

Abstract
ATP sulfurylases (ATPSs) are ubiquitous enzymes that catalyse the primary step of intracellular sulfate activation: the reaction of inorganic sulfate with ATP to form adenosine-5′-phosphosulfate (APS) and pyrophosphate (PPi). With the crystal structure of ATPS from the yeast Saccharomyces cerevisiae, we have solved the first structure of a member of the ATP sulfurylase family. We have analysed the crystal structure of the native enzyme at 1.95 Å resolution using multiple isomorphous replacement (MIR) and, subsequently, the ternary enzyme product complex with APS and PPi bound to the active site. The enzyme consists of six identical subunits arranged in two stacked rings in a D3 symmetric assembly. Nucleotide binding causes significant conformational changes, which lead to a rigid body structural displacement of domains III and IV of the ATPS monomer. Despite having similar folds and active site design, examination of the active site of ATPS and comparison with known structures of related nucleotidylyl transferases reveal a novel ATP binding mode that is peculiar to ATP sulfuryl-ases.
Keywords: adenosine-5′-phosphosulfate/ATP sulfurylase/crystal structure/sulfate activation/sulfur amino acid biosynthesis
Introduction
ATP sulfurylase (ATPS) (adenylsulfuryltransferase/ATP:sulfate adenylyltransferase; E.C. 2.7.7.4) is the first catalytic enzyme in a cascade of proteins that performs the activation of inorganic sulfate (SO42–) and its incorporation into organic compounds. ATPS catalyses adenylylsulfate synthesis, the first intracellular reaction in sulfate assimilation. Sulfate is activated with ATP to give adenosine-5′-phosphosulfate (APS) and inorganic pyrophosphate (PPi) according to the scheme:
MgATP + SO42– → MgPPi + APS
This reaction yields a molecule with a high energy mixed phosphoric–sulfuric acid anhydride bond, which is used for further sulfate activation and reduction in the cell. Thus, ATPS plays an important role in sulfate activation as a component of the sulfur amino acid (methionine/cysteine) biosynthesis pathway. This constitutes part of a global regulatory circuit, which controls the expression of enzymes that function both in the acquisition of sulfur from the environment and its assimilation (Marzluf, 1997). In sulfate-reducing bacteria, ATPS plays a key role not only in amino acid metabolism, but also in anaerobic dissimulation, where ATP production is linked to APS reduction to AMP and sulfite, which is further reduced to hydrogen sulfide (Gavel et al., 1998; Sperling et al., 1998).
In bacteria such as Escherichia coli, ATPS is a tetramer of heterodimers. Each heterodimer of K-12 is composed of two subunits, CysD and CysN (Leyh et al., 1988). The GTPase subunit CysN activates the catalytic subunit CysD by allosteric interactions (Wei et al., 2000). ATPS from sulfate-reducing bacteria and eukaryotic organisms such as yeast (Karamohamed et al., 1999), filamentous fungi (Foster et al., 1994) and plants (Leustek et al., 1994; Logan et al., 1996) have been described as monomers or homo-oligomeric complexes, which do not require GTP for activation. However, in higher eukaryotes, like the marine worm (Rosenthal and Leustek, 1995) and mammalian species (Li et al., 1995; Venkatachalam et al., 1998), gene fusion has occurred; ATPS and the APS kinase, which performs the final step of the synthesis of the high-energy sulfate donor PAPS (3′-phosphodenosine- 5′-phosphosulfate) by 3′-phosphorylation of APS, are arranged as subdomains on a single polypeptide chain. Together they compose the bifunctional PAPS synthetase, which in mammalian cells, besides its role in amino acid metabolism, is also important for the sulfation of proteins, carbohydrates, lipids, drugs and xenobiotics. The fusion of ATPS and APS kinase into one molecule represents an evolutionary trend towards more functional efficiency in sulfate activation and amino acid metabolism in higher organisms, providing a mechanism for channelling the unstable APS intermediate into the adjacent kinase reaction.
ATPS of the yeast S.cerevisiae is encoded by the MET3 gene on chromosome X (Cherest et al., 1987). In contrast to the related nucleotide-binding kinases, which catalyse the removal of the γ-phosphate of an NTP, it can be classified into the superfamily of α/β phosphodiesterases (pyrophosphorylases, ATP PPases) due to its catalytic activity described above. Similarities in its sequence, like the typical HxxH motif, also bring the ATPS into close proximity with the superfamily of nucleotidylyl transferases, such as the prototypical Bacillus subtilis cytidylyltransferase TagD/GCT (CTP:glycerol-3-phosphate cytidylyltransferase) (Park et al., 1997; Weber et al., 1999) or the related class I aminoacyl-tRNA synthetases (for review see Delarue and Moras, 1993), of which several structures are known. All these functionally characterized enzymes cleave the α-β-phosphate bond of an NTP and transfer the NMP to the second substrate.
Figure 1 shows the alignment of yeast ATPS with homologues from Penicillium chrysogenum, Archaeoglobus fulgidus and the C-terminal ATPS domains of maize (Zea mays), mouse and human PAPS synthetase isoforms, all of which show strong homologies. In particular, the correspondence of 66% of the amino acids from yeast ATPS to ATPS from the filamentous fungi P.chrysogenum, which is mainly evident for the first 400 amino acids, is notable. The C-terminal region of yeast ATPS, however, is very distinct from the fungal ATPS, which shows strong sequence homologies to the APS kinases, indicating that they may have evolved from APS kinase (MacRae and Segel, 1997). Although the higher eukaryotic PAPS synthetases from maize and mammals show sequence similarities of up to 70% with yeast ATPS, they do not have the C-terminal 100-amino-acid extensions of yeast and fungal ATPS.
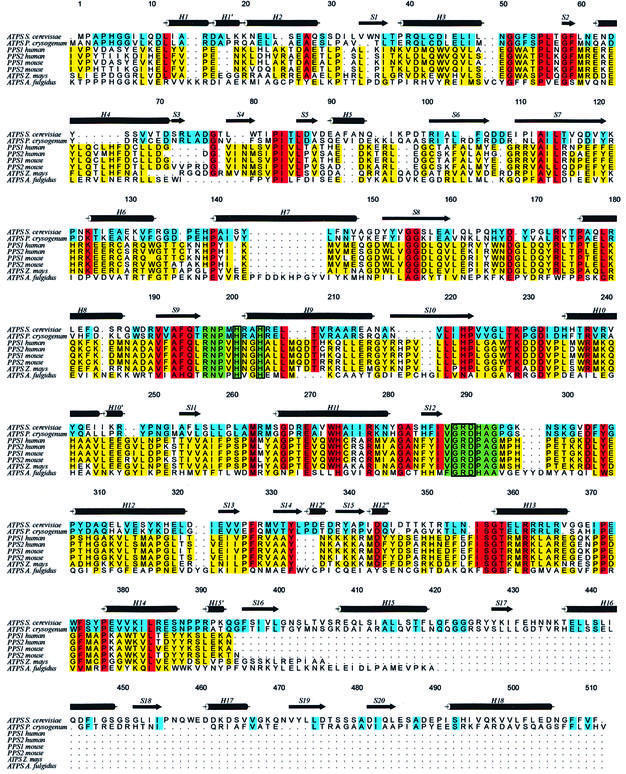
Fig. 1. Sequence alignment of ATP sulfurylase from S.cerevisiae with ATPS of P.chrysogenum, Z.mays and A.fulgidus, and the ATP sulfurylase subdomains of the two isoforms of mouse and human PAPS synthetase. Sequence identities between yeast and fungi ATPS are coloured in blue, and those between yeast ATPS and all others are in red. Less conserved regions are marked in yellow. The highly conserved RNP and GRD motifs are highlighted in green boxes, with the histidines of the HxxH motif drawn in additional rectangles. The secondary structure symbols are set according to the presented ATPS structure.
The identities of yeast ATPS to the PAPS synthetases of both mouse and human isoforms are 28% in all four cases, and still 20% to the less-related archaebacterium A.fulgidus. In spite of this, in all the presented sequences prominent motifs can be highlighted: the HxG/AH motif and the subsequent Leu207 of the ATPS sequence are present together with the preceding residues 190VxAFQxRNP, which are also strictly conserved in all the aligned sequences. Also, the module 288VGRDHAG, which from now on will be referred to as the GRD-loop, is highly conserved. It is known to play a crucial role in substrate binding, analogous to the PP-loop motifs of other nucleotidylyl transferases. Many site-directed mutagenesis studies with ATPS and PAPS synthetases of several organisms (Deyrup et al., 1999; Venkatachalam et al., 1999), as well as with the related cytidylyltransferases (Park et al., 1997), demonstrate that alanine mutants of the above specified residues produce either reduced activity or inactive enzymes. This strongly indicates that nucleotide binding as well as sulfurylase activity involves residues of these motifs. Additionally, a fourth highly conserved motif can be found: the 356ISGTxxR363 module is widespread among the aligned eukaryotic species, but has not yet been investigated for its detailed function.
