

Abstract
The retinoblastoma (pRb)-related p130 pocket protein is a regulator of cell growth and differentiation, and a candidate tumour suppressor. Both pRb and p130 operate through interactions with cellular proteins, including the E2F transcription factors. While such interactions are controlled by phosphorylation of multiple sites of pRb, regulation of p130 remains poorly understood. We now identify 22 in vivo phosphorylation sites of p130, targeted by diverse kinases, and present evidence for three cyclin-dependent kinase 4(6) [Cdk4(6)] specific phosphorylations, which appear critical for controlling the growth-restraining activity of p130. When expressed in U2OS cells, the phosphorylation-deficient mutant p130ΔCdk4, in which the Cdk4 specific sites were mutated to alanine residues, imposed a more sustained G1 arrest than a constitutively active pRbΔCdk, known to repress all cellular E2F activity. Experiments using p130ΔCdk4 and another phosphorylation-deficient mutant, p130PM19A, with 19 phosphorylation sites mutated, revealed that the p130-imposed G1 block reflects cooperative growth-suppressive effects of phosphorylation-regulated E2F binding and phosphorylation-independent sequestration of cyclin E(A)–Cdk2 through the N-terminal cyclin binding motif of p130.
Keywords: cyclins/E2F4/G1 arrest/p130/phosphorylation
Introduction
The retinoblastoma family of ‘pocket proteins’ includes the tumour suppressor pRb and the related p107 and p130, multifunctional proteins implicated in regulation of the cell cycle, differentiation, cellular senescence and cell death (Wang, 1997; Dyson, 1998; Grana et al., 1998; Lipinski and Jacks, 1999). The pocket proteins exert their functions through interactions with cellular proteins, such as the transcription factors E2F1–5, which control expression of genes involved in cell cycle progression (Bernards, 1997; Dyson, 1998). pRb and its relatives have a mainly negative impact on the E2F-regulated promoters, either due to direct inhibition of E2F activity, or recruitment of transcriptional repressors such as histone deacetylase, which precludes transcription through local chromatin remodelling (Kouzarides, 1999). Both redundant and unique properties among the pocket proteins are emerging from biochemical studies and phenotypes of mice lacking genes for one or more family members (Cobrinik et al., 1996; LeCouter et al., 1998; Lipinski and Jacks, 1999; Vooijs and Berns, 1999), generally showing that p107 and p130 are more closely related to each other than to pRb (Qin et al., 1992; Livingston et al., 1993; Lee et al., 1998). Whereas pRb is commonly targeted in cancer and can bind E2F1–4, p107 and p130 interact preferentially with E2F4 and E2F5 (Weinberg, 1995; Dyson, 1998) and their N-terminal domains, and the extended spacer regions have properties distinct from pRb, including binding to cyclin E–Cdk2 and cyclin A–Cdk2 complexes (Hannon et al., 1993; Lacy and Whyte, 1997; Woo et al., 1997; Castano et al., 1998; Coats et al., 1999). A feature that distinguishes p130 and p107 is their expression at distinct times during the cell cycle, with the former dominating in quiescence (G0), and the latter induced as cells approach G1/S transition (Beijersbergen et al., 1995; Moberg et al., 1996; Smith et al., 1998).
The pocket-protein interactions, which underlie the multiple functions of pRb, p130 and p107, are largely controlled by phosphorylation. For pRb, the identification of the serine and threonine residues phosphorylated by the Cdks and exploration of phosphorylation-deficient mutants provided new insights into its biochemical and biological properties. During G1, cyclin D–Cdk4(6) trigger pRb phosphorylation initiating derepression of E2F target genes, while Cdk2 in complex with cyclin E or A phosphorylates pRb at G1/S and in S phase (Lees et al., 1991; Kitagawa et al., 1996; Knudsen and Wang, 1996; Connell-Crowley et al., 1997; Zarkowska and Mittnacht, 1997; Mittnacht, 1998; Harbour and Dean, 2000). In cycling cells, the activation of these kinases in mid to late G1 contributes to the passage through the restriction point (Weinberg, 1995; Bartek et al., 1997), and hyperphosphorylation of pRb from late G1 until mitosis appears essential for continuous expression of E2F target genes and coordination of the S/G2/M events (Chew et al., 1998; Knudsen et al., 1998; Lukas et al., 1999a; Meraldi et al., 1999). Whereas cycles of phosphorylation and dephosphorylation of pRb dominate in cycling cells, quiescent cells harbour p130–E2F4 as the major pocket protein–E2F complex, responsible for repression of cell cycle-regulatory genes like E2F1-3, cdc25A, cdc2 and p107 (Johnson, 1995; Tommasi and Pfeifer, 1995; Zhu et al., 1995; Hurford et al., 1997; Sears et al., 1997; Smith et al., 1998; Furukawa et al., 1999; Iavarone and Massagué, 1999). When cells are stimulated to enter the cell cycle, p130 becomes phosphorylated and, like for pRb, Cdk4(6) and Cdk2 are believed to be involved (Mayol et al., 1995; Grana et al., 1998). In contrast with pRb, and despite the accumulating evidence that phosphorylation regulates the diverse functions of the pocket proteins (Knudsen and Wang, 1996; Brown et al., 1999; Harbour et al., 1999), no information is presently available on the identity of phosphorylation sites regulating the activities of p107 or p130.
We chose to study p130 for its postulated key roles in the induction and/or maintenance of quiescence, regulation of differentiation (Lipinski and Jacks, 1999), and because of its cancer-associated defects, which make p130 a candidate tumour suppressor (Helin et al., 1997; Claudio et al., 2000). To understand the regulation and function of p130 better, we decided to study its in vivo phosphorylation. Here we report the mapping of the in vivo phosphorylated sites of human p130, identification of sites probably targeted by cyclin D–Cdk4(6), cyclin E–Cdk2 and non-Cdk kinases, construction of the first phosphorylation-deficient mutants of p130, the role of phosphorylation in interactions of p130 with E2F4 and cyclin E(A)–Cdk2, and the relative contributions of these functions to the cell-cycle-inhibitory effects of this largest member of the pocket protein family.
Results
Phosphopeptide mapping of p130
To explore p130 phosphorylation in vivo, the T98G human glioma cell line was used, which expresses all three pocket proteins (Mayol et al., 1995) and arrests in G1 when grown in 0.1% serum for 48 h, reflected by low cyclin D-dependent kinase activity and a low percentage of bromodeoxyuridine (BrdU)-positive cells compared with cycling cells (Figures 1A and 2A). Serum restimulation releases T98G cells from the G1 arrest, accompanied by induction of cyclin D1- and cyclin E-associated kinase activities, which culminate at 8 and 10–12 h after serum addition, respectively (Figure 1A and B). The endogenous pRb and p130 become phosphorylated with similar dynamics, as revealed by the appearance of their slower migrating, hyperphosphorylated forms (Figure 1C).
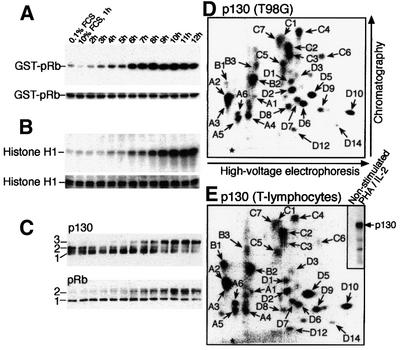
Fig. 1. Dynamics of the pocket protein phosphorylation. (A) Time course of cyclin D1-associated kinase activity in T98G cells restimulated with serum, using glutathione S-transferase–pRb as substrate (top); Coomassie Blue-stained loading control (bottom). (B) The same T98G cell extracts examined for cyclin E-associated kinase activity towards histone H1 (top) versus Coomassie Blue-stained control (bottom). (C) Time course immunoblots of immunoprecipitated p130, or total cell lysates (pRb), with the differentially phosphorylated forms of p130 and pRb marked (left). (D) Phosphopeptide map of 32P-labelled p130 gel purified upon immunoprecipitation from exponentially growing T98G cells. (E) Analogous phosphopeptide map of p130 from stimulated T lymphocytes. Insert, exposure of the membrane before cutting out the labelled p130 for phosphopeptide mapping. An asterisk in (D) and (E) indicates the sample application point.
