

Abstract
Although structures of single-stranded (ss)DNA-binding proteins (SSBs) have been reported with and without ssDNA, the mechanism of ssDNA binding in eukarya remains speculative. Here we report a 2.5 Å structure of the ssDNA-binding domain of human replication protein A (RPA) (eukaryotic SSB), for which we previously reported a structure in complex with ssDNA. A comparison of free and bound forms of RPA revealed that ssDNA binding is associated with a major reorientation between, and significant conformational changes within, the structural modules—OB-folds—which comprise the DNA-binding domain. Two OB-folds, whose tandem orientation was stabilized by the presence of DNA, adopted multiple orientations in its absence. Within the OB-folds, extended loops implicated in DNA binding significantly changed conformation in the absence of DNA. Analysis of intermolecular contacts suggested the possibility that other RPA molecules and/or other proteins could compete with DNA for the same binding site. Using this mechanism, protein–protein interactions can regulate, and/or be regulated by DNA binding. Combined with available biochemical data, this structure also suggested a dynamic model for the DNA-binding mechanism.
Keywords: crystal structure/DNA binding/OB-fold/replication protein A/single-stranded DNA
Introduction
The eukaryotic single-stranded (ss)DNA-binding protein (SSB), replication protein A (RPA), plays a central role in replication, recombination and repair. Human RPA is a heterotrimer with three subunits of ∼70, 32 and 14 kDa, which are referred to as RPA70, RPA32 and RPA14, respectively. In DNA-processing events, RPA also interacts with many additional nuclear proteins, and this interaction both regulates, and is regulated by, an interaction with ssDNA (for reviews see Wold, 1997; Iftode et al., 1999).
RPA undergoes a conformational change upon ssDNA binding, as demonstrated by biochemical methods and electron microscopy. The pattern of RPA digestion with trypsin is different in the presence and absence of ssDNA (Gomes et al., 1996). Electron microscopy revealed three different molecular shapes: globular, elongated contracted and elongated extended (Blackwell et al., 1996). ssDNA does not seem to wrap around RPA, as deduced for bacterial SSB (Raghunathan et al., 1997, 2000), but rather RPA internally reorients and elongates along the DNA. The molecular details and the functional role of the proposed conformational change remain speculative.
RPA binds to DNA with a specific polarity and has at least two, and maybe three, major binding modes. The first mode, which is considered to be a major one, is characterized by an occluded binding site of ∼30 nucleotides (nt) per trimer (Kim et al., 1992). This binding mode exhibits high affinity and low cooperativity. The second mode, which is less stable and may be a precursor for the 30 nt mode, has an 8–10 nt binding site and exhibits a lower affinity and a higher cooperativity (Blackwell and Borowiec, 1994). The transition from the 8 to the 30 nt mode is thought to be a functionally important event implicated in DNA unwinding (Blackwell and Borowiec, 1994). An intermediate 13–14 nt binding mode has recently been reported by Lavrik et al. (1999). In this mode, RPA contacts DNA exclusively through the RPA70 subunit. The functional importance of this mode remains to be investigated.
The major ssDNA-binding activity of RPA is located in the central part of the RPA70 subunit [amino acids (aa) 181–422; RPA70181–422] (Gomes and Wold, 1996; Pfuetzner et al., 1997). Structural analysis of this fragment in complex with a (dC)8-oligonucleotide revealed two structurally similar copies of a structural domain (Bochkarev et al., 1997) known as an OB (oligonucleotide/oligosaccharide binding)-fold (Murzin, 1993). The two DNA-binding domains (DBDs) of RPA70, DBD-A (aa 181–290) and DBD-B (300–422), contact ssDNA in tandem. Each domain directly contacts 3 nt, with 2 nt filling the space between domains.
RPA has a hierarchy of OB-folds (Philipova et al., 1996); in addition to DBD-A and DBD-B, several more OB-folds are present in the protein. These are the N-terminal domain of RPA70 (aa 1–110; RPA70-NTD) (Jacobs et al., 1999), whose function is to interact with other proteins (Braun et al., 1997); the central domain of RPA32 (aa 43–171), which possesses a weak ssDNA-binding activity (DBD-D) (Bochkareva et al., 1998); and RPA14, a subunit with as yet unknown functions (Bochkarev et al., 1999). The C-terminal domain of RPA70 (aa 432–616) is also a DNA-binding domain (DBD-C), which has been hypothesized to contain another OB-fold (Brill and Bastin-Shanower, 1998).