To support and extend the extensive biological and biochemical studies, structural investigations were initiated to elucidate the architecture and functional features of ATPS, with the aim of identifying the active site and the catalytic residues involved in sulfate activation. In this work we present the purification, crystallization and determination of the three-dimensional (3D) structure of ATPS from S.cerevisiae.
Results and discussion
We have developed a procedure to purify 5–10 mg of ATPS from 500 g of yeast cells to electrophoretic homogeneity. The molecular weight of the purified protein was determined to be ∼58 kDa for the protomer. Further analysis has shown that ATPS is a homo-oligomer with a molecular weight of 350–400 kDa, suggesting a homohexameric assembly. Crystallization experiments with purified ATPS yielded trigonal crystals that belong to the spacegroup R32. The protein crystallizes reproducibly within 2 days from solutions containing Cd2+ as an additive, with average dimensions of 0.5 × 0.5 × 0.3 mm3, and with crystals diffracting up to 1.95 Å (native enzyme) and 2.6 Å (product complex), respectively. For the native crystals, the asymmetric unit contains one subunit of ATPS, resulting in a Matthew’s coefficient of 3.38 A3/Da (Matthews, 1968) and a solvent content of 64%. As shown in Table II, the crystals of the enzyme– product complexes contain two independent subunits in the asymmetric unit in the same trigonal R32 spacegroup as the native crystals, indicating a significant change in the crystallographic packing.
Table II. Refinement statistics.
| Native | APS complex | APS–PPi complex | |
|---|---|---|---|
| Space group | R32 | R32 | R32 |
| Cell constants | |||
| a | 187.1 | 185.9 | 186.0 |
| b | 187.1 | 185.9 | 186.0 |
| c | 115.9 | 223.7 | 223.7 |
| α | 90 | 90 | 90 |
| β | 90 | 90 | 90 |
| γ | 120 | 120 | 120 |
| Resolution range (Å) | 20.0–1.95 | 25.0–2.6 | 25.0–2.8 |
| Reflections in working set | 55 315 | 45 468 | 35 298 |
| Reflections in test set | 2835 | 2337 | 1801 |
| Rcryst (%) | 19.7 | 17.6 | 19.3 |
| Rfree (%) | 23.2 | 22.7 | 24.4 |
| Protein atoms (non-H) | 4077 | 8154 | 8154 |
| Nucleotide atoms | – | 54 | 63 |
| Heterogen atoms (non-H) | 73 | 120 | 120 |
| Solvent molecules | 566 | 763 | 600 |
| Average B-factor (Å2) | 35.4 | 35.1 | 35.9 |
| R.m.s.ds | |||
| bond lengths (Å) | 0.0104 | 0.0097 | 0.0068 |
| bond angles | 1.49 | 1.56 | 1.32 |
| ΔB (through bonds) (Å2) | 1.83 | 1.72 | 1.42 |
Rcryst free = Σ|Fobs – Fcalc|/Σ|Fobs|
The 3D structure of ATPS was determined by multiple isomorphous replacement (MIR) using tantalum bromide, mercury (thiomersal) and uranyl acetate as heavy-atom derivatives (Table I). An initial poly-alanine model was built in the MIR density at 2.7 Å resolution. By phase combination of the model and the MIR phases, and their expansion to 1.95 Å, the model could be completed using the amino acid sequence of S.cerevisiae ATPS from the strain S288C/FY1679 (M.de Haan et al., 1995). The resulting native structure contains all 510 residues, without the initial methionine, 11 metal ions, two sulfates, 11 acetate ions and one Tris molecule as ligands. The final R-factor for the native model is 19.6% (Rfree 23.1%). The com- plex with APS was subsequently solved using molecular replacement techniques and refined at 2.6 Å resolution with an R-factor of 17.6% (Rfree 22.7%). Analysis of the stereochemistry with PROCHECK (Laskowski et al., 1993) revealed that 91.2% of the dihedral φ/ψ angles lie in the most favoured regions of the Ramachandran plot, 8.4% in the additional allowed regions and 0.5% in the generously allowed regions. Further details are given in Materials and methods and in Table II.
Table I. Concentration, soaking time and data collection statistics.
| |
Native |
TaBr |
Hg1 |
URA |
APS |
PPi |
|---|---|---|---|---|---|---|
| Reactant | Ta12Br14 | Thiomersal | UO2Ac2 | MgATPγs | MgATPγs/KClO4 | |
| Concentration (mM) | 5 | 10 | 5 | 5 | 5/10 | |
| Soaking time (h) | 12 | 12 | 3 | 3 | 3 | |
| Asymmetric monomers | 1 | 1 | 1 | 1 | 2 | 2 |
| Wavelength (Å) | 1.04 | 1.2539 | 1.54 | 1.54 | 1.54 | 1.54 |
| Limiting resolution (Å) | 1.95 | 2.2 | 2.5 | 2.7 | 2.6 | 2.8 |
| Unique reflections | 55 315 | 35 064 | 26 716 | 21 297 | 45 486 | 35 298 |
| Last shell (Å) | 1.95–2.07 | 2.2–2.37 | 2.5–2.64 | 2.7–2.85 | 2.6–2.76 | 2.8–2.98 |
| Completeness (%) | 98.3 (98.6) | 96.3 (96.3) | 98.8 (98.8) | 99.6 (99.9) | 99.5 (97.3) | 95.5 (97.1) |
| Mean redundancy | 3.8 (3.7) | 8.5 (8.3) | 3.8 (3.7) | 5.3 (5.3) | 6.0 (5.6) | 4.1 (3.8) |
| Mean I/σI | 9.3 (2.7) | 7.4 (1.5) | 6.5 (1.1) | 5.6 (1.8) | 6.4 (1.8) | 5.8 (1.6) |
| Rmerge | 5.1 (28.0) | 6.8 (40.2) | 9.6 (64.6) | 12.2 (40.5) | 11.0 (41.3) | 12.8 (42.8) |
| Phasing power | – | 1.04 | 0.32 | 0.89 | – | – |
| Sites | – | 1 | 1 | 1 | – | – |
Rsym = Σ|I(h)i – <I(h)>|/Σ<I(h)>
Quaternary structure
The quaternary arrangement of ATPS is generated by the D3 symmetry of the R32 spacegroup, resulting in a homohexameric assembly, which had been suggested for the homologue of ATPS from P.chrysogenum. Its molecular weight of ∼350 kDa is in accordance with earlier solution studies. In the D3 symmetric structure, two trimeric rings assemble in a staggered configuration with a twist of 60°. ATPS exhibits overall dimensions of 150.6 × 133.3 × 81.6 Å3. Side and top views of the particle are shown in Figure 2A–C.
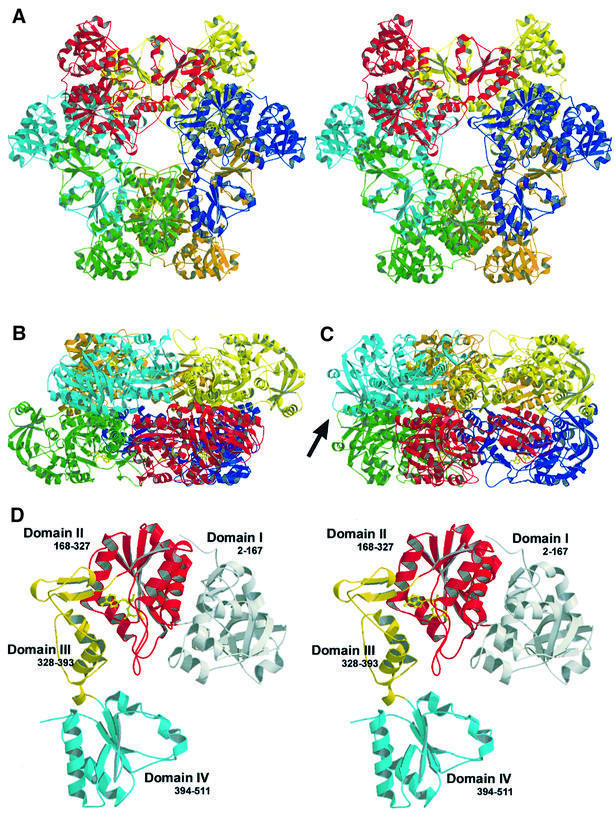
Fig. 2. (A) Stereo ribbon plot of the ATPS homohexamer, top view, and (B) side view. (C) The side view rotated 30° around the 3-fold axis vertical in the paper plane, with a black arrow marking the non-equivalence of the rings in the complex structure. (D) Stereoview of the ATPS protomer in ribbon presentation (top view). The four individual domains are labelled in grey, red, yellow and cyan (from domain I to IV). Domain II is shown with an APS molecule (yellow, ball-and-stick) in the active site.
