

Abstract
As part of a broader effort to identify postreceptor signal regulators involved in specific diseases or organ adaptation, we used an expression cloning system in Saccharomyces cerevisiae to screen cDNA libraries from rat ischemic myocardium, human heart, and a prostate leiomyosarcoma for entities that activated G protein signaling in the absence of a G protein coupled receptor. We report the characterization of activator of G protein signaling (AGS) 8 (KIAA1866), isolated from a rat heart model of repetitive transient ischemia. AGS8 mRNA was induced in response to ventricular ischemia but not by tachycardia, hypertrophy, or failure. Hypoxia induced AGS8 mRNA in isolated adult ventricular cardiomyocytes but not in rat aortic smooth muscle cells, endothelial cells, or cardiac fibroblasts, suggesting a myocyte-specific adaptation mechanism involving remodeling of G protein signaling pathways. The bioactivity of AGS8 in the yeast-based assay was independent of guanine nucleotide exchange by Gα, suggesting an impact on subunit interactions. Subsequent studies indicated that AGS8 interacts directly with Gβγ and this occurs in a manner that apparently does not alter the regulation of the effector PLC-β2 by Gβγ. Mechanistically, AGS8 appears to promote G protein signaling by a previously unrecognized mechanism that involves direct interaction with Gβγ.
Keywords: accessory protein, signal adaptation, hypoxia
Signal processing via heterotrimeric G proteins (Gαβγ) is one of the most widely used systems for information transfer across the cell membrane. Dysfunctional processing of signals through these transducing systems results in organ maladaptation and is characteristic of various disease states. In addition to G protein-coupled receptor (GPCR)-mediated activation of G protein signaling, nature has evolved other modes of signal input to G proteins that either act independent of a GPCR or modulate signal transfer from receptor to G protein. Recent studies also indicate a role of Gα and Gβγ in the processing of signals within the cell distinct from those that they mediate following activation by a cell-surface GPCRs (1–10). In such situations, the G protein subunits (Gα and Gβγ) may actually be complexed with alternative binding partners independent of the typical heterotrimeric Gαβγ.
A variety of approaches (protein interactions strategies, genetic screens, functional screens) have led to the identification of several nonreceptor proteins that indeed influence G protein signaling. Such proteins include the family of RGS proteins that accelerate the GTPase activity of Gα and various entities that influence nucleotide binding properties and/or subunit interactions (2). The latter group of proteins includes activator of G protein signaling (AGS) 1–4, which were identified in a yeast-based functional screen for receptor-independent activators of G protein signaling as an extension of earlier biochemical characterization of such bioactivity in cell and tissue extracts (1–3, 11–13). The interaction of AGS1, AGS3, and AGS3-related proteins with heterotrimeric G proteins has subsequently been shown to regulate a diverse set of functions including asymmetric cell division, addictive behavior, and the circadian rhythm (1, 3–7, 14–18).
Such regulatory mechanisms are of particular interest because they may allow subtle alterations in signal processing as required by the cell as it meets various challenges placed upon it by alterations in its microenvironment. At the same time, such regulatory proteins may also be involved in the disease process itself by participating in the resetting of signaling set points as tissues “overcompensate” in various situations. As part of a broader approach to define signal regulators involved in the remodeling of G protein signaling networks that occurs in response to physiological or pathophysiological challenges, we used a functional screen to identify receptor-independent AGSs up-regulated in the face of such a challenge.
Here, we report the identification of a hypoxia-inducible, receptor independent AGS (AGS8) that is induced as a part of cardiomyocyte adaptation to an ischemic challenge. AGS8 is a representative member of a subgroup of AGS proteins that directly interact with Gβγ. The specific up-regulation of AGS8 in response to hypoxia or ischemia likely represents a component of signal remodeling that occurs during myocyte adaptation to physiological and pathological challenges. Mechanistically, AGS8 appears to promote G protein signaling by a previously unrecognized mechanism for G protein regulation that involves direct interaction with Gβγ.