In the course of DNA-processing events, RPA interacts with many nuclear proteins. In documented cases of RPA–protein interactions, where more detailed information is available, RPA, the target protein or both have at least two distinct regions that contact another protein. This is the case with XPA, whose N-terminal domain interacts with the C-terminal domain of RPA32 (Stigger et al., 1998) and the central domain with an RPA70 fragment containing aa 1–327 (Ikegami et al., 1998). A fragment of UNG2 containing aa 6–18 interacts with an unidentified target in RPA70 and another fragment (aa 67–85) with the C-terminal domain of RPA32 (Otterlei et al., 1999). Binding to p53 requires the N-terminal third of RPA70 with some contribution from the C-terminal third (Lin et al., 1996). Also, two-point interaction has been reported for Rad51 (Golub et al., 1998), RAD52 (Hays et al., 1998), DNA polymerase α/primase (DNA-pol α) and SV40 large T-antigen (Lee and Kim, 1995; Braun et al., 1997). One region is usually located in the N-terminus of the RPA70 subunit, and another either in the C-terminus of this (p53, DNA-pol α), or somewhere in the RPA32 subunit (XPA, UDG2, Rad51 and Rad52, TAg).
In this study, we report a crystal structure of the ssDNA-binding domain of RPA70, whose structure we previously reported as part of a complex with ssDNA. A comparison of free and bound forms of this SSB protein revealed dramatic conformational changes induced by DNA binding. The structure also suggests a possible mechanism of RPA–ssDNA and RPA–protein interaction.
Results and discussion
General description of the structure
A structure of RPA70181–422 in complex with (dC)8 was reported earlier (Bochkarev et al., 1997). A slightly longer RPA70 fragment (residues 181–432, RPA70181–432) was crystallized without ssDNA and the structure was solved using a seleno-methionine (Se-Met)-containing crystal by the multiwave anomalous dispersion (MAD) method. The structure has been refined at 2.5 Å resolution to a working R-factor 21.5% and free R-factor 28.2%. For more details see Materials and methods.
The crystals contain two independent molecules per asymmetric unit, termed molecule I and molecule II (Figure 1A and B, respectively). Each molecule has two independent functional motifs, DBD-A (aa 181–290) and DBD-B (aa 300–426), connected by an extended interdomain linker (aa 290–300). The individual DBDs have a central OB-fold (comprising aa 196–290 for DBD-A and 315–405 for DBD-B) and an N-terminal extension (181–196 and 300–315, respectively). Additionally, DBD-B is flanked by a C-terminal helix (405–420). A similar organization of structural elements is conserved in RPA14 and RPA32 core domains. In molecule II, the linker area is disordered and not included in the final model. In the RPA70181–432 crystals, the conformation of molecule I is stabilized mostly by intermolecular contacts with a symmetry-related molecule I, and molecule II with a symmetry-related molecule II. Most of the contacts between molecules I and II are contained within a hydrophobic region between the C-terminal helices of two DBD-Bs.