Monomer structure
As illustrated in Figure 2D, the U-shaped protomer consists of four easily distinguishable domains and is characterized by a high ratio of regular secondary structure elements.
Domain I is built by the N-terminal part of the peptide chain (residues 2–167) and comprises as a main motif a β-barrel, which is formed by five antiparallel strands in a 1-3-4-5-2 topology. It is topologically similar to the pyruvate kinase β-barrel domain (Figure 3A), but in contrast to the pyruvate kinase β-barrel, which is characterized by an all-β conformation, ATPS shows a more complex folding topology with several helical insertions such as an α–turn–α module (helices H6 and H7) and an additional short β-hairpin motif formed by S3 and S4 after helix H4, making it more similar to an α/β protein. The inner cage of the β-barrel of ATPS is filled with hydrophobic residues to build a large hydrophobic network. Domain I and the adjacent domain II face each other with a large planar surface of mostly polar or charged residues. These interactions are predominantly mediated by hydrogen bonds and salt bridges. Contacts between the residues of helix H3 and the coiled region preceding H11 of the interdomain show a more hydrophobic character.
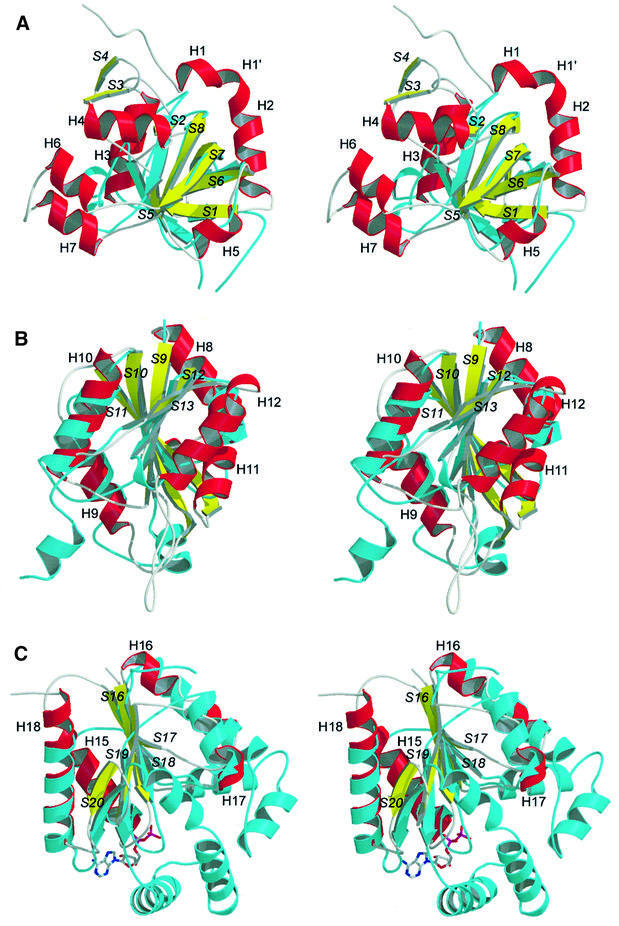
Fig. 3. Stereoviews of ATPS domains I, II and IV (helices, red; strands, yellow; coiled regions or turns, grey) in ribbon presentation showing a structural comparison of the typical fold motifs of the single domains with the best-fitting members of the related superfamilies (cyan). (A) Domain I superimposed with the pyruvate kinase β-barrel domain. (B) Superimposition of domain II with GCT, a typical member of the superfamily of nucleotidylyl transferases. Both polypeptides show the typical α/β-fold of the nucleotidylyl transferases. (C) Overlay of the structure of domain IV with yeast uridylate kinase, complexed with ADP. Both polypeptides show the typical nucleotide and nucleoside kinase fold. All ATPS secondary structure elements are labelled.
Domain II (residues 168–327) comprises the active site and the substrate binding pocket. It shows a typical right-handed α/β-fold in three layers (Rossmann-like fold; Rossmann et al., 1974), which is shared by members of the superfamilies of adenine nucleotide α hydrolases and of nucleotidylyl transferases, e.g. GMP synthetase or the class I aminoacyl-tRNA synthetases and cytidylyltransferases. Five twisted parallel β-strands (S9–S13) with a 3-2-1-4-5 topology are flanked by five α-helices to form the typical α/β/α supersecondary structure. Figure 3B shows a ribbon plot superimposition of ATPS domain II with the structure of the CTP:glycerol-3′-phosphate cytidylyltransferase from B.subtilis (Weber et al., 1999), a typical member of the nucleotidylyl transferase α/β phosphodiesterase superfamily. Using TOP (Lu, 2000) for structure similarity search, CGT was found to fit best with a root mean square deviation (r.m.s.d.) of 1.57 Å (matching residues). But, in addition to these common elements, ATPS exhibits a large loop motif of β-turns between S12 and the helix H12 (residues 288–306). Domain II is packed against domain III mainly by charged and hydrophilic residues of H9 and H10, which are hydrogen bonded to extended parts of the whole domain III.
Domain III consists of the residues 328–393 and is a short domain of 65 amino acids. It links the C-terminal domain IV with domains I and II, and its fold shows no similarities to known protein families (Figure 2D). Residues 327–331 participate in the formation of the upper part of the ATPS substrate binding pocket. The highly conserved 327PFR motif between strands S13 and S14 displays some flexibility when the enzyme binds ATP, causing a conformational change of only a few amino acids, which is associated with the rigid body motion of the entire domains III and IV. Flexibility is also manifested in the electron density of the coiled region after the antiparallel strands S14 and S15 (residues 347–352), which shows weakly defined spheres in the native structure, with temperature factor values of ∼75 Å2, e.g. for Lys350, but is well defined in the product complex.
The C-terminal domain IV again displays an α/β-fold (residues 394–511) with five twisted parallel β-strands (S16–S20), but with a 2-3-1-4-5 topology. This structural classification reveals its close relationship to the superfamily of P-loop-containing nucleotide triphosphate hydrolases and the family of nucleotide kinases, indicating a common evolutionary origin with APS kinase. Typical members of this family are yeast guanylate and uridylate kinase. Despite their common fold topology, the superimposition of domain IV with the structure of uridylate kinase from S.cerevisiae (Figure 3C) shows that the kinase chain possesses additional extensions, such as a multiple α-helix motif between the strands S19 and S20. This large region participates in ATP binding and the formation of the kinase reaction centre. The lack of this module, which is also present but disordered in the APS kinase structure (MacRae et al., 2000), provides striking evidence for the absence of PAPS binding capability and a putative kinase activity of ATPS. The C-terminus at Phe511 binds to Pro250 by hydrophobic side-chain interactions, and with its carboxylate group to Arg248-Nζ of the neighbouring protomer in the same ring.
Within the homohexamer, interactions of the protomers are predominantly mediated through numerous hydrogen bonds and salt bridges, including a broad network of solvent molecules. In addition, we found a number of metal ions contributing to the formation of the monomer– monomer interfaces. They are all characterized by clearly defined high electron density spheres, short distances to the coordinating ligands and high coordination numbers. Because of the presence of 25 mM cadmium in the crystallization medium, these metal ions were interpreted as cadmiums. Cadmium Cd515, for example, which connects one protomer of the upper ring with a subunit of the lower ring of the same particle, is coordinated to Glu182-Oε and to Gly171-O from the upper subunit, which makes further hydrogen bonds to Lys174-Nε. Both residues are linked to the acetate Acy533 from the protomer of the lower ring. Metal ions also connect interfaces to neighbouring subunits of the same ring, such as Cd516, which is 6-fold coordinated by the carboxylate oxygens of two acetates, Acy527 and Acy534, to the Asp189 and finally to His494-Nε, which is contributed from the symmetry-related subunit. Some of these metal sites may play a role in physiological conditions, whereby calciums replace the cadmium ions. Two more metal sites are located on the surface of the molecule. Due to its coordination to the carboxylate oxygens of the highly conserved Asp168, as well as to a sulfate ion, and to His235-Nε and His236-Nε at distances of 2.42 and 2.37 Å, respectively, the first metal Cd512 was interpreted as a cadmium that may be zinc or cobalt under physiological conditions, in accordance with previously described zinc binding sites in ATPS of sulfur-reducing bacteria (Gavel et al., 1998). Being located far away from the active site cleft, it does not seem to fulfil any role in the catalytic mechanism or in substrate binding, but it may contribute to the structural integrity of the monomer and stabilize the complex structure. Additionally, the Cys43-S and the Pro39-O serve, along with four water molecules, as ligands for cadmium Cd513 to form a slightly distorted octahedral complex. Cadmium Cd514, however, mediates interactions between the hexamers in the crystallographic packing and is coordinated to the carboxylate oxygens of Glu22, to Leu18-N, as well as to the acetate Acy526 and to one water molecule. From the other subunit, His319-Nε serves as a ligand completing the octahedral coordination sphere. Other metals found in the density are supposed to be smaller cations like sodium, calcium or magnesium, compensating negative charges on the protein surface. Nevertheless, they are inconsistent throughout the examined structures and, therefore, not described in detail.