Results and Discussion
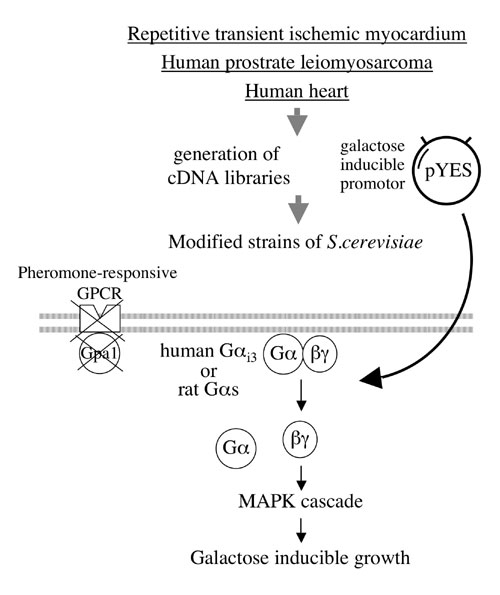
Identification of Receptor-Independent AGSs. Building on the initial strategy that led to the identification of AGS1–3 (12, 13), we undertook a broad screen of cDNA libraries for additional AGS proteins. This screen uses an expression cloning strategy in Saccharomyces cerevisiae to select for receptor-independent activators of the pheromone response pathway in which heterotrimeric G proteins regulate a mitogen-activated protein kinase cascade controlling yeast mating and growth (Fig. 5, which is published as supporting information on the PNAS web site) (12, 13, 19). Our strategy involved the use of a modified yeast strain lacking the pheromone receptor and containing mammalian Gα (Gαi3,Gαs, or Gα16) in place of the yeast Gα subunit. The yeast strain was further modified to respond to activation of the G protein regulated signaling cascade with a readout of growth. This modification allowed us to rapidly screen mammalian cDNA libraries constructed in a galactose-inducible vector for activators of this signaling cascade by selecting for those promoting growth in a galactose-specific manner (12, 13, 19). cDNAs isolated with this strategy that required the presence of heterotrimeric G protein for bioactivity were termed AGS proteins and thus are functionally defined rather than based on any conserved protein sequences.
As part of the goal to identify postreceptor signal regulators involved in organ adaptation or disease processes, we screened cDNA libraries from an animal model of cardiac ischemia, human heart, and a prostate leiomyosarcoma (Table 1 and Fig. 5) using modified yeast strains expressing Gαi3, Gαs, or Gα16. Sixty-five cDNAs encoding nine distinct proteins were isolated (Gαs strain-18; Gαi3 strain-47; Gα16 strain-0) with the three cDNA libraries. Using simple growth assays on selective media containing glucose (no cDNA induction) or galactose (cDNA induction) and yeast strains with deletions or modifications of their pheromone signaling pathways we could determine whether the bioactivity of these cDNAs required Gβγ or acted downstream of G protein to influence pathway activation (Figs. 1 and 5) (12, 13, 19). In such epistasis experiments, nine of the cDNAs required Gβγ for their activity and thus were consistent with the definition of a receptor-independent activator of G protein signaling (Table 1). Three of the nine cDNAs encoded the previously characterized AGS1, AGS3, and AGS4 (12, 13, 20). AGS5 and AGS6 encoded truncated versions of the previously characterized proteins LGN and RGS12 respectively and these truncated versions each contained the G protein regulatory (GPR) motif(s) found in these two proteins. The GPR motif inhibits GDP dissociation from Gαi, transducin, and Gαo (1, 2, 13, 21, 22). AGS7 encodes the C-terminal region of TRIP13 (CAG33025). AGS9 encodes Rpn10, a component of the 26S proteasome, and AGS10 encodes Gαo a major Gα protein found in brain. AGS8 represented a truncated version of an uncharacterized mRNA and is described in detail below.
Table 1. AGS cDNAs isolated in functional screens for receptor-independent AGS.
| Tissue used to generate cDNA libraries for functional screen*
|
||||
|---|---|---|---|---|
| AGS | Protein or gene names in databases and literature | Repetitive transient cardiac ischemia (rat) | Heart (human) | Prostate Leiomyosarcoma (human) |
| AGS1 | DexRas1, RASD1 | + | - | - |
| AGS3 | GPSM1 (C-terminal segment with 3-4 GPR motifs) | + | + | + |
| AGS4 | G18.1b, GPSM3 (three GPR motifs) | + | - | + |
| AGS5 | LGN, GPSM2 (C-terminal 322 amino acids, four GPR motifs) | - | - | + |
| AGS6 | RGS12 (C-terminal 365 amino acids, one GPR motif) | - | - | + |
| AGS7 | TRIP13† | - | - | + |
| AGS8 | KIAA 1866 (C-terminal 372 amino acids) | + | - | - |
| AGS9 | Rpn10 | + | + | - |
| AGS10 | Gαo | + | - | - |
AGS refers to cDNAs isolated in the functional screen and they are numbered according to the order in which they were isolated. Unless indicated otherwise, isolated cDNAs encoded the entire coding sequence. GPR, G protein regulatory motif (13). The rat heart cDNA library contained 3.1 × 1011 colony-forming units (cfu) with an average insert size of 2.0 kb. The human heart cDNA library contained 2.4 × 107 with an average insert size of 1.6 kb. The human prostate leiomyosarcoma cDNA library contained 1.5 × 107 cfu with average insert size of 1.3 kb. Number of transformants screened for each library: Repetitive transient ischemic heart (rat), 3.1 × 105; prostate leiomyosarcoma (human), 4.3 × 106; normal heart (human), 1.0 × 106.
cDNA libraries were screened in yeast strains CY1141 (Gαi3), CY8342 (Gαs), and CY9603 (Gα16) (12, 13).
C-terminal sequence, unpublished data.
Fig. 1.