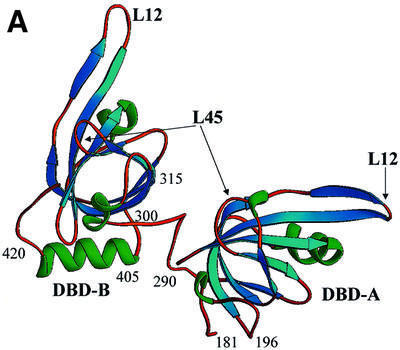
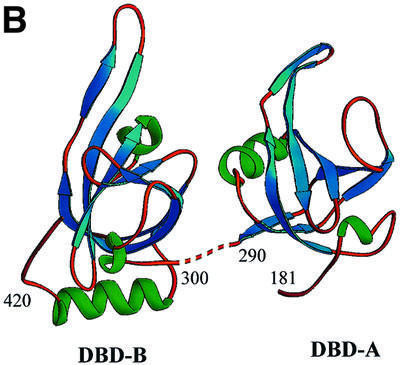
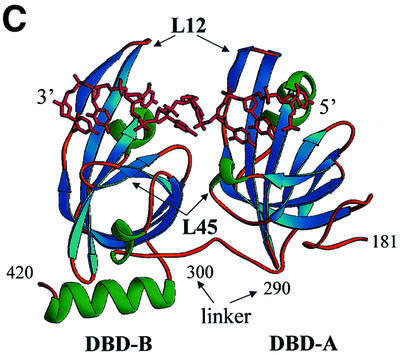
Fig. 1. Comparison of the structures of the RPA70 ssDNA-binding domain in the presence and absence of ssDNA. Ribbon diagrams of two crystallographically independent apo RPA70181–432 molecules, molecule I (A) and molecule II (B), and the RPA70181–422–ssDNA complex structure (C). The β-strands are colored in blue, α-helices in green, and loops in gold. Positions of important amino acids and structural elements are indicated. 3′- and 5′-ends of ssDNA are designated. Structures are aligned in such a way that an orientation of DBD-B is approximately the same in all panels. The drawing was generated using the RIBBONS program (Carson, 1997).
The flexible nature of the major ssDNA-binding domain of RPA in the absence of ssDNA
In the RPA70181–422–ssDNA complex structure, the ssDNA-binding domains DBD-A and DBD-B lie in tandem (Figure 1C). DBD-A contacts the 5′-end, and DBD-B the 3′-end of (dC)8. DNA tunnels through a narrow channel in the upper part of the DBDs and makes numerous stacking and hydrogen bond contacts with two extended loops, termed L12 and L45.
Although the basic structural elements of the free protein are the same, major conformational changes were found to accompany DNA binding. In the absence of DNA, domains A and B adopt different conformations in the two independent molecules in the unit cell; these conformations also differ from those in the DNA-containing complex. The tandem orientation of DBD-A and DBD-B in the RPA70181–422–ssDNA complex appears to be stabilized exclusively by interaction with DNA. Without DNA, the two OB-folds do not interact. The interdomain linker, which bridges the two domains, adopts different conformations in the two independent molecules, suggesting that it is flexible in the absence of DNA. If the linker (290–300) is excluded, the minimal distance between DBD-A and DBD-B in molecule I is ∼13 Å, and that in molecule II is ∼8 Å. The experimental map contained no electron density that could be interpreted as the interdomain linker in the second molecule. We attributed this effect to the flexible nature of this linker. In contrast, the electron density for the linker was well defined in molecule I (Figure 2).
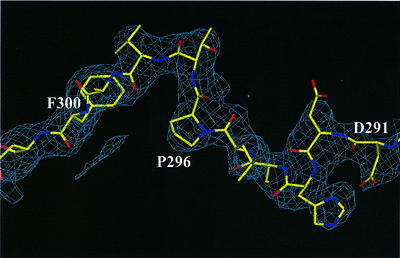
Fig. 2. A representative example of electron density in the solvent-flattened 2.7 Å experimental map calculated with SHARP/SOLOMON. A region that corresponds to the interdomain linker (aa 290–300) in molecule I is shown. The protein density is shown in blue. Superimposed on the map is the refined model of the protein. Contours are drawn at the 1.25σ level.
Taken together, our data indicate that the linker is flexible in solution, and two DBDs can adopt a range of conformations without DNA. We cannot exclude the possibility that flexibility of the interdomain linker may be reduced in the context of the trimer. However, the proteolytic digestion experiments for the RPA701–442 fragment and the full-size RPA trimer in the presence and absence of ssDNA reported by Gomes et al. (1996) are consistent with the interpretation that the flexible part of RPA70 is located in the interdomain linker.
There is strong evidence that two interdomain linkers flanking the major DNA-binding domain of RPA70 are also flexible. One linker connects the N-terminal domain of RPA70 (RPA70-NTD) with DBD-A, and the other DBD-B with the DBD-C. A flexibility of the linker between RPA70-NTD and DBD-A was demonstrated by both biochemical methods and NMR (Gomes et al., 1996; Jacobs et al., 1999). In light of recent characterization of DBD-C as an ssDNA-binding domain containing aa 433–616 (Bochkareva et al., 2000), an interpretation of results reported earlier by Gomes et al. (1996) may be done in more detail. These provide evidence that, without DNA, the linker between DBD-B and DBD-C is also flexible, and, like the linker between DBD-A and DBD-B, is stabilized upon DNA binding.