The active site and nucleotide binding
As shown in Figure 4, the active site lies as a deep, bowl-like cleft in the centre of the ATPS protomer. It consists mainly of the domain II secondary structure elements H9, S9, S10 and S12, as well as the H9 preceding RNP-loop from Phe194 to His201 and the GRD-loop, which are both highly conserved. Moreover, the domain III residues Pro327, Phe328 and Arg329, together with Met330 and Val331, contribute to the formation of the cleft, which is open to the top of the molecule but faces the inner compartment of the U-shaped protomer (Figure 5A). It reaches maximum dimensions of 19.5 Å in length and 17.5 Å in width, and has, unlike some other nucleotidylyl transferases, no contact with symmetry-related subunits. The funnel directing into the cleft is lined with a bundle of positively charged residues to build an anion hole for the incoming nucleotide. While the bottom and side walls of the cleft are mainly formed by either hydrophilic or positively charged residues, which are supposed to play a crucial role in phosphate/sulfate binding or even in the catalytic mechanism, the back of the pocket is hydrophobic in character, consisting of the highly conserved residues Phe194, Leu207, Ile287, Val288, Gly289, His292, Phe328 and Met330. These residues interact together with the uncharged residues Val191–Ala193 as a network in the inner compartment of the domain to compose the pocket for the nucleosyl moiety of the ATP/APS.
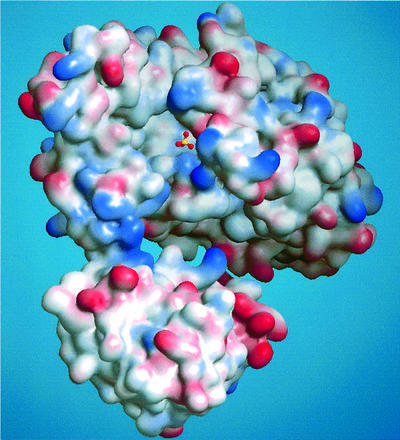
Fig. 4. Electrostatic surface potential plot of the ATPS protomer (positive, blue; negative, red) showing the entrance to the active site of the native molecule with a sulfate located in the substrate binding pocket. The funnel leading into the bowl-like cleft is mainly positively charged.
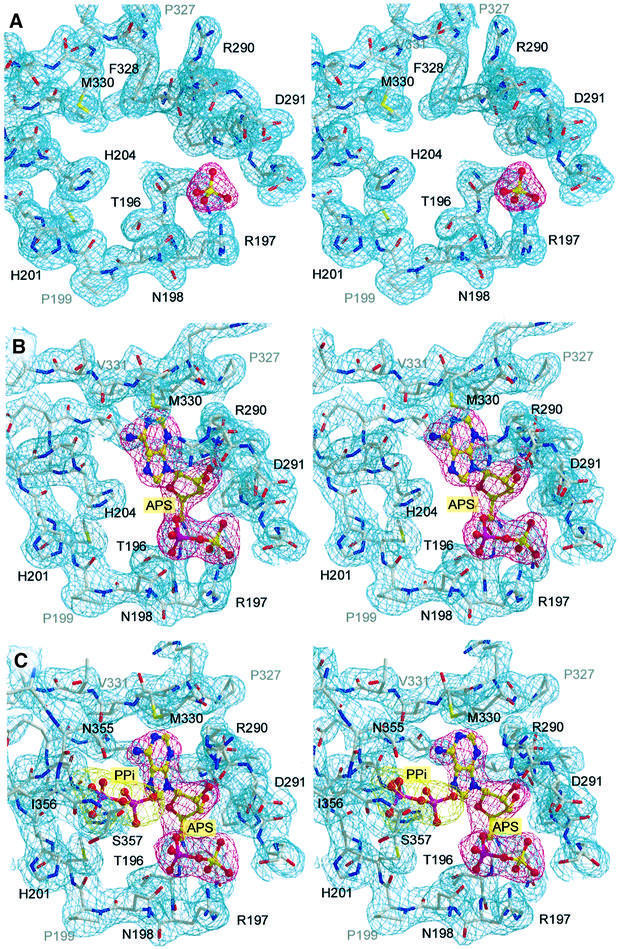
Fig. 5. Active site residues of the apo-protomer liganded with sulfate at 1.95 Å (A), the binary enzyme product complex with APS at 2.6 Å (B), and the ternary product complex with an additional pyrophosphate at 2.8 Å (C). All atoms are shown with the final 2Fo – Fc electron density contoured at 1.0σ.
The crystal structure of ATPS was initially solved in the absence of nucleotide; however, the crystallization required 25 mM CdSO4 as an essential additive. Due to the presence of sulfate in the crystallization buffer, a sulfate, which is one of the substrates of ATPS, along with several water molecules were found in the active site (Figures 4 and 5A). With an average B-factor of 37 Å2, the sulfate ion exhibits well-defined density and is coordinated by three amino acids and several solvent molecules: O1 is hydrogen bonded only to WAT779, whereas O2 is linked to Ala293-N and a water molecule. The Arg197-Nζ as well as one water molecule interacts with O3, whereas the oxygen O4 shows a hydrogen bond to Gln195-Nε. Thus, the negative charge of the sulfate appears well balanced and we assume that the sulfate occupies the regular substrate binding site.
The adenylylsulfate complex
In order to obtain further information about substrate binding and the mechanism of sulfate activation, we attempted to determine the structure of ATPS complexed with its second substrate ATP. We performed soaking experiments with the apo-crystals and the ATP analogues, MgATPγs and MgATPαs, which are both unhydrolysable or at least very slowly hydrolysed to AMP or ADP, respectively. Surprisingly, we obtained identical results from both trials: the 2Fo – Fc Fourier map, calculated using the phases from the refined model of ATPS, shows strong positive density in the active site cleft, which can clearly be fitted with an adenosyldinucleotide molecule lacking the γ-phosphate (Figure 5B). Two possible explanations for this observation could be the spontaneous hydrolysis of both ATP analogues in solution, or disorder of the γ-phosphate in the crystal packing. But, as shown in Figure 5B and in the schematic drawing (Figure 6), the nucleotide is well coordinated by the surrounding enzyme residues so that the pocket does not allow free rotation of the γ-phosphate. Also, the hydrolysis of the relatively stable ATP analogues in solution or by enzymatic catalysis does not seem to be a very plausible explanation for this observation, especially since ATPS has no phosphorylase activity. It is more likely that the enzymatic reaction has occurred in the crystal and that the density represents the product APS. An average B-factor of 31.3 Å2 for the whole APS molecule compared with a main chain B-factor of 32.1 Å2 indicates strong binding. The APS adopts an open L-shaped conformation, with the adenosyl moiety protruding deeply into the space between helix H9 and the β-strands S9 and S12. The sulfate group, oriented to the right side of the α-phosphate, is coordinated to Gln195- Nε, Arg197-Nζ and Ala293-N in the same pocket, as described above for the native state. The α-phosphate is fixed between the residues Thr196, Arg197 and Asn198. Whilst Arg197 coordinates to the α-phosphate oxygen via its peptide nitrogen, Thr196 and Asn198 form hydrogen bonds with their side-chain hydroxyl groups. The conserved Gly289 of the GRD-loop allows close approach of the main chain to the APS ribose moiety, with the contacts to the ribose unit being formed by backbone interactions of Gly289-N and His292-O to the 3′-oxygen and of Arg290-O to the 2′-oxygen, respectively. Additionally, the adenyl moiety is associated with Val331-N and Met330-S by hydrogen bonds and shows van der Waals contacts to numerous other residues. Further details of these interactions are depicted schematically in Figure 6.