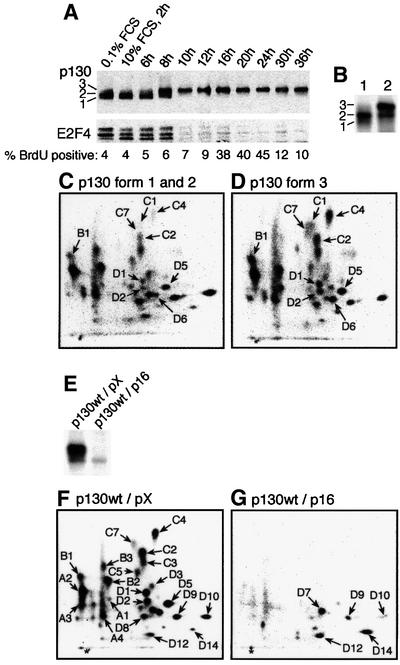
Fig. 2. Phosphorylation-dependent release of E2F4 from p130. (A) Serum-starved T98G cells were restimulated with serum for the indicated time before immunoprecipitation of p130 and immunoblotting for p130 and E2F4 as indicated. The percentage of BrdU-positive cells (1 h pulse) is indicated below the blots. (B) Serum-starved or restimulated (8 h) T98G cells were labelled with [32P]orthophosphate (4 h), before immunoprecipitation and phosphopeptide mapping of p130. The combined forms 1 and 2, and form 3 (from lane 2) were processed for phosphopeptide analysis as shown in (C) and (D), respectively. (E) T98G cells were electroporated with expression vectors for HA-tagged p130wt combined with an excess of expression plasmid for p16ink4a or empty vector (pX) as indicated, and labelled with [32P]orthophosphate. p130 was immunoprecipitated with antibody 12CA5 and the proteins separated by SDS–PAGE, electroblotted to nitrocellulose and exposed. Samples were processed for phospho peptide mapping as indicated in (F) and (G), respectively.
To assess p130 phosphorylation in vivo, exponentially growing T98G cells were labelled with [32P]orthophosphate and endogenous p130 was immunoprecipitated. The number of phosphorylated peptides from a tryptic digest of 32P-labelled p130 was >25. To facilitate the presentation, the map was divided into four sections, referred to as A–D (clockwise), and the phosphopeptides in each section were numbered (Figure 1D). The phosphopeptide map of p130 isolated from T98G cells was reproduced in experiments with proliferating normal human T lymphocytes (Figure 1E), demonstrating that the phosphorylation events that regulate p130 in T98G and normal cells are analogous, although minor quantitative differences exist between individual phosphopeptide spots (only phosphopeptides observed reproducibly are numbered). In contrast to strong labelling of p130 from the proliferating lymphocytes, no signal was detected in similar amounts of p130 immunoprecipitated from non-stimulated lymphocytes (Figure 1E, insert), indicating that the phosphate turnover on p130 in quiescent cells is slow or absent.
Contribution of cyclin D–Cdk4(6) and cyclin E–Cdk2 to phosphorylation of p130
Phosphorylation of p130 during the G0/G1–S phase progression is reflected by the electrophoretic mobility shifts from the so-called forms 1 and 2 (in G0 and early G1) to form 3, the hyperphosphorylated p130 protein with the lowest affinity for E2F4 seen at G1/S transition (Figure 2A; Mayol et al., 1995; Smith et al., 1996). The appearance of hyperphosphorylated p130 and the release of E2F4 (Figure 2A) correlated with the increased activities of cyclin D1–Cdk4(6) and E–Cdk2 (Figure 1A and B), supporting the candidacy of these kinases as regulators of the p130–E2F4 interaction in vivo (Beijersbergen et al., 1995; Mayol et al., 1995). To examine which phosphorylation events may underlie the transition of p130 to form 3, we labelled T98G cells with [32P]orthophosphate between 8 and 12 h after serum stimulation, immunoprecipitated p130, and separated forms 1 and 2 (pooled together) from form 3 by cutting the corresponding bands from the membrane upon exposure (Figure 2B, lane 2). As indicated in Figure 2C and D, phosphopeptides B1, C1, C2, C4, C7, D1, D2, D5 and D6 were over-represented in the p130 form 3 relative to forms 1 and 2. These results indicate that the shift to form 3 is probably mediated by a complex series of phosphorylations on p130.
To reveal phosphorylations on p130 that may be mediated by Cdk4(6) and Cdk2, we co-electroporated exponentially growing T98G cells with expression vectors encoding p130wt with either p16ink4a [a potent inhibitor of Cdk4(6); Sherr and Roberts, 1999], or the empty pX vector. Cells were labelled with [32P]ortho phosphate and the p130wt protein was immunoprecipitated via the haemagglutinin (HA) tag and analysed by autoradiography upon SDS–PAGE separation, followed by two-dimensional phosphopeptide mapping. The capacity of the endogenous kinases was sufficient to phosphorylate the bulk of the ectopic p130wt protein (Figure 2E, lane 1), while co-expression of p16ink4a prevented the hyperphosphorylation of p130 to form 3 (Figure 2E, lane 2). Phosphopeptide mapping confirmed that the phosphorylation pattern of ectopic p130wt (Figure 2F) recapitulates the pattern of endogenous p130 in T98G cells and primary T lymphocytes (Figure 1D and E). Co-expression of p16ink4a imposed a G1 arrest (data not shown), reflecting inhibition of both Cdk4(6) (direct) and Cdk2 (indirect, through redistribution of the Kip/Cip Cdk inhibitors from cyclin D–Cdk complexes to cyclin E– Cdk2; Sherr and Roberts, 1999). As can be deduced from a comparison of phosphopeptide patterns without (Figure 2F) and with ectopic p16ink4a (Figure 2G), >80% of all phosphorylations were Cdk mediated and/or Cdk dependent. In contrast, inhibition of Cdk2 had only a small effect as long as Cdk4(6) was active, since only the C3 phosphopeptide was clearly affected by co-transfection with dominant-negative dnCdk2 or by using the Cdk2 inhibitor roscovitine (data not shown). In repeated experiments, some phosphorylations of p130wt (D7, D9, D10, D12 and D14) were resistant to inhibition of G1 cyclin–Cdk complexes (Figure 2G), suggesting that such phosphorylations are performed by non-Cdk kinases.
The question of whether there was an absolute requirement for cyclin D–Cdk4(6) activity in the Cdk-mediated phosphorylation of p130 was addressed by using the pRb-negative human SAOS-2 cells, which lack cyclin D–Cdk4(6) activity due to high levels of endogenous p16ink4a and low levels of cyclin D (Lukas et al., 1995). In contrast to T98G cells, p130 never reached the fully shifted form 3 in SAOS-2 cells (Figure 3A) (Cheng et al., 2000), and after serum starvation and restimulation of SAOS-2, E2F4 was gradually released from p130 upon phosphorylation to form 2b (Figure 3B). In vivo [32P]orthophosphate labelling of ectopic or endogenous p130 revealed the lack of phosphorylation of p130 peptides C4, C7, D1 and D2 when cyclin D–Cdk4(6) activity is absent (SAOS-2) as compared with T98G cells with active cyclin D-dependent kinases (Figure 3D and G; and data not shown). Since Cdk2 is active in SAOS-2 cells, these results suggested that in the absence of cyclin D–Cdk4(6) activity, Cdk2 may phosphorylate p130 at most sites, resulting in delayed yet significant release of E2F4, compatible with cell proliferation (compare the different kinetics of BrdU incorporation into DNA and p130–E2F4 co-immunoprecipitation in T98G versus SAOS-2 cells after serum addition; Figures 2A and 3B). To confirm that the majority of phosphorylations on p130 that took place in SAOS-2 cells can be explained by the activity of Cdk2, we electroporated SAOS-2 cells with p130wt together with either p27kip1(T187A) (a stable inhibitor of Cdk2; Sheaff et al., 1997) or dnCdk2, either of which eliminates the Cdk2 activity. Co-expression of either p27kip1(T187A) or dnCdk2 almost completely eliminated the phosphorylation of p130wt (Figure 3C), stressing the importance of Cdk2 in phosphorylation of p130 in cells compromised in Cdk4(6) kinase activity. In contrast, restoration of Cdk4(6) activity by ectopic cyclin D1 resulted in full phosphorylation of p130 to form 3, even in SAOS-2 cells (Figure 3C), and confirmed the specificity of Cdk4(6) for phosphopeptides C4, C7, D1 and D2 (compare Figure 3D with E), which Cdk2 is apparently unable to phosphorylate in vivo. To corroborate this specificity, we co-expressed p130wt together with p16ink4a and cyclin E in T98G cells, a scenario in which Cdk4(6) is directly inhibited by p16ink4a, but Cdk2 remains active since excess of cyclin E prevents the indirect inactivation of Cdk2 otherwise imposed by p16ink4a. The resulting phosphopeptide maps (Figure 3G and H) showed that under conditions permissive for Cdk2 activity alone (Figure 3H), the pattern seen in T98G cells mimicked the spectrum of peptides phosphorylated in SAOS-2 cells, thereby confirming that the phosphorylations on phosphopeptides C4, C7, D1 and D2 are Cdk4(6) specific. Furthermore, the absence of phosphorylations of these peptides correlated with less retarded migration of p130wt in SDS–PAGE (Figure 3F), similar to what was observed in SAOS-2 cells.