Internal flexibility of the OB-fold
DNA binding induces large conformational changes in individual OB-folds. All Cα atoms comprising DBD-A (residues 183–289) in molecule I were superimposed with their equivalents derived from the RPA70181–422–ssDNA complex structure using the LSQ_EXPLICIT function in program O (Jones et al., 1991). This initial alignment was then improved using the LSQ_IMPROVE function, which removed outliers. A similar procedure was repeated with DBD-B. The improvement procedure excluded from superposition (as outliers) a fragment containing aa 214–219 in DBD-A, and two fragments, 330–342 and 385–390, in DBD-B. The root mean square deviation (r.m.s.d.) for the remaining Cα atoms was 0.75 and 0.71 Å for DBD-A and DBD-B, respectively (Figure 3).
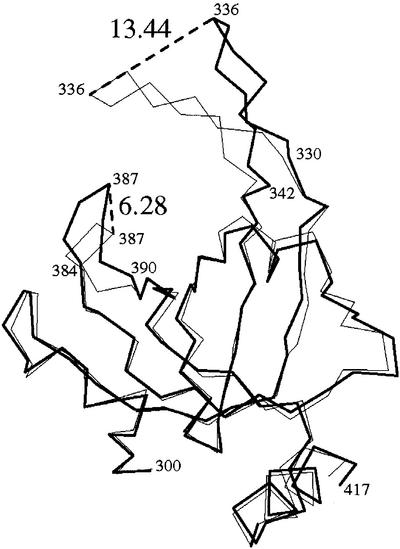
Fig. 3. Conformational change in DBD-B induced by ssDNA binding. A superposition of free and bound DBD-B was generated as discussed in the text. Shown is the Cα trace of the free (bold line) and bound (thin line) domain. Important amino acids are labeled. Maximal shifts of the L12 and L45 loop are indicated with dashed lines, and the size of the shift is indicated in Å (large numbers).
The excluded fragments were centered at the tips of the two extended loops, L12 and L45, which play a central role in DNA binding. The maximal shift between the bound and free conformations of the L12 loop was 7.7 Å in DBD-A and 13.4 Å in DBD-B. Another large shift (6.3 Å) was observed in the conformation of the L45 loop of DBD-B, but not DBD-A. Although structural analysis did not detect a significant conformational change in the L45 loop of DBD-A, we imagine that this is a crystal packing effect and, in solution, that this loop is also flexible. In short, experimental data confirm the hypothesis, which we formulated earlier, that both the L12 and L45 loops are flexible in the absence of DNA.
Surprisingly, despite a global structural difference between molecules I and II in the crystals, pairs of equivalent DBDs have very similar conformations. Two DBD-As from the two independent molecules can be superimposed, all Cα atoms against all, with an r.m.s.d. of only 0.35 Å. The r.m.s.d. for the two DBD-Bs, with all Cα atoms included, is 0.38 Å. We attributed this result to specific intermolecular contacts, which are discussed in the next section.
Analysis of intermolecular contacts suggests a model for RPA–protein interaction
Although the overall conformation of molecules I and II is different, the details of their intermolecular contacts are strikingly similar. In the unit cell, the two molecules are packed in such a way that the conformation of molecule I is stabilized mostly by intermolecular contacts with a symmetry-equivalent molecule I, and that molecule II is stabilized by a contact with the symmetry-related molecule II. In both molecules, DBD-A forms a pseudo-dimer with DBD-B coming from a symmetry-equivalent molecule (DBD-A–DBD-Bsym; Figure 4A). Taken as one molecule, the pseudo-dimer derived from molecule I can be superimposed with that derived from molecule II with an r.m.s.d. of 0.43 Å for 232 Cα atoms. Interestingly, in the crystals, a non-crystallographic (NC) 2-fold symmetry associates DBD-A–DBD-Bsym pairs from molecules I and II. Obviously, there is no NC symmetry that relates molecules I and II; the two have different conformations.