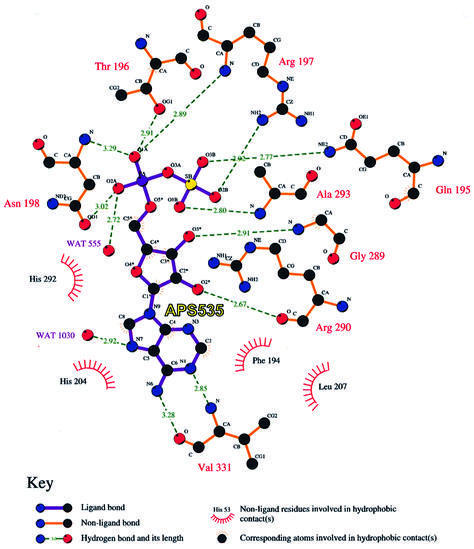
Fig. 6. Schematic drawing of the coordination of APS in the binding pocket, its hydrogen bonding and hydrophobic interactions with solvent molecules and the residues of ATPS. Bond distances are given in Å.
Nucleotide binding causes a significant rigid body displacement within the ATPS hexameric assembly. This rearrangement of protomer subdomains and, accordingly, of the entire enzyme complex seems to be caused by one single amino acid of the 327PFR motif (Figure 7). In the absence of a nucleotide, the phenyl ring of Phe328, which marks a hinge point for the domain movements, is stuck deeply in the hydrophobic adenyl binding site. When ATP binds, the phenyl ring is displaced from its position in the unliganded molecule and is pushed out to the top of the pocket. The flipping of the Phe328 by 90° induces a reorientation of the backbone and side chains of the neighbouring Pro327 and Arg329, with residue Met330 being shifted by ∼5 Å and creating, together with the side chain of Arg290, a binding site for the adenyl group of ATP in the pocket. From these residues onwards, domains III and IV move by ∼2–3 Å as rigid entities towards domains I and II (r.m.s.d. of all atoms 2.8 Å). This displacement mechanism may serve for substrate recognition but also has an effect on the protomers in the two rings of the hexamer. Although both protomers show electron density for the APS molecule, the extent of the conformational change is slightly different, with an r.m.s.d. of all atoms of 0.97 and 0.33 Å for the Cα-trace. This means that they are no longer crystallographically equivalent (Figure 2C, black arrow) and that two subunits now constitute the asymmetric unit, which almost leads to a doubling of the third cell axis (Table II). The crystallographic dyad axis relates two stacked hexamers. This non-equivalence may be caused by asymmetric ligation of ATP/APS similar to PPAT (Izard and Geerlof, 1999). However, this possible effect of half site occupied hexamers is very small and only indicated by the raised B-factors of the APS molecule in one of the two subunits in the asymmetric unit.
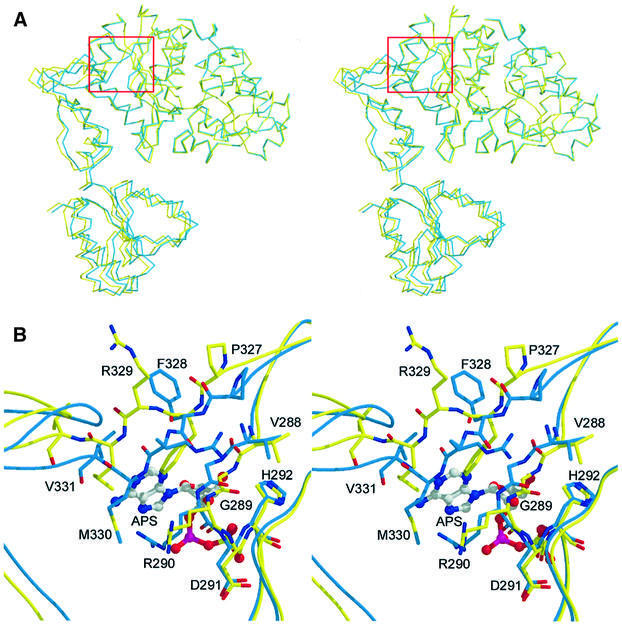
Fig. 7. (A) Superimposition of the native (yellow) and a complex (blue) protomer showing the rigid body displacement of the domains III and IV. The red box marks the centre of the hinge point. (B) Close-up stereoview of (A) showing the reaction centre with a nucleotide in the active site. Phe328 flipping by 90° causes the displacement of Pro327, Met330, Val331 and all following residues of the molecule.
To obtain the enzyme–substrate analogue complex, further X-ray investigations using 10 mM potassium chlorate, which has been considered to be a competitive inhibitor for ATPS activity (Schriever et al., 1997), were carried out, mixed with 5 mM MgATPαS in the buffer. The resulting electron density resembles that of the binary complex, with the exception of new density in the active site cleft (Figure 5C), which can clearly be interpreted as a pyrophosphate molecule. Forming the ternary complex E⋅APS⋅PPi, it is localized in the middle of the bowl. The APS and PPi phosphates are at a distance of 6.2 Å from each other. We could also observe additional weak electron density at His201-Nε, which could be interpreted as a chlorate molecule, which could be hydrogen bonded or even covalently bound to the nitrogen. Thus, the inhibition could be an effect of blocking His201 and thereby preventing the exit of the PPi molecule. The PPi is well hydrogen bonded to APS-N6 and -N7, and to His204-Nε. On the other side it binds to N355-Oδ and -Nδ, and to Ile356-O, which belongs to the conserved ISGT motif. This module builds a partial lid on the top of the active site bowl and may contribute to additional pyrophosphate binding and release during the reaction cycle. This new observation supports the suggestion of the presence of APS in the enzyme complex.
Conserved motifs and comparison with other nucleotidylyl transferases
As mentioned above, the active site domain II of ATPS exhibits distinct features, like typical fold topologies and active site design, resembling that of other nucleotidylyl transferases. Superimposition of the structure of the ATPS binary product complex with the structure of TagD/GCT from B.subtilis reveals strong similarities, particularly in the active site region between H9 and S9, and also in the 327PFR motif-containing loop region after S13 (Figures 5A–C and 8A). As a result, the nucleotidylyl moiety of APS in the ATPS complex structure possesses the same orientation in the pocket as CTP in the active site of GCT (Figure 8A). Although all nucleotidylyl transferases exhibit α/β-pyrophosphorylase activity, the enzymes of these families and superfamilies serve different biological purposes and have, for this reason, a broad range of further substrates for metabolic conversion with nucleoside triphosphates. Consequently, ATPS displays neither the P-loop GxxGxGKT/S motif of the related nucleotide and nucleoside kinases (Saraste et al., 1990; Bork and Koonin, 1994), nor the typical PSB-loop motif TYPKSGT of the sulfotransferase family (Kakuta et al., 1997; Bidwell et al., 1999), which are all known to bind nucleotides like ATP or PAPS. Additionally, ATPS exhibits no homology to the fingerprint motifs of the related cytidylyltransferases, like the 113RTEGISTT, 63RYVDEVI or 8GTFDLL motifs, the latter preceding the HWGH-box of GCT from B.subtilis (Weber et al., 1999). Instead of this, ATPS possesses, apart from the typical HxxH-box, its own fingerprint motifs, such as the highly conserved 197RNP-, 289GRD- and 327PFR-loops, including the 356IRSGT region in the intermediate domain III. Due to these special modules, the architecture of the active site and the substrate specificity of ATPS become unique and a considerably different nucleotide binding mode is observed. Accordingly, not only the phosphate/sulfate binding of APS in ATPS is highly modified. For example, in ATPS as well as in GCT, an arginine residue, which is invariant in many nucleotidylyl transferases, is coordinated to the respective nucleotide (Figure 8). But, while Arg113 of the extended 113RTEGISTT-loop fulfils this function in the GCT structure, in the ATPS structure, the structural counterpart Arg329, a residue of the conserved 327PFR motif, is exposed to the solvent and is not involved in nucleotide binding. Instead, Arg290 from the 289GRD-loop binds to the nucleotidylyl moiety of APS and may also promote transition state stabilization by binding to the β-phosphate of ATP. Besides this, the large loop from Asp291 to Gly306 is not present in the GCT structure (Figures 3B and 8A). Directing towards the active site, this loop is also conserved in the PAPS synthetases and, due to its positively charged character, may have a function in channelling the reaction product APS to the APS kinase reaction centre (Figure 4).