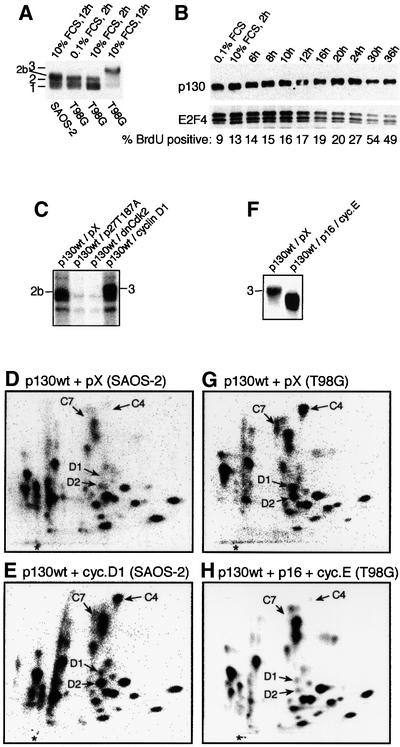
Fig. 3. p130 phosphorylation in the absence of cyclin D–Cdk4 activity. (A) Starved SAOS-2 and T98G cells were stimulated with serum (FCS), and p130 immunoprecipitated and visualized by blotting. Note that the most phosphorylated form of p130 in SAOS-2 cells (form 2b) is different from the fully phosphorylated form 3 in T98G cells. (B) Time course dissociation of E2F4 (bottom) from p130 (top) immunoprecipitated from serum-restimulated SAOS-2 cells, with parallel monitoring of BrdU incorporation (1 h pulse). (C–E) SAOS-2 cells were electroporated with the indicated expression plasmids, labelled with [32P]orthophosphate and processed for immuno precipitation of p130wt (C), and phosphopeptide mapping (D and E). (F–H) T98G cells were electroporated with the indicated expression plasmids. The [32P]orthophosphate-labelled p130wt was processed for phosphopeptide mapping as indicated (G–H).
Identification of phosphorylation sites in p130
The precise identity of the phosphorylation sites corresponding to the individual phosphopeptides was elucidated by phosphoamino acid analysis and cycle sequencing of isolated tryptic phosphopeptides from in vivo labelled p130, combined with site-directed mutagenesis. As a representative example from the large body of data, we chose the phosphopeptide D5, since this represents the site homologous to serine 795 (S795) in pRb, essential for the neutralization of pRb’s interaction with E2F (Connell-Crowley et al., 1997). Phosphoamino acid analysis revealed that D5 was phosphorylated on serine (Figure 4A, insert) and cycle sequencing released a 32P-labelled amino acid in cycle 3 (Figure 4A). As the charge of D5 is +1 at pH 1.9, there exist four candidate tryptic peptides in p130 containing an SP site that could represent D5. Site-directed mutagenesis of S1044 to an alanine resulted in the specific loss of D5 when this mutant was ectopically expressed in T98G cells and labelled by [32P]orthophosphate (Figure 4B), proving that phosphorylation of S1044 gave rise to the tryptic phosphopeptide D5: TGSPR. Figure 4B further illustrates tryptic phosphopeptide maps of four additional serine/threonine to alanine single mutants of p130, and should be considered as confirmation for data obtained by cycle sequencing and phosphoamino acid analysis. In all cases except threonine 986 (T986) and S966, a single mutation of a serine or threonine to an alanine residue had no impact on other phosphorylations. Table I shows the list of all mapped phosphorylations, and Figure 5 illustrates their localization in p130. It is striking that the 22 mapped phosphorylation sites cluster in four regions: three sites localize close to the border between the N-terminal part and the A-pocket, six in the spacer region (flanking the RLF cyclin binding motif), another six in the C-terminal region and seven sites in the B-pocket, within a sequence of p130 non-conserved in pRb and p107. The latter region is possibly linked to some unique function of p130, and it is also this region that contains the only sites whose mutations affect phosphorylations of other residues. Thus, phosphorylation of S966 (D7; an SP site) seems to be required for the phosphorylation of S962 (D9/D10; a non-SP/TP site: D10 is a partial cleavage product of D9; Figure 4B), and phosphorylation of T986 (A2; a TP site) for the phosphorylation of S982 and S973 (both non-SP/TP sites contained in the A2 phosphopeptide; data not shown). Furthermore, S966 still gets phosphorylated when p16ink4a is co-expressed with p130wt, while the phosphorylation of T986 was blocked by p16ink4a co-expression. These observations, and the fact that several phosphorylation sites lack the proline residue at position +1, indicate that other, non-Cdk kinases participate in the phosphorylations in this part of p130.
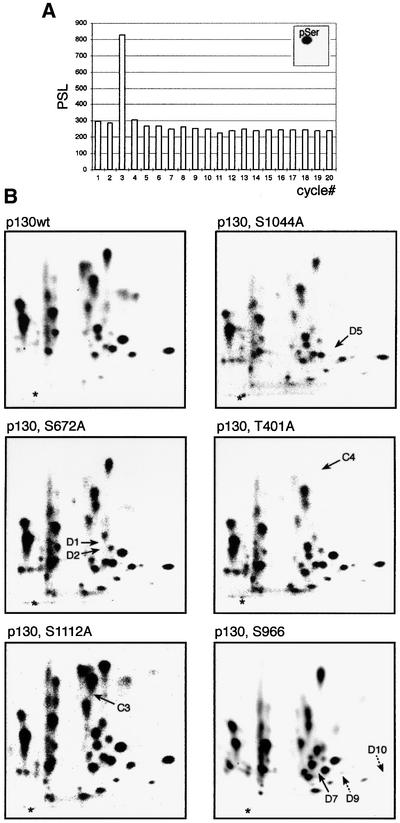
Fig. 4. Cycle sequencing and confirmation of phosphorylation sites. (A) Phosphopeptide D5 was eluted from a TLC plate and processed for Edman degradation and phosphoamino acid analysis (insert). The histogram shows the amount of radioactivity (PSL values obtained from a PhosphorImager) released from phosphopeptide D5 in individual cycles. (B) Examples of phosphopeptide maps of the indicated p130 single-point mutants expressed as HA-tagged proteins in T98G cells. Arrows point to a missing phosphopeptide (see Table I for an overview).
Table I. Summary of identified in vivo phosphorylation sites in p130.
| Phosphorylation site | T401 | S413 | T417 | S639 | T642 | S662 | S672 | S688 | T694 | S948 | S952 | S962 | S966 | S973 | S982 | T986 | S1035 | S1044 | S1068 | S1080 | T1097 | S1112 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Coordinate | C4 | A1 | A1 | B1 | B1 | C2 | D1/D2 | C5 | C5 | D8 | D8 | D9/D10 | D7 | A2 | A2 | A2 | C7 | D5 | D3 | C1 | D6 | C3 |
| Phosphorylated in T lymphocytes | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Phosphorylated in T98G cells | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Phosphorylated in SAOS-2 cells |
– |
+ |
+ |
+ |
+ |
+ |
– |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
+ |
– |
+ |
+ |
+ |
+ |
+ |
| Sites phosphorylated by Cdks | + | + | + | + | + | + | + | + | + | – | + | – | – | + | + | + | + | + | + | + | + | + |
| Cdk4(6)-specific phosphorylations | + | – | – | – | – | – | + | – | – | – | – | – | – | – | – | – | + | – | – | – | – | – |
| Phosphorylated by a kinase different from Cdksa | – | – | – | – | – | – | – | – | – | + | – | + | + | + | + | – | – | – | – | – | – | – |
| Dependent on Cdk pre-phosphorylationb | – | – | – | – | – | – | – | – | – | + | – | – | – | + | + | – | – | – | – | – | – | – |
Listed are only phosphopeptides, indicated by letters and numbers, where we have identified the site(s) of phosphorylation. Upper part, phosphorylations (+) on endogenous p130 in the indicated cell types. Lower part, kinases responsible for p130 phosphorylations.
aPhosphorylations (+) that still occur when p16 is co-expressed with p130wt, or sites different from the SP/TP Cdk minimal consensus.
bPhosphorylation sites (+) that are not SP or TP sites and depend on pre-phosphorylation by Cdk on another site.