Bioactivity of AGS8 in the yeast-based functional assay. In A–C, data are presented in a series of three panels to illustrate viability of the transformed yeast and the galactose-dependent growth under the selective pressure of exclusion of histidine from the media (12, 13). Galactose media promotes expression of individual mammalian cDNAs in the pYES-DEST52 vector containing the GAL1 promoter. In each experiment, the same panel of independent yeast transformants are spotted in parallel on glucose plus histidine (Left, no selection), glucose minus histidine (Center, selection without induction), and galactose minus histidine (Right, selection plus induction). The histidine analog aminotriazole is included in the media lacking histidine and this confers selective growth when the pheromone response pathway is activated. (A) Growth promoting activity of cDNA 1–16. Transformants in yeast strains expressing rat Gαs and in yeast strains lacking Gα,Gβ, or downstream signaling molecules (ΔGα, yeast lacking Gα; ΔGβ, yeast lacking Gβ; ΔSte20, yeast lacking p21 activated kinase; ΔSte5, yeast lacking the kinase scaffolding protein). (B) Effect of AGS8 (cDNA 1–16) in yeast expressing various types of Gα. This assay was repeated with two independent AGS8 (cDNA 1–16) clones. (C) Effect of AGS8 (cDNA 1–16) in yeast expressing Gαi2-G204A. Yeast strains containing Gαi2 or Gαi2-G204A was transfected with empty vector and AGS1 or AGS8 (cDNA 1–16). AGS1, which does not function in yeast expressing Gαi2-G204A, was used as a positive internal control (12, 13).
Identification and Bioactivity of AGS8. AGS8 was isolated from the cDNA library generated from a model of repetitive transient ischemia of the heart. In this model, myocardial ischemia was induced by inflation of a pneumatic snare (40 s every 20 min for 8-h periods per day) implanted around the proximal left anterior descending artery (LAD), which leads to marked coronary collateral development in 10 days (E.T. and W.M.C., unpublished observations, and ref. 23). The ischemic area of the left ventricle perfused by the LAD was isolated as “ischemic area” at day 3 and used to generate a cDNA library for screening in the yeast-based expression cloning system described above. This screen yielded six distinct cDNAs: AGS1, AGS3, AGS4, AGS8, or cDNA 1–16, AGS9, and AGS10 (Table 1). AGS8 (cDNA 1–16) encoded the C-terminal 372 aa of an uncharacterized protein KIAA1866 (GenBank accession no. XM_217792.2) plus 621 nt of 3′ untranslated region. Mutation of the first methionine within this terminal 372-aa fragment of the AGS8 cDNA to alanine eliminated its bioactivity in the functional screen, confirming that the reading frame defined within KIAA1866 confers the bioactivity (M.S., W.M.C., and S.M.L. unpublished observations).
As has been reported for AGS1–3 (12, 13, 24), the yeast-based assay system allows rapid determination of the basic mechanisms of action for isolated cDNAs. As mentioned above, epistasis experiments indicated that within the G protein-regulated mitogen-activated protein kinase cascade conferring growth in this yeast strain, AGS8 functioned at the level of G protein itself as its activity was not observed in yeast strains lacking Gβ, Ste20 (p21 activated kinase) or the scaffolding protein Ste5 (Fig. 1 A). AGS8 (cDNA 1–16) was active in yeast strains expressing Gαs, Gαi3, Gα16 and Gpa1 suggesting that its interaction with G protein may involve Gβγ, which is common to each of the yeast strains (Fig. 1B). AGS8 (cDNA 1–16) was also active in yeast strains expressing a mutant of Gαi2 (Gαi2-G204A), which is postulated to be “locked” in a GDP bound state (12, 13, 25), suggesting that AGS8 is influencing subunit interactions independent of nucleotide exchange (Fig. 1C) (1, 2).
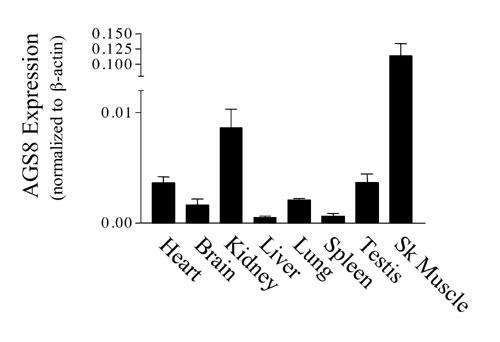
KIAA1866 is a predicted protein from the rat genome containing 4 fibronectin type 3 protein domains, which are found in a large number of intracellular proteins and cell surface receptors (cd00063, www.ncbi.nlm.nih.gov/Structure/cdd/cddsrv.cgi?uid=cd00063; SM00060, www.ncbi.nlm.nih.gov/Structure/cdd/cddsrv.cgi?uid=smart00060; pfam00041, www.ncbi.nlm.nih.gov/Structure/cdd/cddsrv.cgi?uid=pfam00041) (Fig. 2A). Full-length AGS8 (GenBank accession no. DQ256268), encoding 1,730 aa, was cloned from a rat heart cDNA library by RT-PCR. The rat cDNA exhibits 67% amino acid identity with the human predicted protein NP_115921 and 86% amino acid identity with the mouse predicted protein XP_354975 (Fig. 2 A). RT-PCR analysis indicated that AGS8 mRNA was expressed in several tissues (Fig. 6, which is published as supporting information on the PNAS web site).