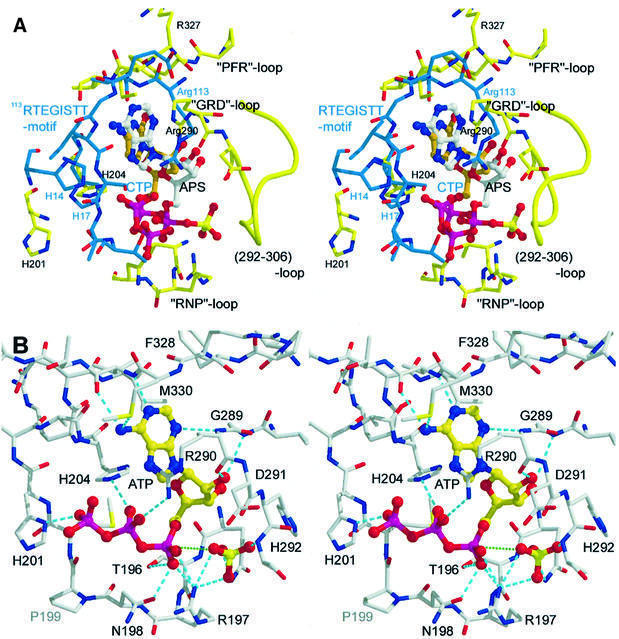
Fig. 8. (A) Stereoview of the superimposition of the active site residues of GCT (blue) and ATPS (yellow). The carbons of CTP are marked in orange. (B) Stereoview of the active site of ATPS with sulfate and a modelled ATP molecule in the typical horseshoe conformation. Hydrogen bonds are shown in cyan; the direction of the nucleophilic attack of the sulfate is marked in green.
Catalytic residues, mechanism and stereoselectivity
The active site residues of ATPS, Thr196, Arg197 and Asn198 are well coordinated to the α-phosphate of APS by hydrogen bonds (Figure 6). However, the two histidines of the HRAH motif, which bind to the α- and β-phosphates of a nucleotide in many nucleotidylyl transferases, are located in the ATPS product complex far away from the α-phosphorus of the APS, with a distance of 5.7 Å (Figures 5B and C, and 8A). This suggests that His201 and His204 of ATPS are not directly involved in the cleavage of the α-β-phosphodiester bond, but play a role in pyrophosphate binding and stabilization of a productive ATP conformation, as described for the related class I aminoacyl-tRNA synthetases (Leatherbarrow et al., 1985; Perona et al., 1993). Based on the orientation of the adenosyl moiety in the active site of the product complex and the sulfate position in the native structure, we modelled an energy-minimized ATP molecule showing the typical U-shaped (horseshoe) conformation resembling that of numerous nucleotidylyl transferases (Figure 8B). But in sharp contrast to the coordination of CTP in the active site of GCT, ATP is hydrogen bonded with its β-phosphate-O1 to His204-Nδ and with -O3 to Arg290-Nζ, whereas His201-Nδ coordinates to O4 of the γ-phosphate. As typical for ATP-binding enzymes, a Mg2+ ion could coordinate to the β- and γ-phosphate groups (Garcia et al., 1990) but has not been identified in the crystal structure. Due to the horseshoe-like conformation of ATP in the pocket, this model allows an ‘in-line’ nucleophilic attack of the adjacent sulfate (Figure 8B). In support of the kinetic and stereochemical investigations of Zhang (1999), our structural data confirm a substitution mechanism with stereochemical inversion at the α-phosphorus leading directly to the formation of APS, with PPi as leaving group and without covalent adenylyl– enzyme intermediates (Izard and Geerlof, 1999; D’Angelo et al., 2000). The enzyme provides suitable binding sites for its substrates, ATP and sulfate, brings the reactive groups in close contact and promotes the stabilization of the nucleotide conformation most favourable for the ‘in-line’ attack of the sulfate. During the reaction cycle, it stabilizes the pentavalent transition state of the reactants. Unlike numerous other nucleotidylyl transferases, such as GCT or the class I aminoacyl-tRNA synthetases, where an active proton-donating contribution of the HxxH histidines to the α-β-phosphate hydrolysis mechanism has been discussed, our model confirms that the histidines of the HRAH motif of ATPS bind and stabilize the pyrophosphate group before and during hydrolysis of the α-β-phosphodiester bond, and finally assist in the dissociation process.
Finally, the structure of the homohexameric ATPS is the first structure of a member of the ATP sulfurylase family. According to the fold of the active site domain II and its catalytic activity, ATPS can be classified as a new member of the nucleotidylyl transferase α/β phosphodiesterase superfamily. But, containing three independent folding topologies in one peptide chain, this unique composition of the protomer may suggest a new class of α/β proteins. The structure and location of domain IV within the ATPS protomer confirms its evolutionary relationship to the related APS kinase, but raises questions about its function in ATPS. The structure of the ternary E⋅APS⋅PPi complex rationalizes many features of the nucleotide binding and gives clues to a deeper insight into the catalytic mechanism of ATPS. Furthermore, this work presents a starting point for structural and functional studies on the more complex bifunctional PAPS synthetase, where PAPS synthesis is performed in a concerted reaction system that synthesizes APS and channels it to the APS kinase domain for the final PAPS formation.
Materials and methods
Purification, isolation and characterization
ATPS was natively purified from S.cerevisiae using a modified version of a previously described protocol (Groll et al., 1997). Five hundred grammes of cells were suspended in buffer A (20 mM Tris–HCl pH 7.5, 1 mM EDTA and 1 mM NaN3) at a 1:1 ratio to give a creamy suspension. For cell lysis, the suspension was mixed with an equivalent volume of glass beads (diameter 0.5 mm) and subjected to a cell disintegrator (Biomatik, München) in 100 ml aliquots. After 10 min of cell breakage (4°C, 5000 r.p.m.), the suspension was filtered to remove the beads and then centrifuged (4°C, 10 000 g, 60 min). The supernatant was ultracentrifuged at 4°C and 140 000 g for 60 min. The centrifugation yielded a clear, deep yellow solution containing the soluble protein. After combining the extract, ATPS was purified to homogeneity in a four-step procedure using ion exchange and affinity chromatography as well as gel filtration. The extract was applied to a 300 ml Q-Sepharose column equilibrated with buffer B (buffer A with 250 mM NaCl). The column was washed with buffer B and proteins were eluted with a linear gradient of 250–800 mM NaCl in a total volume of 1 l (4°C, flow rate 10 ml/min, 10 ml fractions). ATPS eluted at 450 mM NaCl as monitored by 12% SDS–PAGE. Fractions containing ATPS were pooled and loaded on a 100 ml ceramic hydroxyapatite column (Biorad, München) equilibrated with 10 mM potassium phosphate pH 7.5. After washing the column with 20 mM potassium phosphate pH 7.5, ATPS eluted at 100 mM phosphate in a 1 l gradient (10–500 mM K2HPO4/KH2PO4, 4°C, flow rate 4 ml/min, 10 ml fractions). The combined fractions were dialysed against buffer C (20 mM Tris–HCl pH 7.5, 10 mM MgCl2) and concentrated to 10–20 mg/ml by ultrafiltration using a 100 kDa polysulfone membrane (Sartorius, Weinheim).
The concentrated sample was loaded on a 30 ml affinity column (Reactive Red, Sigma), pre-equilibrated at 4°C with buffer C. The column was washed with buffer C and elution performed with a linear 200 ml gradient of 0–1000 mM NaCl. ATPS eluted at 500 mM sodium chloride. The flow rate was 1.5 ml/min and 10 ml fractions were collected. The fractions were combined, dialysed against buffer A and concentrated to 20 mg/ml by ultrafiltration using 10 kDa Centripreps and Centricons (Amicon, Hellsdorf). The protein solution was then loaded on a Superose 6 column (Pharmacia) pre-equilibrated with buffer A. The gel filtration was performed at an FPLC station (ÄKTA, Pharmacia) at room temperature with a flow rate of 0.5 ml/min. ATPS was manually collected and stored at 4°C. The size of the protomer was determined by 12% SDS–PAGE, and that of the protein complex by native PAGE and gel filtration.