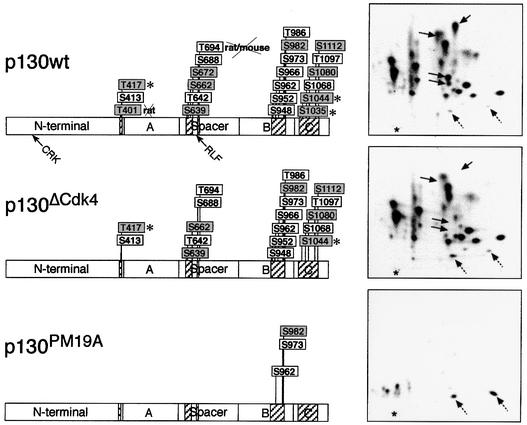
Fig. 5. In vivo phosphorylation sites in p130. Schematic summary of the 22 mapped in vivo phosphorylation sites. Shaded boxes, residues conserved in p107; an asterisk indicates residues conserved in pRb. A, B and C and spacer refer to domains of p130. All residues except T401 and T694 are conserved between human and mouse and/or rat p130. In p130ΔCdk4, the three Cdk4-specific residues, T401, S672 and S1035, were mutated to alanine residues, and in the p130PM19A mutant all except three mapped sites (non-Cdk sites) were mutated to alanine. Phosphopeptide maps of the wild type and the two phosphorylation-deficient mutants expressed in U2OS cells are also shown (right); solid arrows point to the normal position of phosphopeptides represented by the mutated residues and dashed arrows indicate the position of D12 and D14. The relative positions of the two cyclin A and E binding motifs (CRK or RLF) are indicated in the p130wt construct by arrows.
Repression of E2F and growth arrest by phosphorylation-deficient p130
According to the order in which we identified the p130 phosphorylation sites, a phosphorylation-deficient mutant p130PM19A was created, in which 19 of the 22 mapped phosphorylation sites were mutated to alanine (Figure 5; only three non-Cdk sites were left as wild type). Furthermore, we created a mutant in which only the three Cdk4(6)-specific phosphorylation sites, T401, S672 and S1035, were mutated to alanines (p130ΔCdk4). Derivatives of p130PM19A and p130ΔCdk4, with the N-terminal cyclin binding motif ACRK (Castano et al., 1998) mutated to AAAA [p130PM19A(CRK/AAA) and p130ΔCdk4(CRK/AAA)] or the spacer RLF cyclin binding motif substituted by AAA [p130PM19A(RLF/AAA) and p130ΔCdk4(RLF/AAA)], were also made, alone or combined. The abilities of these p130 mutants to bind E2F4 and repress E2F activity, sequester cyclin E(A)–Cdk2 and affect cell proliferation were assessed by transient transfection and immunoprecipitation, flow cytometry and reporter assays (Figures 6 and 7). E2F4 bound relatively poorly to wild-type p130 under these conditions, reflecting efficient phosphorylation by endogenous kinases. Furthermore, supporting our data, which implicate cyclin D–Cdk4(6) and cyclin E–Cdk2 in phosphorylation of p130 (Figures 2 and 3), co-expression of cyclin D1 resulted in an almost complete shift of p130wt to form 3 and increased release of E2F4 (Figure 6B), while co-electroporation of p16ink4a prevented the phosphorylation of p130 to form 3 and resulted in an increased binding of E2F4. Both the p130ΔCdk4 and p130PM19A mutants co-immunoprecipitated significant amounts of E2F4 (p130PM19A being the strongest interactor), indicating that critical phosphorylation sites for the release of E2F4 from p130 had been mutated. Unexpectedly, E2F1 was also co-immunoprecipitated with p130, in amounts comparable to (p130ΔCdk4) or even higher (p130PM19A) than those complexed to pRbΔCdk (Figure 6B).
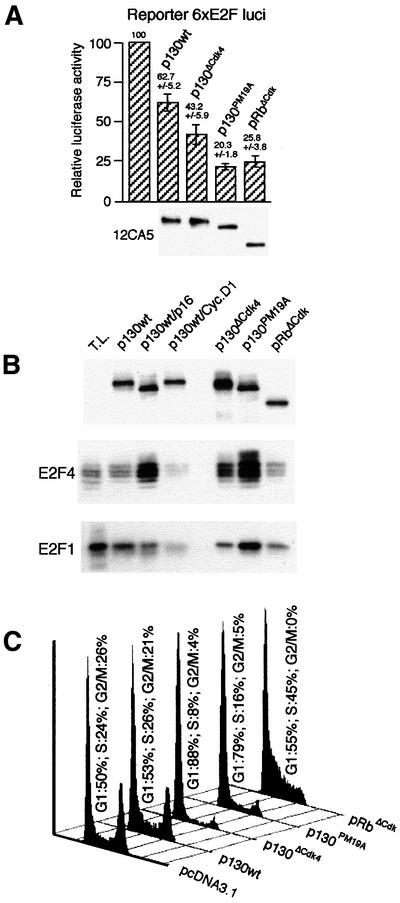
Fig. 6. Repression of E2F and G1 arrest by p130 mutants. (A) Summary of luciferase reporter assays in extracts of U2OS cells transfected with fixed amounts of expression plasmids for the indicated pocket proteins (data represent mean values ± SD from three experiments). Bottom, HA-tag immunoblots of immunoprecipitates from one experiment, to indicate the relative amounts of the pocket proteins. (B) HA-tagged p130 was immunoprecipitated 36 h after electroporation of the indicated plasmids into U2OS cells, and the precipitates immunoblotted for p130, E2F4 and E2F1. T.L., total lysate (30 µg protein) from exponentially growing U2OS cells. (C) U2OS cells were transfected with the indicated p130 expression plasmids or empty plasmid control (pcDNA3.1) in combination with CD20 surface marker. Thirty-six hours later, transfected cells were sorted (CD20-positive cells) and analysed for their DNA content by fluorescence activated cell sorting.
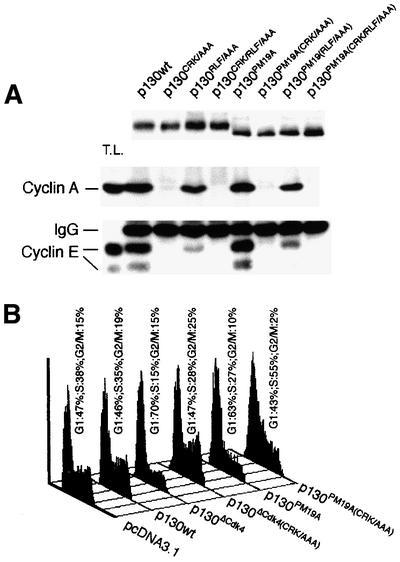
Fig. 7. The effects of cyclin binding motifs. (A) U2OS cells were electroporated with the indicated HA-tagged p130 constructs in which either of the two cyclin binding motifs were mutated alone or in combination. Two days after electroporation, lysates were processed for anti-HA immunoprecipitations in the presence of saturating amounts of lysate from exponentially growing non-electroporated U2OS cells and analysed by immunoblotting for cyclin A or E. Anti-HA blotting of the upper part of the gel indicated that equal amounts of expressed p130 constructs were loaded. (B) U2OS cells were transfected with the indicated expression plasmids and analysed by FACS as in Figure 6C.