Fig. 2.

AGS8 coding region and regulation of AGS8 expression in the heart. (A) Schematic diagram of the sequence AGS8/KIAA1866 and related proteins in mouse and human. The line above the rat sequence refers to cDNA 1–16 isolated in the yeast-based functional screen. (B) Expression of AGS8 in various rat models of cardiac dysfunction. The expression level of AGS8 was analyzed by real-time PCR. Left ventricle samples were separated into “nonischemic” and “ischemic” areas as described in Experimental Procedures. Control refers to saline infused or sham operated rat. Data are presented as the mean ± SEM of five (ischemic protocol) or three (tachycardia, hypertrophy, heart failure) rats for each group. *, P < 0.01 vs. nonischemic area. (C) Hypoxic induction of AGS8 in different cell types. Cultured adult left ventricular cardiomyocytes (LVCM), rat aortic smooth muscle cells (SMC), rat aortic endothelial cells (EC), and rat cardiac fibroblasts (CF) were exposed to normoxia or 0% oxygen for 1 h. RNA was immediately extracted, and the expression of AGS8 was determined by real-time PCR. Data are expressed as the percent of change of level of AGS8 compared to cells cultured in normoxia. AGS8/β-actin (normoxia control): LVCM, 4.5 × 10–4; SMC, 8.7 × 10–4; EC, 4.5 × 10–3; CF, 1.2 × 10–6. Data are from four independent preparations. *, P < 0.01 vs. normoxia control. Data are presented as the mean ± SEM.
Regulation of Expression of AGS8 by Ischemia/Hypoxia. Although AGS8 cDNA was isolated in the yeast screen by using a cDNA library prepared from ischemic myocardium, it was unclear whether it was constitutively expressed or whether it was actually induced in response to the ischemic challenge. Thus, we asked whether AGS8 is induced in the ischemic myocardium. AGS8 mRNA expression was up-regulated 3.5 times in the ischemic area of the left ventricle compared to the nonischemic area in the repetitive transient ischemia model (Fig. 2B). This difference was not observed in the corresponding regions of the heart in nontreated control rats (data not shown). We next asked whether this induction was specific for the ischemic challenge or whether it represented a more general response to myocardial insult. Interestingly, the induction of AGS8 was specific for the transient ischemia model because it was not observed in other cardiac dysfunction models, including sustained tachycardia, hypertrophy, and heart failure (Fig. 2B).
Of the AGS cDNAs isolated in the screen of the repetitive transient ischemia animal model library (AGS1, AGS3, AGS4, AGS8, AGS9, AGS10), only AGS8 mRNA was up-regulated in the ischemic myocardium (M.S., W.M.C., and S.M.L. unpublished observations). We next asked whether the AGS8 mRNA induction observed in the ischemic myocardium reflected changes within the myocyte itself. The hypoxic induction (0% O2 for 1 h) of AGS8 was evaluated in cultured adult left ventricle myocytes, cardiac fibroblasts, rat aortic smooth muscle cells, and rat aortic endothelial cells. The hypoxic induction of AGS8 mRNA was only observed in cultured adult left ventricle myocytes (Fig. 2C). Thus, the induction of AGS8 mRNA is specific for the myocyte and the type of stress placed upon the heart.
AGS8 and G Protein Regulation. The cardiac- and ischemic-specific induction of AGS8 and its ability to influence G protein signaling in the functional screen suggests that AGS8 may influence G protein signaling mechanisms as part of the myocyte adaptation to hypoxia. To provide a foundation to address this hypothesis, we asked basic questions regarding the interaction of AGS8 with G proteins. The interaction of the bioactive AGS8 cDNA (cDNA 1–16; amino acids A1359-W1730; termed AGS8-C) with G proteins was examined in GST pull-down assays using purified G protein subunits and rat brain lysates. The “Input” lanes contain one-tenth of the mixture used in individual interaction assays. AGS8-C effectively bound purified brain Gβγ but not purified Gαi1 (Fig. 3A Left). In brain lysates, AGS8-C pulled down Gβγ, but not Gαi1/2, Gαi3, Gαo, or Gαs (Fig. 3A Right). The absence of Gα in the pull downs from brain lysate suggest that AGS8 is either influencing subunit interactions or that there is a population of Gβγ that exists free of Gα. AGS8-C actually exhibits a preference for interaction with Gβ1γ2 as compared to Gβ2γ2 further indicating the specificity of the interaction (Fig. 3B). The importance of this specific interaction with a Gβ subtype in the action of AGS8 is not clear, but it is one of the few situations of such selectivity for binding partners or effectors of Gβγ.
Fig. 3.