Crystallization was carried out at 18°C using the hanging drop vapour diffusion method. The protein concentration was 20 mg/ml in buffer A. After mixing protein and reservoir solution (0.5 M NaOAc, 50 mM HEPES pH 7.5, 25 mM CdSO4) in a 2:1 ratio, crystals were obtained after 2 days. For cryo experiments, crystals were transferred from the crystallization drop to the reservoir buffer with 20% glycerol as cryo protectant. After 3 h of soaking, crystals were flash-frozen at 100 K in a nitrogen Oxford cryo system using a nylon loop. Heavy-atom derivative search was performed at 18°C by dissolving heavy-atom reagents in the cryoprotectant buffer. After 3 h of soaking, crystals could be mounted in the cryo stream. Additional soaking experiments with ATP analogues (5 mM MgATPγs/MgATPαs) and potassium chlorate (10 mM) were carried out under the same conditions as described above.
Crystallographic data collection
All X-ray datasets were collected at 100 K, in-house on a 345 MAR Research Imaging Plate detector, using a Rigaku RU200 rotating-anode X-ray generator (λ = 1.54 Å), and at the wiggler beamline BW6 at DESY/Hamburg with synchrotron radiation of different wavelengths, using the MAR Research CCD detector (Marresearch, Hamburg). Both systems are supplied with a nitrogen Oxford cryo system. The diffraction data were processed with the MOSFLM program package (Leslie, 1991). Further data reduction and truncation of structure factors were performed with the CCP4 suite (CCP4, 1994). Statistics for all used datasets are given in Table I.
Structure determination and refinement
The native structure was solved by MIR using heavy-atom derivatives of the enzyme of tantalum bromide, mercury (ethyl mercury thiosalicylate/thiomersal) and uranyl acetate. For crystallographic data calculation, the CCP4 program suite (CCP4, 1994) was used. Initial heavy-atom sites of tantalum cluster focal point at 4 Å were determined by difference Patterson maps, vector verification and difference Fourier maps using the program RSPS (CCP4, 1994). Mercury and uranium sites were determined and verified by cross phasing with the localized tantalum cluster position. Heavy-atom positions, occupancies, B-factors and initial phases were calculated, including the anomalous diffraction data of all three derivative datasets. The refinement of all parameters and the first phases were calculated with MLPHARE (CCP4, 1994) and resulted in an overall figure of merit of 0.38. The initial 2.7 Å Fourier map was improved by solvent flattening using SOLOMON (CCP4, 1994). The first secondary structure elements could be built in this map as an incomplete poly-alanine model. All building steps were carried out with MAIN (Turk, 1992). With this first model and the derivative phases, an improved phase combined Fourier map could be calculated, from which the initial amino acid side chains and thus the peptide sequence could be derived. A successive cyclic procedure of manual model building and phase combining of MIR and model phases, combined with phase expansion to 1.95 Å, led to the first complete model of the ATPS protomer. The asymmetric unit contains one molecule of ATPS with a solvent content of 64%. This molecule was used as the starting model for the refinement and density modification using 2Fo – Fc and Fo – Fc maps at 1.95 Å resolution, which were calculated with the CNS program package V1.0 (Brunger et al., 1998). The rebuilding was alternated with positional refinement, simulated annealing, and positional and individual B-factor refinement of the model. After finishing the refinement of the protein model, metal ions, ligands and water molecules were added, the latter automatically using CNS applications and final visual inspection. The refinement steps included the optimization of the weighing factors of the X-ray term and led to a final R-factor of the native molecule of 19.6% (Rfree 23.1%).
The diffraction data of the enzyme–product complex with APS and with PPi were processed in an identical manner to the apo-crystal described above. Initial phases of the complex were obtained by molecular replacement. Only a truncated version (residues 1–330) of the refined coordinates of the native molecule was applied as search model for Patterson search techniques MOLREP (CCP4, 1994). The translation function yielded one solution with 26.9σ above the mean, clearly superior to the next peak (13.9σ). From the calculated translation function, the position of the first monomer was obtained (correlation coefficient 22.3%, R-factor 54.4%). The rotation function yielded the position of the second monomer in the asymmetric unit (final correlation coefficient 39.5%, final R-factor 49.9%). After manual completion of the two molecules, the initial model was improved by rigid body refinement, positional refinement and simulated annealing at 2.6 Å resolution using CNS applications, and the Engh and Huber parameters (Engh and Huber, 1991). The model of the APS was built in the density using MAIN. Manual rebuilding and further refinement steps, including the completion of the model with metal ions, ligands and 763 water molecules, resulted in a final R-factor of 17.6% (Rfree 22.7%). The additional electron density of the potassium chlorate soaking experiments was fitted with a pyrophosphate molecule. A summary of the refinement statistics is shown in Table II. The model with the substrate ATP was built using SYBYL (Tripos Inc., St Louis, MO). The atomic coordinates and structure factor amplitudes for the native ATPS structure, the binary and the ternary product complex have been deposited in the PDB with the entry codes 1G8F, 1G8G and 1G8F.
Figure preparation
Figure 1 was prepared with ALSCRIPT (Barton, 1993) using a CGC sequence pile-up. Figures 2, 3, 7 and 8 were prepared with MOLSCRIPT (Kraulis, 1991) and rendered with RASTER3D (Merritt and Murphy, 1994). Figure 4 was produced with WEBLABVIEWER (www.msi.com); Figure 5 was prepared with BOBSCRIPT (Esnouf, 1997) and rendered with RASTER3D (Merritt and Murphy, 1994); and Figure 6 was produced with LIGPLOT (Wallace et al., 1995).
Acknowledgments
Acknowledgements
The authors wish to thank Gleb P.Bourenkov and Hans Bartunik for excellent support at the BW6 Beamline (Hamburg), and acknowledge Hans-Georg Beisel for assisting the substrate modelling and John Richardson for helpful discussions and final reading of the manuscript.
References
- Barton G.J. (1993) ALSCRIPT: a tool to format multiple sequence alignments. Protein Eng., 6, 37–40. [DOI] [PubMed] [Google Scholar]
- Bidwell L.M., McManus,M.E., Gaedigk,A., Kakuta,Y., Negishi,M., Pedersen,L. and Martin,J.L. (1999) Crystal structure of human catecholamine sulfotransferase. J. Mol. Biol., 293, 521–530. [DOI] [PubMed] [Google Scholar]
- Bork P. and Koonin,E.V. (1994) A P-loop-like motif in a widespread ATP pyrophosphatase domain: implications for the evolution of sequence motifs and enzyme activity. Proteins, 20, 347–355. [DOI] [PubMed] [Google Scholar]
- Brunger A.T. et al. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Cherest H., Kerjan,P. and Surdin-Kerjan,Y. (1987) The Saccharomyces cerevisiae MET3 gene: nucleotide sequence and relationship of the 5′ non-coding region to that of MET25. Mol. Gen. Genet., 210, 307–313. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project No. 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- D’Angelo I., Raffaelli,N., Dabusti,V., Lorenzi,T., Magni,G. and Rizzi,M. (2000) Structure of nicotinamide mononucleotide adenylyltransferase: a key enzyme in NAD(+) biosynthesis. Structure, 8, 993–1004. [DOI] [PubMed] [Google Scholar]
- deHaan M., Smits,P.H.M. and Grivell,L.A. (1995) Amino acid sequence of ATP sulfurylase from S. cerevisiae. Direct submission to the EMBL/GenBank/DDBJ databases.