In situ staining of U2OS cells expressing the p130ΔCdk4 and p130PM19A mutants or wild-type p130 constructs confirmed their nuclear localization (data not shown), and reporter assays using the plasmid 6×E2F-luc (Figure 6A) showed that both p130ΔCdk4 and p130PM19A repressed the endogenous E2F activity more efficiently than p130wt and similarly to pRbΔCdk. It should be noted that p130ΔCdk4 was as stable as the wild-type p130 protein, while the p130PM19A mutant was significantly less stable (data not shown). Therefore, the input plasmid concentration was adjusted accordingly to obtain equal protein expression levels of the pocket protein constructs (Figure 6A, anti-HA immunoblot).
Co-immunoprecipitation experiments using U2OS cells expressing ectopic p130wt, p130(CRK/AAA), p130(RLF/AAA) or the combined mutant p130(CRK/RLF/AAA), and the same cyclin binding mutations on a phosphorylation-deficient p130 background (p130PM19A) showed that the majority of cyclin E–Cdk2 and A–Cdk2 interact with the N-terminal ACRK motif of p130 (Figure 7A). The RLF/AAA mutation in the spacer only affected cyclin A binding marginally, while cyclin E binding was more significantly affected. In contrast to the inability of the ectopic wild-type p130 to bind E2Fs efficiently (Figure 6B, lane 2), this largely hyperphosphorylated protein (due to the endogenous kinases) formed abundant complexes with cyclins E and A (Figure 7A, lane 2). As the amounts of cyclin E(A) bound to such phosphorylated p130wt were virtually identical to the amounts of cyclins co-immunoprecipitated with the phosphorylation-deficient p130PM19A mutant (Figure 7A, compare lane 2 with 6), sequestration of cyclin E(A)–Cdk2 by p130 appears largely independent of p130 phosphorylation status. These distinct modes of regulating the interactions of p130 with E2Fs and cyclin E(A), respectively, are also reflected by a decrease in E2F4, but an increase in cyclin E–Cdk2, bound to p130 at 10–12 h post-serum stimulation of T98G cells, coinciding with the shift of p130 to form 3 in late G1 (Figure 2A; Supplementary figure 1, available at The EMBO Journal Online.)
Since both E2F and Cdk activities are required for G1/S transition and cell proliferation (Lukas et al., 1997; Chew et al., 1998; Knudsen et al., 1998), we tested the ability of the individual p130 constructs to arrest cells in G1. Flow cytometry analysis of transfected U2OS cells showed that wild-type p130 had only a minor effect on cell cycle progression, while expression of either p130ΔCdk4 or p130PM19A imposed an efficient block in G1 (Figure 6C). The G1 arrest imposed by these phosphorylation-deficient mutants was clearly more sustained than the effect of pRbΔCdk, which caused only a transient G1 delay with subsequent accumulation of cells in S phase (Figure 6C) (Lukas et al., 1999b). Interestingly, removal of the ability of the phosphorylation-deficient p130 mutants to sequester cyclin A/E allowed the transfected cells to escape the G1 block. In the case of the p130ΔCdk4(CRK/AAA) mutant the cells regained the full potential to proliferate, while the p130PM19A(CRK/AAA) mutant cells accumulated in S phase (Figure 7B), analogous to the effect of pRbΔCdk (Figure 6C).
Discussion
Work from several laboratories over the last decade identified some 10–12 of the 16 candidate Cdk consensus sites of pRb as targets of cyclin D–Cdk4 and/or cyclin E–Cdk2 (Lees et al., 1991; Knudsen and Wang, 1996; Mittnacht, 1998). Here we have mapped 22 serine/threonine residues of p130 phosphorylated in vivo and a few more remain to be identified, indicating that the regulation of p130 by phosphorylation is significantly more complex than that of pRb. Unexpectedly, only three of these mapped residues are conserved in pRb, while 10 are conserved in p107, and 12 sites appear unique to p130 (Figure 5).
At least three classes of kinases cooperate to phosphorylate p130 in human cells: cyclin D–Cdk4(6), cyclin E(A)–Cdk2, and so far unidentified non-Cdk kinases. The non-Cdk kinases probably target those sites efficiently phosphorylated, even in cells arrested by p16ink4a (S962, S966 and the sites represented by peptides D12 and D14), but also S948, S973 and S982 in the B-pocket (Figure 5). The latter three residues are not SP/TP sites targeted by Cdks, yet their phosphorylation is prevented by p16ink4a, probably due to dependency on pre-phosphorylation of other residues by Cdks. Such sequential modification is supported, for instance, by the inability of S973 and S982 to become phosphorylated in vivo when T986 (a TP site) is mutated to alanine (data not shown). Thus, multiple kinase activities converge to phosphorylate p130, including Cdk4(6), Cdk2 and non-Cdk kinases. We imagine a temporal sequence of phosphorylation events regulating p130 function initiated by non-Cdk kinases in early G1, represented by those sites still phosphorylated in the presence of p16ink4a, followed by cyclin D–Cdk4(6), cyclin E–Cdk2 and finally by non-Cdk kinases, which require pre-phosphorylation of p130 by Cdks (Table I).
The identity of cellular substrates of Cdk4(6) versus Cdk2 has recently attracted considerable attention, reflecting the key roles of Cdks in cell cycle control and their deregulation in cancer, which inspired a search for novel Cdk-inhibitory drugs (Garrett and Fattaey, 1999). Here we show that among the 22 mapped phosphorylation sites of p130, most appear to be targeted by Cdks and three specifically by Cdk4(6). All three residues selectively targeted by Cdk4(6), T401 (N-terminus), S672 (spacer region) and S1035 (C-terminus), are conserved in mouse p130, and have homologues in p107, but only S1035 is conserved in pRb. Interestingly, S795 of human pRb, whose phosphorylation appears critical for E2F release (Connell-Crowley et al., 1997), was suggested to be targeted by both Cdk4(6) and Cdk2 (Kitagawa et al., 1996), in agreement with our data for the homologous site in p130, S1044. Kitagawa et al. (1996) also found that S780 of pRb was a good substrate for Cdk4(6), but not for Cdk2, consistent with our finding that the homologous S1035 in p130 is one of the three sites targeted specifically by Cdk4(6). The similarities between pRb and p130 are limited, however, with the vast majority of their phosphorylation sites being non-conserved. Moreover, despite phosphorylation of S567 of pRb by Cdk2 (at least in vitro) appears essential for release of E2F (Harbour and Dean, 2000), we have so far been unable to show phosphorylation of the homologous site in p130 (S603; non-conserved in p107). However, we cannot rule out that S603 of p130 might be phosphorylated under certain conditions in vivo, and further studies are needed to clarify this issue.
Mapping in vivo phosphorylation sites allows the restriction of mutagenesis to only those sites of physiological relevance and assessment of their impact on biochemical and biological functions of the pocket proteins. The phosphorylation-deficient mutants of p130 constructed here represent the first attempt to explore constitutively active alleles of a non-Rb pocket protein. The properties of p130PM19A suggest that we have largely achieved this goal, at least as judged from repression of E2F4, the major transcription factor regulated by p130 in vivo, and from the impact of this mutant on cell proliferation (Figure 6C). Thus, E2F4 remained bound to p130PM19A in serum-stimulated cells, despite the active endogenous cyclin D- and E-dependent kinases, and even upon experimental elevation of either cyclin D1 or cyclin E (data not shown). Our data show that both Cdk4(6) and Cdk2 can affect the p130–E2F4 complex, yet the shift to the fully phosphorylated form 3 of p130, which has the lowest affinity for E2F4, requires active Cdk4(6) and phosphorylation of the Cdk4(6)-specific target sites (see above). The pronounced effects of both p130ΔCdk4 and p130PM19A on E2F-mediated transcription reflect a combination of direct physical sequestration of E2F4 and E2F5 (Sardet et al., 1995), as well as E2F1 by p130 (our data) and transcriptional silencing of E2F1-3 gene promoters by the p130–E2F4 complex (Johnson, 1995; Dyson, 1998; Furukawa et al., 1999).