Biochemical characterization of AGS8. GST pull down assay of AGS8 with purified G protein (A Left), rat brain lysate (A Right) and Sf9 synthesized biotinylated Gβ1γ2 and Gβ2γ2 (B). The C-terminal 372-aa fragment of AGS8 (AGS8-C) was expressed as a GST fusion protein. GST-fusion proteins (300 nM) were incubated with purified G protein subunit Gαi1 (30 nM) (A Lower Left), bovine brain Gβγ (30 nM) (A Upper Left and B) or rat brain lysate (1% Nonidet P-40 lysate, 1 mg protein) (A Right) or biotinylated Gβγ (30 nM) (B) in a total volume of 250 μl at 4°C. Proteins were then adsorbed to a glutathione matrix and retained G protein subunits identified by immunoblotting after gel electrophoresis. Membrane transfers of bound proteins were first probed with specific antisera followed by stripping and reprobing of the membrane transfer with GST antisera to validate equal amounts of fusion proteins. The arrows to the right indicate the migration of indicated proteins. The “Input” lanes contain one-tenth of the mixture used in individual interaction assays. The data are representative of four (A) and three (B) independent experiments.
These data suggest that AGS8 promotes G protein signaling in the functional screen by interacting with Gβγ rather than Gα. AGS8 may achieve this by interacting with heterotrimer and promoting subunit dissociation independent of nucleotide exchange, interacting with Gβγ during basal cycling of the G protein through its activation–deactivation cycle, or complexing with Gβγ before it ever has a chance to form a heterotrimer with Gα. If AGS8 indeed complexes with Gβγ, then either it dissociates before interaction of Gβγ with its effector or its interaction with Gβγ does not impede Gβγ binding to and activation of its effector, providing an unexpected mode of activation for G protein signaling systems.
The influence of AGS8 on Gβγ-mediated signaling was further examined in mammalian cells by using PLC-β2 as a representative Gβγ sensitive effector. In COS-7 cells transfected with the Gβγ effector PLC-β2, cotranfection with Gβγ increases inositol phosphate production, and this effect is blocked by coexpression of Gαi3, which binds to Gβγ and prevents its interaction with effector (Fig. 4A Left). In contrast, coexpression of AGS8-C did not alter the increase in inositol phosphates observed with Gβγ transfection∥ and actually reversed the inhibitory effect of Gαi3 (Fig. 4A Left). A similar functional role of AGS8 appears operative in the yeast-based assay. Gβγ interfaces to the mitogen-activated protein kinase cascade in S. cerevisiae, and this action is “antagonized” by Gα, which sequesters Gβγ, precluding Gβγ interaction with its effector. As observed in the mammalian system, AGS8 also reversed the inhibitory action of expressed Gα in the functional screen in yeast (Fig. 4A Right). Full-length AGS8 also did not antagonize the ability of Gβγ to elicit an increase in inositol phosphates (Fig. 4B). The elevated level of coexpressed Gβγ and AGS8 may reflect a greater stability of the proteins when complexed with each other. These data suggest that AGS8 interacts with Gβγ in a manner that still allows Gβγ interaction with specific effectors. This concept is supported by in vitro data using purified Gα, Gβγ and the Gβγ effector PLC-β2 (C. J. Yuan, S.M., S.M.L., and A.V.S., unpublished data). This finding is of particular note, because mammalian Gβγ interacting proteins such as Gα, PLC-β2, GRK2, adenylyl cyclase type II, and ion channels generally share a common interface on Gβγ (26).
Fig. 4.

Influence of AGS8 on Gβγ sensitive signaling pathway. (A) (Left) Influence of AGS8-C on the generation of inositol phosphates. COS-7 cells were transfected in 12-well plates with control vectors or cDNAs as indicated (0.3 μg of pcDNA3::PLC-β2, 0.3 μg of pcDNA3::Gαi3, 0.2 μg of pcDNA3::Gβ1 plus 0.2 μg of pcDNA3::Gγ2,1 μg of pcDNA3::AGS8-C). The amount of DNA transfected was adjusted to 2 μg per well with pcDNA3 vector. Data are expressed as the percent of inositol phosphates in cells transfected with vector alone (1,002.5 ± 138.5 cpm). (Right) Effect of AGS8 in yeast lacking Gα. Yeast strain CY1316 containing empty vector (pRS424) or pRS424::Gαs(E10K) were transformed with empty vector (pYES2) or pYES2::AGS8-C. Data are presented as described in Fig. 1, and are representative of three independent experiments. (B) Influence of full-length AGS8 on the generation of inositol phosphates. COS-7 cells were transfected in 12-well plates with control vectors or cDNAs as indicated (0.3 μg of pcDNA3::PLC-β2, 0.2 μg of pcDNA3::Gβ1 plus 0.2 μg of pcDNA3::Gγ2, 1.3 μg of pcDNA3::AGS8). The amount of DNA transfected was adjusted to 2 μg per well with pcDNA3 vector. Data are expressed as the percent of inositol phosphates in cells transfected with vector alone (1,055.5 ± 163.2 cpm). Data are presented as the mean ± SEM of five (A Left) or eight (B) experiments with duplicate or triplicate determinations. In both A and B, expression of transfected cDNAs was determined by immunoblotting of 10 μg of whole cell lysates. *, P < 0.05.
