Curr. Opin. Cell Biol., 10, 205–219. [DOI] [PubMed] [Google Scholar]
- Jacobs D., Glossip,D., Xing,H., Muslin,A.J. and Kornfeld,K. (1999) Multiple docking sites on substrate proteins from a modular system that mediates recognition by ERK MAP kinase. Genes Dev., 13, 163–175. [PMC free article] [PubMed] [Google Scholar]
- Kallunki T., Deng,T., Hibi,M. and Karin,M. (1996) c-Jun recruits JNK to phosphorylate dimerization partners via specific docking interactions. Cell, 87, 929–939. [DOI] [PubMed] [Google Scholar]
- Kyriakis J.M. and Avruch,J. (1996) Protein kinase cascades activated by stress and inflammation. BioEssays, 18, 567–577. [DOI] [PubMed] [Google Scholar]
- Lim Y.M., Nishizawa,K., Nishi,Y., Tsuda,L., Inoue,Y.H. and Nishida,Y. (1999) Genetic analysis of rolled, which encodes a Drosophila mitogen-activated protein kinase. Genetics, 153, 763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall C.J. (1995) Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell, 80, 179–185. [DOI] [PubMed] [Google Scholar]
- Millward T.A., Zolnierowicz,S. and Hemmings,B.A. (1999) Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem. Sci., 24, 186–191. [DOI] [PubMed] [Google Scholar]
- Muda M., Boschert,U., Dickinson,R., Martinou,J.C., Martinou,I., Camps,M., Schlegel,W. and Arkinstall,S. (1996) MKP-3, a novel cytosolic protein-tyrosine phosphatase that exemplifies a new class of mitogen-activated protein kinase phosphatase. J. Biol. Chem., 271, 4319–4326. [DOI] [PubMed] [Google Scholar]
- Muda M. et al. (1998) The mitogen-activated protein kinase phosphatase-3 N-terminal noncatalytic region is responsible for tight substrate binding and enzymatic specificity. J. Biol. Chem., 273, 9323–9329. [DOI] [PubMed] [Google Scholar]
- New L., Jiang,Y., Zhao,M., Liu,K., Zhu,W, Flood,L.J., Kato,Y., Parry,G.C. and Han.J. (1998) PRAK, a novel protein kinase regulated by the p38 MAP kinase. EMBO J., 17, 3372–3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- New L., Zhao,M., Li,Y., Bassett,W.W., Feng,Y., Ludwig,S., Padova,F.D., Gram,H. and Han,J. (1999) Cloning and characterization of RLPK, a novel RSK-related protein kinase. J. Biol. Chem., 274, 1026–1032. [DOI] [PubMed] [Google Scholar]
- Nishida E. and Gotoh,Y. (1993) The MAP kinase cascade is essential for diverse signal transduction pathways. Trends Biochem. Sci., 18, 128–131. [DOI] [PubMed] [Google Scholar]
- Pawson T. (1995) Protein modules and signalling networks. Nature, 373, 573–580. [DOI] [PubMed] [Google Scholar]
- Pawson T. and Nash,P. (2000) Protein–protein interactions define specificity in signal transduction. Genes Dev., 14, 1027–1047. [PubMed] [Google Scholar]
- Pierrat B., Correia,J.S., Mary,J.L., Tomas-Zuber,M. and Lesslauer,W. (1998) RSK-B, a novel ribosomal S6 kinase family member, is a CREB kinase under dominant control of p38α mitogen-activated protein kinase (p38αMAPK). J. Biol. Chem., 273, 29661–29671. [DOI] [PubMed] [Google Scholar]
- Pinna L.A. and Ruzzene,M. (1996) How do protein kinases recognize their substrates? Biochim. Biophys. Acta, 1314, 191–225. [DOI] [PubMed] [Google Scholar]
- Pritchard C.A., Samuels,M.L., Bosch,E. and McMahon,M. (1995) Conditionally oncogenic forms of the A-Raf and B-Raf protein kinases display different biological and biochemical properties in NIH 3T3 cells. Mol. Cell. Biol., 15, 6430–6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulido R., Zuniga,A. and Ullrich,A. (1998) PTP-SL and STEP protein tyrosine phosphatases regulate the activation of the extracellular signal-regulated kinases ERK1 and ERK2 by association through a kinase interaction motif. EMBO J., 17, 7337–7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricardo M., Biondi,R.M.F., Cheung,P.C., Casamayor,A., Deak,M., Currie,R.A. and Alessi,D.R. (2000) Identification of a pocket in the PDK1 kinase domain that interacts with PIF and the C-terminal residues of PKA. EMBO J., 19, 979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.J. and Cobb,M.H. (1997) Mitogen-activated protein kinase pathways. Curr. Opin. Cell Biol., 9, 180–186. [DOI] [PubMed] [Google Scholar]