When expressed in human cells proficient in Cdk4(6) and Cdk2 activities, the phosphorylation-deficient mutants p130ΔCdk4 and p130PM19A both induced a sustained G1 arrest, considerably more pronounced than the effect of a constitutively active mutant pRbΔCdk, which only delays G1/S transition (Lukas et al., 1999b; Figure 6C). Since p130PM19A inhibits E2F activity to an extent comparable to pRbΔCdk, the differential impact of these two mutant pocket proteins on G1/S progression strongly suggests that the p130 mutant inhibits the cell cycle through an additional mechanism, independent of E2F. A candidate for such a function of p130, not conserved in pRb, has recently been identified in the ability of p130 and p107 to physically bind cyclin E–Cdk2 and cyclin A–Cdk2 complexes, thereby inhibiting and/or subverting their activity towards other substrates (Grana et al., 1998). Of the two sequences responsible for the interaction of p130 with cyclinE(A)–Cdk2, the more N-terminal ACRK motif appears to account for the bulk of p130-bound cyclin E and A (Figure 7A; Castano et al., 1998). Mutating the ACRK motif to alanines within the context of p130PM19A gave rise to a protein [p130PM19A(CRK/AAA)], whose ability to arrest cells in G1 was considerably diminished (Figure 7B), indicating that the ability to sequester the cyclins indeed contributes to the G1-inhibitory effect of p130. The binding of cyclin E/A to the major cyclin binding domain (the N-terminal ACRK motif) of p130 occurs independently of phosphorylation, as revealed by the fact that the almost completely hyperphosphorylated p130wt protein binds equal amounts of cyclin E and A as compared with the hypophosphorylated p130PM19A mutant (Figure 7A), and by efficient binding of the endogenous, hyperphosphorylated form 3 of p130 to cyclin E–Cdk2 in late G1 cells, at the time when most E2F4 is released due to p130 phosphorylation (Figure 2A; Supplementary figure 1). On the other hand, the less pronounced interaction of cyclin E(A)–Cdk2 with the spacer region of p130 might be regulated by phosphorylation (currently under investigation), consistent with the fact that six phosphorylation sites flank the RLF cyclin binding motif (Figure 5). In any case, our data clearly suggest that the stable G1 arrest obtained with the phosphorylation-deficient mutants of p130 reflects the cooperation of at least two distinct functions of p130: E2F binding, which is regulated by phosphorylation, and sequestration of cyclin E(A)–Cdk2 to the major cyclin binding site in the N-terminus, which is phosphorylation independent. Such a differential control over the two G1-inhibitory functions may help p130 to regulate timely entry into S phase in two steps. First, derepression of E2F in mid-G1, following phosphorylation of the pocket proteins by cyclin D-dependent kinases, would activate the S phase-promoting transcriptional programme, possibly coinciding with the G1 restriction point. Secondly, the N-terminus-mediated sequestration of cyclin E(A)–Cdk2 by p130 remains active in late G1, regardless of the phosphorylation events that release E2F, and may titrate the accumulating activity of cyclin E(A)–Cdk2 kinases until several hours after E2F derepression, when threshold levels of Cdk2 and possibly other activities optimal for initiation of DNA synthesis are achieved.
The present demonstration that the phosphorylation-deficient mutants of p130 (p130ΔCdk4 and p130PM19A) both imposed a more sustained G1 arrest than constitutively active pRb is unexpected, given the widely held view that only the latter pocket protein qualifies as a tumour suppressor (Lipinski and Jacks, 1999). Taken together with the recent identification of tumour-associated mutations of p130 in human cancers (Helin et al., 1997; Claudio et al., 2000), our data are consistent with the candidacy of p130 for a tumour suppressor. Most recently, cells from mice with simultaneous inactivation of p130 and p107 (Bruce et al., 2000) or E2F4 and E2F5 (Gaubatz et al., 2000) were shown to be resistant to p16-mediated G1 arrest, implying that in addition to pRb, the p130/p107 pocket proteins provide an essential regulatory function in G1, and further support the key role of cyclin D-dependent kinases, whose critical contribution to p130 phosphorylation and E2F4 release is demonstrated in our present study. The mechanistic basis of the more potent inhibition of G1/S transition by p130 than by pRb may reflect the ability of p130 to inhibit the events downstream of E2F4/5, such as transcription of genes encoding E2F1-3, cdc25A, Cdc2, p107, etc. (Johnson, 1995; Tommasi and Pfeifer, 1995; Zhu et al., 1995; Hurford et al., 1997; Sears et al., 1997; Smith et al., 1998; Furukawa et al., 1999; Iavarone and Massagué, 1999), and to sequester the S-phase-promoting cyclin E(A)–Cdk2 (Lacy and Whyte, 1997; Woo et al., 1997; Castano et al., 1998). While inhibition of E2F is shared by pRb (Lukas et al., 1997), pRb’s ability to affect cyclin E and thus cyclin E–Cdk2 activity is only limited, eventually allowing cells exposed to non-phosphorylatable pRb to enter S phase (Chew et al., 1998; Knudsen et al., 1998; Lukas et al., 1999a; this study). As it seems that only the functions mediated by the large A/B pocket of p130, such as E2F binding, are regulated by phosphorylation and thus negated by the cyclin D- dependent kinases, the second G1-inhibitory function of p130, the ability to sequester cyclin E–A–Cdk2 through the N-terminal CRK motif, must be neutralized by other mechanisms. One possibility is that the protein turnover of p130 becomes accelerated at G1/S transition due to its targeting for the proteasome-mediated degradation (Smith et al., 1998), thereby also eliminating the N-terminal, phosphorylation-independent activity of p130. From a wider perspective, our data on the regulation of p130 provide the necessary basis for detailed functional analyses of the phosphorylation-dependent and -independent activities of p130, including its roles in the regulation of fundamental biological processes such as the induction and maintenance of quiescence, cellular differentiation and prevention of oncogenic transformation.
Materials and methods
Cell culture and synchronization
Human T98G glioblastoma, SAOS-2 and U2OS osteosarcoma cell lines were grown in DME medium supplemented with 10% fetal calf serum (FCS). Subconfluent cells were arrested in G1 by 48 h culture in 0.1% FCS, restimulated with 10% FCS, and labelled for 4 h in phosphate-free DME with 20 mM HEPES pH 7.2, 10% dialysed phosphate-free FCS and 2 mCi/ml [32P]orthophosphate (PBS43; Amersham). Primary human T lymphocytes were purified using Lymphoprep (Nycomed Pharma), seeded in RPMI medium (Gibco-BRL) with 40 mM HEPES pH 7.2, 10% FCS, 5 µg/ml phytohaemagglutinin A (PHA-L) and interleukin 2 (IL-2; 100 IU/ml) and 65 h post-stimulation labelled with [32P]orthophosphate as described above.
Site-directed mutagenesis and gene transfer
Human p130 gene was mutagenized using the QuikChange Site-Directed Mutagenesis kit (Stratagene) and primers of ∼27 nucleotides. For in vivo labelling experiments, cells were transfected using the calcium phosphate method (1 ml transfection mix containing 7 µg of specific plasmids and carrier empty vector to 16 µg final DNA for a 10 cm dish) or electroporated (Bio-Rad gene pulser) using 1.5 × 106 cells and 7 µg of plasmid DNA.
Antibodies and immunochemical methods
Antibodies to p130 (SC317), E2F4 (SC866), cyclin A (SC751), E2F1 (SC193) and anti-HA tag (SC805) were from Santa Cruz Biotechnologies. Antibody 12CA5 against the HA tag was produced as mouse ascites. Monoclonal antibodies DCS11 and HE172 were used to immunoprecipitate cyclin D1 and E, respectively. Immunoprecipitation, kinase assays and immunoblotting procedures have been described previously (Lukas et al., 1997, 1999b). In experiments shown in Figures 6B and 7A, lysate from exponentially growing U2OS cells was added in order to have cyclins and E2Fs in excess of the ectopically expressed p130 protein.
Electrophoresis and two-dimensional phosphopeptide mapping
SDS–PAGE gels were run in Bio-Rad Protean II xi system. Two-dimensional phosphopeptide mapping and phosphoamino acid analysis were performed as described (Boyle et al., 1991; Blume-Jensen et al., 1995; Hansen et al., 1996). Tryptic phosphopeptides were separated on a Hunter Thin Layer Electrophoresis System (HTLE-7000), at 2000 V for 30 min (14°C). The second dimension chromatography was performed in isobuturic acid buffer for 16 h.