The current data with AGS8 in both the yeast and mammalian systems along with the recent reports regarding direct regulation of Gβγ (27, 28) and diversity of Gβγ binding partners (29, 30) suggest that Gβγ may possess additional protein binding sites for signal processing (28, 31). Such Gβγ binding partners may serve as effectors for mammalian Gβγ, influence receptor coupling to Gαβγ or localize Gβγ within a larger signaling complex, where it can regulate specific signaling pathways independent of the classical heterotrimeric Gαβγ.
Perspective. Adaptation of the myocardium to ischemia involves alteration of several signaling pathways (i.e., ERK1/2, AKT, protein kinase C, and/or phospholipase C) involved in collateral growth (32, 33), apoptosis (33, 34), energy metabolism (33, 35), and the development of a “preconditioned” myocardium. Gβγ signaling is clearly involved in many aspects of such signaling pathways (36–39). AGS8 provides an unexpected mechanism for regulation of the G protein activation cycle and is specifically induced in the heart in response to transient repetitive ischemia and/or hypoxia. Thus, the receptor-independent regulation of the key signaling cassettes involving Gα and Gβγ may be a very powerful component of organ adaptation and adjustment of the general signaling set point characteristic of individual cells.
Experimental Procedures
Animal Models. Repetitive transient myocardial ischemia. Repetitive transient myocardial ischemia was achieved in male Wistar rats (-315g) as described (23). Briefly, a pneumatic snare was implanted around the proximal left anterior descending artery (LAD) under isoflurane anesthesia. After 3–4 days of recovery, repetitive transient ischemia was induced by coronary occlusion (40-s occlusion per 20 min over an 8-h period for a total of 24 occlusion periods), and this was repeated for 3 days, after which the animals were killed for tissue extraction. Left ventricles were quickly separated into the “ischemic area” perfused by the LAD and the “nonischemic area” perfused by the left circumflex artery and the right coronary artery. The samples were frozen in liquid nitrogen and stored at –70°C until use.
Cardiac hypertrophy and cardiac tachycardia. Male 8-week-old Sprague–Dawley rats (190–210 g) were separated into three groups: Control, saline-infused (CNT); tachycardia, isoproterenol-infusion (ISO) (3 μg/g per day); and hypertrophy, angiotensin II-infusion (ATII) (0.75 μg/g per day). Rats were anesthetized with pentobarbital sodium (40 μg/g, i.p.) and an osmotic minipump (Model 2002, Altezt, Cupertino, CA) was implanted s.c. Systolic blood pressure and heart rate were measured by tail-cuff method. After 12 days of infusion, the rats were anesthetized, and hearts were rapidly excised. Left ventricles were rapidly frozen in liquid nitrogen and stored at –70°C until use. The heart rate was increased in the isoproterenol-infused group (beats/min, mean ± SEM, ISO 506.7 ± 20.1 vs. CNT 439.3 ± 8.8, P < 0.05). The systolic blood pressure (SBP) and left ventricular/body weight (LV/BW) were increased in the angiotensin II-treated group [SBP (mm Hg): ATII 199.2 ± 5.7 vs. CNT 139.6 ± 12.0, P < 0.01; LV/BW (103): ATII 2.802 ± 0.115 vs. CNT 2.137 ± 0.067, P < 0.01].
Aortocaval fistula (ACF) model. The ACF model of cardiac failure in rat was created as described (40). Left ventricular tissue samples were obtained from animals killed 21 weeks after surgery in the late stages of heart failure.
Generation of cDNA Libraries, Functional Screen in S. cerevisiae. mRNA isolated from the ischemic area of the repetitive transient ischemia model of rat heart was used to synthesize cDNAs using SuperScript II (Invitrogen), which were directionally cloned into pDONR222 (Invitrogen) and swapped into the pYES-DEST52 yeast expression vector by recombination. A human prostate leiomyosarcoma cDNA library in pCMVSport6 (Invitrogen) and ProQuest normalized human heart cDNA library in pEXP-AD502 (Invitrogen) were swapped into pYES-DEST52 (Invitrogen) by recombination. Functional screens and growth assays in modified strains of S. cerevisiae were conducted as described (12, 13, 19).
Miscellaneous Procedures. Additional information on materials, AGS8 cloning, AGS8 mRNA analysis, generation of AGS8 antibody, protein interaction assays, cell culture, and data analysis are provided in Supporting Text, which is published as supporting information on the PNAS web site.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants NS24821 and MH55391 (to S.M.L.). Cell lines were propagated in the Cell and Molecular Imaging Core Facility in the Department of Pharmacology and Center of Excellence in Cardiovascular Research at Louisiana State University Health Sciences Center, supported by the National Institutes of Health Grant P20 RR018766 (S.M.L., program director). S.M.L. is greatly appreciative for support provided by the David R. Bethune/Lederle Laboratories Professorship in Pharmacology and the Research Scholar Award from Yamanouchi Pharmaceutical Company (now named Astellas Pharma).