- Delarue M. and Moras,D. (1993) The aminoacyl-tRNA synthetase family: modules at work. BioEssays, 15, 675–687. [DOI] [PubMed] [Google Scholar]
- Deyrup A.T., Krishnan,S., Singh,B. and Schwartz,N.B. (1999) Activity and stability of recombinant bifunctional rearranged and monofunctional domains of ATP-sulfurylase and adenosine 5′-phosphosulfate kinase. Biochemistry, 38, 6311–6316. [DOI] [PubMed] [Google Scholar]
- Engh R. and Huber,R. (1991) Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallogr. A, 47, 392–400. [Google Scholar]
- Esnouf R.M. (1997) An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J. Mol. Graph., 15, 132–134. [DOI] [PubMed] [Google Scholar]
- Foster B.A., Thomas,S.M., Mahr,J.A., Renosto,F., Patel,H.C. and Segel,I.H. (1994) Cloning and sequencing of ATP sulfurylase from Penicillium chrysogenum. Identification of a likely allosteric domain. J. Bacteriol., 176, 7055–7064. [PubMed] [Google Scholar]
- Garcia G.A., Leatherbarrow,R.J., Eckstein,F. and Fersht,A.R. (1990) Metal ion dependence of phosphorothioate ATP analogues in the Bacillus stearothermophilus tyrosyl-tRNA synthetase reaction. Biochemistry, 29, 1643–1648. [DOI] [PubMed] [Google Scholar]
- Gavel O.Y., Bursakov,S.A., Calvete,J.J., George,G.N., Moura,J.J. and Moura,I. (1998) ATP sulfurylases from sulfate-reducing bacteria of the genus Desulfovibrio. A novel metalloprotein containing cobalt and zinc. Biochemistry, 37, 16225–16232. [DOI] [PubMed] [Google Scholar]
- Groll M., Ditzel,L., Lowe,J., Stock,D., Bochtler,M., Bartunik,H.D. and Huber,R. (1997) Structure of 20S proteasome from yeast at 2.4 Å resolution. Nature, 386, 463–471. [DOI] [PubMed] [Google Scholar]
- Izard T. and Geerlof,A. (1999) The crystal structure of a novel bacterial adenylyltransferase reveals half of sites reactivity. EMBO J., 18, 2021–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakuta Y., Pedersen,L.G., Carter,C.W., Negishi,M. and Pedersen,L.C. (1997) Crystal structure of estrogen sulphotransferase. Nature Struct. Biol., 4, 904–908. [DOI] [PubMed] [Google Scholar]
- Karamohamed S., Nilsson,J., Nourizad,K., Ronaghi,M., Pettersson,B. and Nyren,P. (1999) Production, purification and luminometric analysis of recombinant Saccharomyces cerevisiae MET3 adenosine triphosphate sulfurylase expressed in Escherichia coli. Protein Expr. Purif., 15, 381–388. [DOI] [PubMed] [Google Scholar]
- Kraulis P.J. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Laskowski R., MacArthur,M., Moss,D.S. and Thornton,J. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr., 26, 283–291. [Google Scholar]
- Leatherbarrow R.J., Fersht,A.R. and Winter,G. (1985) Transition-state stabilization in the mechanism of tyrosyl-tRNA synthetase revealed by protein engineering. Proc. Natl Acad. Sci. USA, 82, 7840–7844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie A. (1991) Macromolecular Data Processing. In Moras,D., Podjarny,A.D. and Thierry,J.C. (eds), Crystal. Computing V. Oxford University Press, Oxford, UK, pp. 27–38.
- Leustek T., Murillo,M. and Cervantes,M. (1994) Cloning of a cDNA encoding ATP sulfurylase from Arabidopsis thaliana by functional expression in Saccharomyces cerevisiae. Biochem. J., 301, 349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyh T.S., Taylor,J.C. and Markham,G.D. (1988) The sulfate activation locus of Escherichia coli K12: cloning, genetic and enzymatic characterization. J. Biol. Chem., 263, 12886–12892. [PubMed] [Google Scholar]
- Li H., Deyrup,A., Mensch,J.R.,Jr, Domowicz,M., Konstantinidis,A.K. and Schwartz,N.B. (1995) The isolation and characterization of cDNA encoding the mouse bifunctional ATP sulfurylase-adenosine 5′-phosphosulfate kinase. J. Biol. Chem., 270, 29453–29459. [DOI] [PubMed] [Google Scholar]
- Logan H.M., Cathala,N., Grignon,C. and Davidian,J.C. (1996) Cloning of a cDNA encoded by a member of the Arabidopsis thaliana ATP sulfurylase multigene family. Expression studies in yeast and in relation to plant sulfur nutrition. J. Biol. Chem., 271, 12227–12233. [DOI] [PubMed] [Google Scholar]
- Lu G. (2000) TOP: a new method for protein structure comparisons and similarity searches. J. Appl. Crystallogr., 33, 176–183. [Google Scholar]
- MacRae I. and Segel,I.H. (1997) ATP sulfurylase from filamentous fungi: which sulfonucleotide is the true allosteric effector? Anal. Biochem., 244, 367–373. [DOI] [PubMed] [Google Scholar]
- MacRae I.J., Segel,I.H. and Fisher,A.J. (2000) Crystal structure of adenosine 5′-phosphosulfate kinase from Penicillium chrysogenum. Biochemistry, 39, 1613–1621. [DOI] [PubMed] [Google Scholar]
- Marzluf G.A. (1997) Molecular genetics of sulfur assimilation in filamentous fungi and yeast. Annu. Rev. Microbiol., 51, 73–96. [DOI] [PubMed] [Google Scholar]
- Matthews B.W. (1968) Solvent content of protein crystals. J. Mol. Biol., 33, 491–497. [DOI] [PubMed] [Google Scholar]
- Merritt E.A. and Murphy,M.E.P. (1994) Raster3D Version 2.0. A program for photorealistic molecular graphics. Acta Crystallogr. D, 50, 869–873. [DOI] [PubMed] [Google Scholar]
- Park Y.S., Gee,P., Sanker,S., Schurter,E.J., Zuiderweg,E.R. and Kent,C. (1997) Identification of functional conserved residues of CTP:glycerol-3-phosphate cytidylyltransferase. Role of histidines in the conserved HXGH in catalysis. J. Biol. Chem., 272, 15161–15166. [DOI] [PubMed] [Google Scholar]
- Perona J.J., Rould,M.A. and Steitz,T.A. (1993) Structural basis for transfer RNA aminoacylation by Escherichia coli glutaminyl-tRNA synthetase. Biochemistry, 32, 8758–8771. [DOI] [PubMed] [Google Scholar]
- Rosenthal E. and Leustek,T. (1995) A multifunctional Urechis caupo protein, PAPS synthetase, has both ATP sulfurylase and APS kinase activities. Mol. Cell Biol., 15, 6526–6534. [DOI] [PubMed] [Google Scholar]
- Rossmann M.G., Moras,D. and Olsen,K.W. (1974) Chemical and biological evolution of nucleotide-binding protein. Nature, 250, 194–199. [DOI] [PubMed] [Google Scholar]
- Saraste M., Sibbald,P.R. and Wittinghofer,A. (1990) The P-loop—a common motif in ATP- and GTP-binding proteins. Trends Biochem. Sci., 15, 430–434. [DOI] [PubMed] [Google Scholar]
- Schriever C., Breithardt,G. and Schmidt,A. (1997) Undersulfation of proteoheparan sulfate stimulates the expression of basic fibroblast growth factor and protein synthesis but suppresses replication of coronary smooth muscle cells. Mol. Mar. Biol. Biotechnol., 6, 180–188. [DOI] [PubMed] [Google Scholar]
- Sperling D., Kappler,U., Wynen,A., Dahl,C. and Truper,H.G. (1998) Dissimilatory ATP sulfurylase from the hyperthermophilic sulfate reducer Archaeoglobus fulgidus belongs to the group of homo-oligomeric ATP sulfurylases. FEMS Microbiol. Lett., 162, 257–264. [DOI] [PubMed] [Google Scholar]
- Turk D. (1992) Weiterentwicklung eines Programms für Molekülgraphik und Elecktronendichte-Manipulation und seine Anwendung auf verschiedene Protein-Strukturaufklärungen. PhD Thesis, Technische Universität, München, Germany.
- Venkatachalam K.V., Fuda,H., Koonin,E.V. and Strott,C.A. (1998) Site-selected mutagenesis of a conserved nucleotide binding HXGH motif located in the ATP sulfurylase domain of human bifunctional 3′-phosphoadenosine 5′-phosphosulfate synthase. Biochim. Biophys. Acta, 1429, 284–291. [DOI] [PubMed] [Google Scholar]
- Venkatachalam K.V., Fuda,H., Koonin,E.V. and Strott,C.A. (1999) Site-selected mutagenesis of a conserved nucleotide binding HXGH motif located in the ATP sulfurylase domain of human bifunctional 3′-phosphoadenosine 5′-phosphosulfate synthase. J. Biol. Chem., 274, 2601–2604. [DOI] [PubMed] [Google Scholar]
- Wallace A.C., Laskowski,R.A. and Thornton,J.M. (1995) LIGPLOT: a program to generate schematic diagrams of protein–ligand interactions. Protein Eng., 8, 127–134. [DOI] [PubMed] [Google Scholar]
- Weber C.H., Park,Y.S., Sanker,S., Kent,C. and Ludwig,M.L. (1999) A prototypical cytidylyltransferase: CTP:glycerol-3-phosphate cytidylyl transferase from Bacillus subtilis. Structure, 7, 1113–1124. [DOI] [PubMed] [Google Scholar]
- Wei J., Liu,C. and Leyh,T.S. (2000) The role of enzyme isomerization in the native catalytic cycle of the ATP sulfurylase-GTPase system. Biochemistry, 39, 4704–4710. [DOI] [PubMed] [Google Scholar]
- Zhang H. and Leyh,T.S. (1999) α-Thio-APS: a stereomechanistic probe of activated sulfate synthesis. J. Am. Chem. Soc., 121, 8692–8697. [Google Scholar]