Elution of phosphopeptides from TLC plates for automated Edman degradation
Eluted phosphopeptides (Hansen et al., 1996) were coupled to SequelonTM-AA membrane (Millipore) by use of carbodiimide coupling, according to standard procedures as described by the manufacturer. Edman degradation was performed using an Applied Biosystems gas phase sequencer (Model 477A) as described by Blume-Jensen et al. (1995).
Reporter assays
U2OS cells were seeded at 2.5 × 105 cells per 10 cm dish and transfected by the calcium phosphate method using 2 µg of the E2F reporter construct pGL3TATAbasic-6×E2F (6×E2F-luc) (Müller et al., 1997), 1 µg of CMV–LacZ and expression vectors for p130wt (2 µg), p130ΔCdk4 (2 µg) or p130PM19A (8 µg) (all in pcDNA1), or pECE-pRbΔCdk (2 µg) (Lukas et al., 1997). Empty plasmid was added to a final concentration of 16 µg of DNA per transfection. Cells were harvested 36 h later and luciferase activity measured using the Luciferase Assay System (Promega) and a Berthold Lumat LB95d instrument. β-galactosidase activity was quantitated as an internal control for transfection efficiency. The data on relative luciferase activity represent mean values from three experiments, normalized relative to the β-galactosidase readout.
Flow cytometry analysis
For flow cytometry analysis, CD20 (pCMVCD20; 2 µg) was co-transfected with pcDNA3.1 empty vector control or HA-tagged p130wt (4 µg), p130ΔCdk4 (4 µg), p130ΔCdk4(CRK/AAA) (4 µg), p130PM19A (16 µg), p130PM19A(CRK/AAA) (16 µg) or HA-tagged pRbΔCdk (4 µg) constructs into U2OS cells. Cells were harvested 36 h after replacement of medium, stained with fluorescein isothiocyanate-conjugated anti-CD20 antibodies (Becton-Dickinson), and processed for flow cytometry as described previously on a Becton-Dickinson FACScan machine (Lukas et al., 1995).
Supplementary data
Supplementary data for this paper are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank Christer Wernstedt for cycle sequencing, Carl-Henrik Heldin for support, and Steven Reed, Ed Harlow, Kristian Helin and Michele Pagano for reagents. This work was supported by the Danish Cancer Society, the Nordic Cancer Union and the Danish Medical Research Council.
References
- Bartek J., Bartkova,J. and Lukas,J. (1997) The retinoblastoma protein pathway in cell cycle control and cancer. Exp. Cell Res., 237, 1–6. [DOI] [PubMed] [Google Scholar]
- Beijersbergen R.L., Carlee,L., Kerkhoven,R.M. and Bernards,R. (1995) Regulation of the retinoblastoma protein-related p107 by G1 cyclin complexes. Genes Dev., 9, 1340–1353. [DOI] [PubMed] [Google Scholar]
- Bernards R. (1997) E2F: a nodal point in cell cycle regulation. Biochim. Biophys. Acta, 1333, M33–40. [DOI] [PubMed] [Google Scholar]
- Blume-Jensen P., Wernstedt,C., Heldin,C.-H. and Rönnstrand,L. (1995) Identification of the major phosphorylation sites for protein kinase C in Kit/stem cell factor receptor in vitro and in intact cells. J. Biol. Chem., 270, 14192–14200. [DOI] [PubMed] [Google Scholar]
- Boyle W.J., van der Geer,P. and Hunter,T. (1991) Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods Enzymol., 201, 110–149. [DOI] [PubMed] [Google Scholar]
- Brown V.D., Phillips,R.A. and Gallie,B.L. (1999) Cumulative effect of phosphorylation of pRB on regulation of E2F activity. Mol. Cell. Biol., 19, 3246–3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce J.L., Robert,K., Hurford,R.K.,Jr, Classon,M., Koh,J. and Dyson,N. (2000) Requirements for cell cycle arrest by p16ink4a. Mol. Cell, 6, 737–742. [DOI] [PubMed] [Google Scholar]
- Castano E., Kleyner,Y. and Dynlacht,B.D. (1998) Dual cyclin-binding domains are required for p107 to function as a kinase inhibitor. Mol. Cell. Biol., 18, 5380–5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L., Rossi,F., Fang,W., Mori,T. and Cobrinik,D. (2000) Cdk2-dependent phosphorylation and functional inactivation of the pRb-related p130 protein in pRB(–), p16INK4A(+) tumor cells. J. Biol. Chem., 275, 30317–30325. [DOI] [PubMed] [Google Scholar]
- Chew Y.P., Ellis,M., Wilkie,S. and Mittnacht,S. (1998) pRB phosphorylation mutants reveal role of pRB in regulating S-phase completion by a mechanism independent of E2F. Oncogene, 17, 2177–2186. [DOI] [PubMed] [Google Scholar]
- Claudio P.P. et al. (2000) Mutations in the retinoblastoma-related gene RB2/p130 in lung tumors and suppression of tumor growth in vivo by retrovirus-mediated gene transfer. Cancer Res., 60, 372–382. [PubMed] [Google Scholar]
- Coats S., Whyte,P., Fero,M.L., Lacy,S., Chung,G., Randel,E., Firpo,E. and Roberts,J.M. (1999) A new pathway for mitogen-dependent cdk2 regulation uncovered in p27(Kip1)-deficient cells. Curr. Biol., 9, 163–173. [DOI] [PubMed] [Google Scholar]
- Cobrinik D. et al. (1996) Shared role of the pRB-related p130 and p107 proteins in limb development. Genes Dev., 10, 1633–1644. [DOI] [PubMed] [Google Scholar]
- Connell-Crowley L., Harper,J.W. and Goodrich,D.W. (1997) cyclin D1/Cdk4 regulates retinoblastoma protein-mediated cell cycle arrest by site-specific phosphorylation. Mol. Biol. Cell, 8, 287–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson N. (1998) The regulation of E2F by pRB-family proteins. Genes Dev., 12, 2245–2262. [DOI] [PubMed] [Google Scholar]
- Furukawa Y., Iwase,S., Kikuchi,J., Nakamura,M., Yamada,H. and Matsuda,M. (1999) Transcriptional repression of the E2F-1 gene by interferon-α is mediated through induction of E2F-4/pRB and E2F-4/p130 complexes. Oncogene, 18, 2003–2014. [DOI] [PubMed] [Google Scholar]
- Garrett M.D. and Fattaey,A. (1999) CDK inhibition and cancer therapy. Curr. Opin. Genet. Dev., 9, 104–111. [DOI] [PubMed] [Google Scholar]
- Gaubatz S., Lindeman,G.J., Ishida,S., Jakoi,L., Nevins,J.R., Livingston,D.M. and Rempel,R.E. (2000) E2F4 and E2F5 play an essential role in pocket protein-mediated G1 control. Mol. Cell, 6, 729–735. [DOI] [PubMed] [Google Scholar]
- Grana X., Garriga,J. and Mayol,X. (1998) Role of the retinoblastoma protein family, pRB, p107 and p130 in the negative control of cell growth. Oncogene, 17, 3365–3383. [DOI] [PubMed] [Google Scholar]
- Hannon G.J., Demetrick,D. and Beach,D. (1993) Isolation of the Rb-related p130 through its interaction with CDK2 and cyclins. Genes Dev., 7, 2378–2391. [DOI] [PubMed] [Google Scholar]
- Hansen K., Johnell,M., Siegbahn,A., Rorsman,C., Engström,U., Wernstedt,C., Heldin,C.-H. and Rönnstrand,L. (1996) Mutation of a Src phosphorylation site in the PDGF β-receptor leads to increased PDGF-stimulated chemotaxis but decreased mitogenesis. EMBO J., 15, 5299–5313. [PMC free article] [PubMed] [Google Scholar]
- Harbour J.W. and Dean,D.C. (2000) The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev., 14, 2393–2409. [DOI] [PubMed] [Google Scholar]
- Harbour J.W., Luo,R.X., Dei Santi,A., Postigo,A.A. and Dean,D.C. (1999) Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell, 98, 859–869. [DOI] [PubMed] [Google Scholar]
- Helin K., Holm,K., Niebuhr,A., Eiberg,H., Tommerup,N., Hougaard,S., Poulsen,H.S., Spang-Thomsen,M. and Norgaard,P. (1997) Loss of the retinoblastoma protein-related p130 protein in small cell lung carcinoma. Proc. Natl Acad. Sci. USA, 94, 6933–6938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurford R.K., Cobrinik,D.,Jr, Lee,M.H. and Dyson,N. (1997) pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev., 11, 1447–1463. [DOI] [PubMed] [Google Scholar]