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: AGS, activator of G protein signaling.
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. DQ256268).
Footnotes
Immunoblotting of nondetergent lysates from AGS8-transfected COS cells with AGS8-specific antibody indicated marked enrichment of AGS8 in the pellet fraction obtained by centrifugation at 100,000 × g. AGS8-C and full-length AGS8 generally elicit a small but consistent increase in inositol phosphate production, suggesting that AGS8 is interacting with endogenous G proteins to increase Gβγ-mediated signaling. However, the magnitude of this effect depends on the levels of AGS8 expressed, which is sensitive to the number of plasmids used in the cotransfection experiments.
References
- 1.Blumer, J. B., Cismowski, M. J., Sato, M. & Lanier, S. M. (2005) Trends Pharmacol. Sci. 26, 470–476. [DOI] [PubMed] [Google Scholar]
- 2.Sato, M., Blumer, J. B., Simon, V. & Lanier, S. M., Annu. Rev. Pharmacol., in press. [DOI] [PubMed]
- 3.Cismowski, M. J. & Lanier, S. M. (2005) Rev. Physiol. Biochem. Pharmacol. 155, 57–80. [DOI] [PubMed] [Google Scholar]
- 4.David, N. B., Martin, C. A., Segalen, M., Rosenfeld, F., Schweisguth, F. & Bellaiche, Y. (2005) Nat. Cell Biol. 7, 1083–1090. [DOI] [PubMed] [Google Scholar]
- 5.Wang, H., Ng, K. H., Qian, H., Siderovski, D. P., Chia, W. & Yu, F. (2005) Nat. Cell Biol. 7, 1091–1098. [DOI] [PubMed] [Google Scholar]
- 6.Hampoelz, B., Hoeller, O., Bowman, S. K., Dunican, D. & Knoblich, J. A. (2005) Nat. Cell Biol. 7, 1099–1105. [DOI] [PubMed] [Google Scholar]
- 7.Gotta, M., Dong, Y., Peterson, Y. K., Lanier, S. M. & Ahringer, J. (2003) Curr. Biol. 13, 1029–1037. [DOI] [PubMed] [Google Scholar]
- 8.Le-Niculescu, H., Niesman, I., Fischer, T., DeVries, L. & Farquhar, M. G. (2005) J. Biol. Chem. 280, 22012–22020. [DOI] [PubMed] [Google Scholar]
- 9.Colombo, K., Grill, S. W., Kimple, R. J., Willard, F. S., Siderovski, D. P. & Gonczy, P. (2003) Science 300, 1957–1961. [DOI] [PubMed] [Google Scholar]
- 10.Jamora, C., Yamanouye, N., Van Lint, J., Laudenslager, J., Vandenheede, J. R., Faulkner, D. J. & Malhotra, V. (1999) Cell 98, 59–68. [DOI] [PubMed] [Google Scholar]
- 11.Sato, M., Ribas, C., Hildebrandt, J. D. & Lanier, S. M. (1996) J. Biol. Chem. 271, 30052–30060. [DOI] [PubMed] [Google Scholar]
- 12.Cismowski, M. J., Takesono, A., Ma, C., Lizano, J. S., Xie, X., Fuernkranz, H., Lanier, S. M. & Duzic, E. (1999) Nat. Biotechnol. 17, 878–883. [DOI] [PubMed] [Google Scholar]
- 13.Takesono, A., Cismowski, M. J., Ribas, C., Bernard, M., Chung, P., Hazard, S., 3rd, Duzic, E. & Lanier, S. M. (1999) J. Biol. Chem. 274, 33202–33205. [DOI] [PubMed] [Google Scholar]
- 14.Sanada, K. & Tsai, L. H. (2005) Cell 122, 119–131. [DOI] [PubMed] [Google Scholar]
- 15.Yu, F., Wang, H., Qian, H., Kaushik, R., Bownes, M., Yang, X. & Chia, W. (2005) Genes Dev. 19, 1341–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bowers, M. S., McFarland, K., Lake, R. W., Peterson, Y. K., Lapish, C. C., Gregory, M. L., Lanier, S. M. & Kalivas, P. W. (2004) Neuron 42, 269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yao, L., McFarland, K., Fan, P., Jiang, Z., Inoue, Y. & Diamond, I. (2005) Proc. Natl. Acad. Sci. USA 102, 8746–8751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng, H. Y., Obrietan, K., Cain, S. W., Lee, B. Y., Agostino, P. V., Joza, N. A., Harrington, M. E., Ralph, M. R. & Penninger, J. M. (2004) Neuron 43, 715–728. [DOI] [PubMed] [Google Scholar]