- Schaeffer H.J. and Weber,M.J. (1999) Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol. Cell. Biol., 19, 2435–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.A, Poteet-Smith,C.E., Malarkey,K. and Sturgill,T.W. (1999) Identification of an extracellular signal-regulated kinase (ERK) docking site in ribosomal S6 kinase, a sequence critical for activation by ERK in vivo. J. Biol. Chem., 274, 2893–2898. [DOI] [PubMed] [Google Scholar]
- Sturgill T.W. and Wu,J. (1991) Recent progress in characterization of protein kinase cascades for phosphorylation of ribosomal protein S6. Biochim. Biophys. Acta, 1092, 350–357. [DOI] [PubMed] [Google Scholar]
- Tanoue T., Moriguchi,T. and Nishida,E. (1999) Molecular cloning and characterization of a novel dual specificity phosphatase, MKP-5. J. Biol. Chem., 274, 19949–19956. [DOI] [PubMed] [Google Scholar]
- Tanoue T., Adachi,M., Moriguchi,T. and Nishida,E. (2000) A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nature Cell Biol., 2, 110–116. [DOI] [PubMed] [Google Scholar]
- Treisman R. (1996) Regulation of transcription by MAP kinase cascades. Curr. Opin. Cell Biol., 8, 205–215. [DOI] [PubMed] [Google Scholar]
- Wang Z., Canagarajah,B.J., Boehm,J.C., Kassisa,S., Cobb,M.H., YoungP.R., Abdel-Meguid,S., Adams,J.L. and Goldsmith,E.J. (1997) The structure of mitogen-activated protein kinase p38 at 2.1-Å resolution. Proc. Natl Acad. Sci. USA, 94, 2327–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waskiewicz A.J., Flynn,A., Proud,C.G. and Cooper,J.A. (1997) Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J., 16, 1909–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilsbacher J.L., Goldsmith,E.J. and Cobb,M.H. (1999) Phosphorylation of MAP kinases by MAPK/ERK kinases involves multiple regions of MAP kinases. J. Biol. Chem., 274, 16988–16994. [DOI] [PubMed] [Google Scholar]
- Wilson K.P., Fitzgibbon,M.J., Caron,P.R., Griffith,J.P., Chen,W., McCaffrey,P.G., Chambers,S.P. and Su,M.S. (1996) Crystal structure of p38 mitogen-activated protein kinase. J. Biol. Chem., 271, 27696–27700. [DOI] [PubMed] [Google Scholar]
- Xia Y. and Karin,M. (1998) JNKK1 organizes a MAP kinase module through specific and sequential interactions with upstream and downstream components mediated by its amino-terminal extension. Genes Dev., 12, 3369–3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S.H., Whitmarsh,A.J., Davis,R.J. and Sharrocks,A.D. (1998) Differential targeting of MAP kinases to the ETS-domain transcription factor Elk-1. EMBO J., 17, 1740–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S.H., Galanis,A. and Sharrocks,A.D. (1999) Targeting of p38 mitogen-activated protein kinases to MEF2 transcription factors. Mol. Cell. Biol., 19, 4028–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., Strand,A., Robbins,D., Cobb,M.H. and Goldsmith,E.J. (1994) Atomic structure of the MAP kinase ERK2 at 2.3 Å resolution. Nature, 367, 704–711. [DOI] [PubMed] [Google Scholar]
- Zuniga A., Torres,J., Ubeda,J. and Pulido,R. (1999) Interaction of mitogen-activated protein kinases with the kinase interaction motif of the tyrosine phosphatase PTP-SL provides substrate specificity and retains ERK2 in the cytoplasm. J. Biol. Chem., 274, 21900–21907. [DOI] [PubMed] [Google Scholar]