- Iavarone A. and Massagué,J. (1999) E2F and histone deacetylase mediate transforming growth factor β repression of cdc25A during keratinocyte cell cycle arrest. Mol. Cell. Biol., 19, 916–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D.G. (1995) Regulation of E2F-1 gene expression by p130 (Rb2) and D-type cyclin kinase activity. Oncogene, 11, 1685–1692. [PubMed] [Google Scholar]
- Kitagawa M. et al. (1996) The consensus motif for phosphorylation by cyclin D1–Cdk4 is different from that for phosphorylation by cyclin A/E–Cdk2. EMBO J., 15, 7060–7069. [PMC free article] [PubMed] [Google Scholar]
- Knudsen E.S. and Wang,J.Y. (1996) Differential regulation of retinoblastoma protein function by specific Cdk phosphorylation sites. J. Biol. Chem., 271, 8313–8320. [DOI] [PubMed] [Google Scholar]
- Knudsen E.S., Buckmaster,C., Chen,T.T., Feramisco,J.R. and Wang,J.Y. (1998) Inhibition of DNA synthesis by RB: effects on G1/S transition and S-phase progression. Genes Dev., 12, 2278–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. (1999) Histone acetylases and deacetylases in cell proliferation. Curr. Opin. Genet. Dev., 9, 40–48. [DOI] [PubMed] [Google Scholar]
- Lacy S. and Whyte,P. (1997) Identification of a p130 domain mediating interactions with cyclin A/cdk 2 and cyclin E/cdk 2 complexes. Oncogene, 14, 2395–2406. [DOI] [PubMed] [Google Scholar]
- LeCouter J.E., Kablar,B., Whyte,P.F., Ying,C. and Rudnicki,M.A. (1998) Strain-dependent embryonic lethality in mice lacking the retinoblastoma-related p130 gene. Development, 125, 4669–4679. [DOI] [PubMed] [Google Scholar]
- Lee J.O., Russo,A.A. and Pavletich,N.P. (1998) Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature, 391, 859–865. [DOI] [PubMed] [Google Scholar]
- Lees J.A., Buchkovich,K.J., Marshak,D.R., Anderson,C.W. and Harlow,E. (1991) The retinoblastoma protein is phosphorylated on multiple sites by human cdc2. EMBO J., 10, 4279–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski M.M. and Jacks,T. (1999) The retinoblastoma gene family in differentiation and development. Oncogene, 18, 7873–7882. [DOI] [PubMed] [Google Scholar]
- Livingston D.M., Kaelin,W., Chittenden,T. and Qin,X. (1993) Structural and functional contributions to the G1 blocking action of the retinoblastoma protein. Br. J. Cancer, 68, 264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas J., Parry,D., Aagaard,L., Mann,D.J., Bartkova,J., Strauss,M., Peters,G. and Bartek,J. (1995) Retinoblastoma-protein-dependent cell-cycle inhibition by the tumour suppressor p16. Nature, 375, 503–506. [DOI] [PubMed] [Google Scholar]
- Lukas J., Herzinger,T., Hansen,K., Moroni,M.C., Resnitzky,D., Helin,K., Reed,S.I. and Bartek,J. (1997) cyclin E-induced S-phase without activation of the pRb/E2F pathway. Genes Dev., 11, 1479–1492. [DOI] [PubMed] [Google Scholar]
- Lukas C., Sörensen,C.S., Kramer,E., Santoni-Rugiu,E., Lindeneg,C., Peters,J.M., Bartek,J. and Lukas,J. (1999a) Accumulation of cyclin B1 requires E2F and cyclin-A-dependent rearrangement of the anaphase-promoting complex. Nature, 401, 815–818. [DOI] [PubMed] [Google Scholar]
- Lukas J., Sörensen,C.S., Lukas,C., Santoni-Rugiu,E. and Bartek,J. (1999b) p16INK4a, but not constitutively active pRb, can impose a sustained G1 arrest: molecular mechanisms and implications for oncogenesis. Oncogene, 18, 3930–3935. [DOI] [PubMed] [Google Scholar]
- Mayol X., Garriga,J. and Grana,X. (1995) Cell cycle-dependent phosphorylation of the retinoblastoma-related protein p130. Oncogene, 11, 801–808. [PubMed] [Google Scholar]
- Meraldi P., Lukas,J., Fry,A.M., Bartek,J. and Nigg,E.A. (1999) Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A. Nature Cell Biol., 1, 88–93. [DOI] [PubMed] [Google Scholar]
- Mittnacht S. (1998) Control of pRB phosphorylation. Curr. Opin. Genet. Dev., 8, 21–27. [DOI] [PubMed] [Google Scholar]
- Moberg K., Starz,M.A. and Lees,J.A. (1996) E2F-4 switches from p130 to p107 and pRB in response to cell cycle reentry. Mol. Cell. Biol., 16, 1436–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller H., Moroni,M.C., Vigo,E., Petersen,B.O., Bartek,J. and Helin,K. (1997) Induction of S-phase entry by E2F transcription factors depends on their nuclear localization. Mol. Cell. Biol., 17, 5508–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X.Q., Chittenden,T., Livingston,D.M. and Kaelin,W.G.,Jr (1992) Identification of a growth suppression domain within the retinoblastoma gene product. Genes Dev., 6, 953–964. [DOI] [PubMed] [Google Scholar]
- Sardet C., Vidal,M., Cobrinik,D., Geng,Y., Onufryk,C., Chen,A. and Weinberg,R.A. (1995) E2F-4 and E2F-5, two members of the E2F family, are expressed in the early phases of the cell cycle. Proc. Natl Acad. Sci. USA, 92, 2403–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears R., Ohtani,K. and Nevins,J.R. (1997) Identification of positively and negatively acting elements regulating expression of the E2F2 gene in response to cell growth signals. Mol. Cell. Biol., 17, 5227–5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheaff R.J., Groudine,M., Gordon,M., Roberts,J.M. and Clurman,B.E. (1997) Cyclin E–CDK2 is a regulator of p27Kip1. Genes Dev., 11, 1464–1478. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. and Roberts,J.M. (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev., 13, 1501–1512. [DOI] [PubMed] [Google Scholar]
- Smith E.J., Leone,G., DeGregori,J., Jakoi,L. and Nevins,J.R. (1996) The accumulation of an E2F-p130 transcriptional repressor distinguishes a G0 cell state from a G1 cell state. Mol. Cell. Biol., 16, 6965–6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E.J., Leone,G. and Nevins,J.R. (1998) Distinct mechanisms control the accumulation of the Rb-related p107 and p130 proteins during cell growth. Cell Growth Differ., 9, 297–303. [PubMed] [Google Scholar]
- Tommasi S. and Pfeifer,G.P. (1995) In vivo structure of the human cdc2 promoter: release of a p130–E2F-4 complex from sequences immediately upstream of the transcription initiation site coincides with induction of cdc2 expression. Mol. Cell. Biol., 15, 6901–6913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vooijs M. and Berns,A. (1999) Developmental defects and tumor predisposition in Rb mutant mice. Oncogene, 18, 5293–5303. [DOI] [PubMed] [Google Scholar]
- Wang J.Y. (1997) Retinoblastoma protein in growth suppression and death protection. Curr. Opin. Genet. Dev., 7, 39–45. [DOI] [PubMed] [Google Scholar]
- Weinberg R.A. (1995) The retinoblastoma protein and cell cycle control. Cell, 81, 323–330. [DOI] [PubMed] [Google Scholar]
- Woo M.S., Sanchez,I. and Dynlacht,B.D. (1997) p130 and p107 use a conserved domain to inhibit cellular cyclin-dependent kinase activity. Mol. Cell. Biol., 17, 3566–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarkowska T. and Mittnacht,S. (1997) Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J. Biol. Chem., 272, 12738–12746. [DOI] [PubMed] [Google Scholar]
- Zhu L., Xie,E. and Chang,L.S. (1995) Differential roles of two tandem E2F sites in repression of the human p107 promoter by retinoblastoma and p107 proteins. Mol. Cell. Biol., 15, 3552–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]