- 19.Cismowski, M. J., Takesono, A., Ma, C., Lanier, S. M. & Duzic, E. (2002) Methods Enzymol. 344, 153–168. [DOI] [PubMed] [Google Scholar]
- 20.Cao, X., Cismowski, M. J., Sato, M., Blumer, J. B. & Lanier, S. M. (2004) J. Biol. Chem. 279, 27567–27574. [DOI] [PubMed] [Google Scholar]
- 21.Bernard, M. L., Peterson, Y. K., Chung, P., Jourdan, J. & Lanier, S. M. (2001) J. Biol. Chem. 276, 1585–1593. [DOI] [PubMed] [Google Scholar]
- 22.Peterson, Y. K., Hazard, S., III, Graber, S. G. & Lanier, S. M. (2002) J. Biol. Chem. 277, 6767–6770. [DOI] [PubMed] [Google Scholar]
- 23.Toyota, E., Warltier, D., Brock, T., Ritman, E., Kolz, C., O'Malley, P., Rocic, P., Focardi, M. & Chilian, W. (2005) Circulation 112, 2108–2113. [DOI] [PubMed] [Google Scholar]
- 24.Cismowski, M. J., Ma, C., Ribas, C., Xie, X., Spruyt, M., Lizano, J. S., Lanier, S. M. & Duzic, E. (2000) J. Biol. Chem. 275, 23421–23424. [DOI] [PubMed] [Google Scholar]
- 25.Berghuis, A. M., Lee, E., Raw, A. S., Gilman, A. G. & Sprang, S. R. (1996) Structure (London) 4, 1277–1290. [DOI] [PubMed] [Google Scholar]
- 26.Ford, C. E., Skiba, N. P., Bae, H., Daaka, Y., Reuveny, E., Shekter, L. R., Rosal, R., Weng, G., Yang, C. S., Iyengar, R., et al. (1998) Science 280, 1271–1274. [DOI] [PubMed] [Google Scholar]
- 27.Yoshikawa, D. M., Bresciano, K., Hatwar, M. & Smrcka, A. V. (2001) J. Biol. Chem. 276, 11246–11251. [DOI] [PubMed] [Google Scholar]
- 28.Bonacci, T. M., Ghosh, M., Malik, S. & Smrcka, A. V. (2005) J. Biol. Chem. 280, 10174–10181. [DOI] [PubMed] [Google Scholar]
- 29.Dell, E. J., Connor, J., Chen, S., Stebbins, E. G., Skiba, N. P., Mochly-Rosen, D. & Hamm, H. E. (2002) J. Biol. Chem. 277, 49888–49895. [DOI] [PubMed] [Google Scholar]
- 30.Lukov, G. L., Hu, T., McLaughlin, J. N., Hamm, H. E. & Willardson, B. M. (2005) EMBO J. 24, 1965–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scott, J. K., Huang, S. F., Gangadhar, B. P., Samoriski, G. M., Clapp, P., Gross, R. A., Taussig, R. & Smrcka, A. V. (2001) EMBO J. 20, 767–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsunaga, T., Warltier, D. C., Weihrauch, D. W., Moniz, M., Tessmer, J. & Chilian, W. M. (2000) Circulation 102, 3098–3103. [DOI] [PubMed] [Google Scholar]
- 33.Bernaudin, M., Tang, Y., Reilly, M., Petit, E. & Sharp, F. R. (2002) J. Biol. Chem. 277, 39728–39738. [DOI] [PubMed] [Google Scholar]
- 34.Depre, C., Tomlinson, J. E., Kudej, R. K., Gaussin, V., Thompson, E., Kim, S. J., Vatner, D. E., Topper, J. N. & Vatner, S. F. (2001) Proc. Natl. Acad. Sci. USA 98, 9336–9341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Onody, A., Zvara, A., Hackler, L., Jr., Vigh, L., Ferdinandy, P. & Puskas, L. G. (2003) FEBS Lett. 536, 35–40. [DOI] [PubMed] [Google Scholar]
- 36.Naga Prasad, S. V., Esposito, G., Mao, L., Koch, W. J. & Rockman, H. A. (2000) J. Biol. Chem. 275, 4693–4698. [DOI] [PubMed] [Google Scholar]
- 37.Uchiyama, T., Engelman, R. M., Maulik, N. & Das, D. K. (2004) Circulation 109, 3042–3049. [DOI] [PubMed] [Google Scholar]
- 38.Cohen, M. V., Baines, C. P. & Downey, J. M. (2000) Annu. Rev. Physiol. 62, 79–109. [DOI] [PubMed] [Google Scholar]
- 39.Tong, H., Rockman, H. A., Koch, W. J., Steenbergen, C. & Murphy, E. (2004) Circ. Res. 94, 1133–1141. [DOI] [PubMed] [Google Scholar]
- 40.Wei, C. C., Lucchesi, P. A., Tallaj, J., Bradley, W. E., Powell, P. C. & Dell'Italia, L. J. (2003) Am. J. Physiol. 285, H784–H792. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}