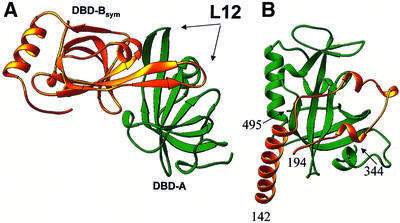
Fig. 4. The OB-fold can fulfill regulatory functions. (A) Pseudo-dimer of DBD-A (green) and symmetry-related DBD-B (DBD-Bsym; gold) (both DBDs derived from molecule I). The L12 loop of DBD-Bsym is in the ssDNA-binding cleft of DBD-A. The DBD-A orientation is approximately the same as in Figure 1C. (B) Functional interaction in the telomeric SSB; an OB-fold of the large subunit (fragment 344–495; green) binds an extended loop (fragment 142–194) of the small subunit (PDB entry 1OTC).
The interaction between DBD-A and DBD-Bsym is symmetrical. In the pseudo-dimer, DBD-B extends its ssDNA-binding loop (L12) into the DNA-binding cleft of DBD-A; the L12 loop of DBD-A, in its turn, is bound in the DNA-binding cleft of DBD-B. To some extent, this interaction is reminiscent of the dimerization mechanism in the gene V protein, in which the L45 loop from one subunit protruded into the putative DNA-binding groove of the other (Skinner et al., 1994). The specificity of molecular packing suggests that the pseudo-dimer, and not the single molecule, was a building block during RPA70181–432 crystal growth. This, in turn, suggests that there should be a strong interaction between DBD-A and DBD-B in solution.
Several lines of evidence suggest that the interdomain interactions that were revealed in this structure are not simply juxtapositions enforced by crystal packing, but rather are associations that are important (directly or indirectly) for RPA functions. First, similar interactions are observed in two independent and conformationally different molecules in the crystals. Secondly, this interaction can be explained from an electrostatic point of view in both global and local scale. Globally, the isoelectric point (pI) for DBD-A is 8.9 and for DBD-B is 4.9. At close to neutral pH, two oppositely charged DBDs should interact, forming a dipole. Locally, in the RPA70–ssDNA complex structure, the L12 loop of DBD provides basic amino acids for interaction with acidic phosphate groups of DNA, and the acidic environment of the DNA-binding cleft interacts with the DNA bases. In the absence of DNA, basic groups from L12 take the place of bases in the binding cleft. Thirdly, distantly similar and functionally important contacts have been reported to stabilize an interaction between two subunits in the telomeric ssDNA-binding protein (Horvath et al., 1998) (Figure 4B) and the gene V protein of bacteriophage f1 (Skinner et al., 1994).
The mechanism for DBD interaction may have regulatory implications. ssDNA binding is known to regulate, and be regulated by, the interaction with other nuclear proteins. In one example, in the absence of ssDNA, RPA binds to and inhibits sequence-specific double-stranded (ds)DNA-binding activity of the tumor suppressor protein p53 (Miller et al., 1997). The presence of ssDNA eliminates the RPA–p53 interaction and activates p53 for sequence-specific dsDNA binding. p53 might extend a loop into one of RPA’s DBDs. Binding of ssDNA to RPA would release p53, thus activating it. The proposed mechanism of protein–protein interaction is not unprecedented. In telomeric ssDNA-binding protein, an extended loop of the small subunit binds reversibly to an OB-fold of the large subunit. This interaction is modulated by protein–DNA binding, which is initiated by other parts of the two subunits (Figure 4B).
Our finding expands the repertoire of known OB-fold functions. Initially, the OB-fold was characterized as an oligonucleotide/oligosaccharide-binding fold. Later, an oligopeptide-binding representative of the OB-fold was described. The term OB then evolved to mean oligonucleotide/oligosaccharide/oligopeptide-binding fold, or for short, oligo-binding fold (Horvath et al., 1998). Here, we have demonstrated that the same OB-fold serves both as a DNA-binding and a protein-binding element. Furthermore, the binding of protein and DNA is mutually exclusive, as both share the same binding site. A competition for this site can, therefore, regulate RPA activity. Thus, we propose that the OB-fold can also fulfill regulatory functions.
In total, there are five, and perhaps six, OB-folds in RPA; potentially, each can participate in protein–protein interactions. Together, these folds provide a basis for an extensive and flexible network of RPA–protein interaction. Three factors can regulate those interactions, namely: direct competition between DNA and proteins for a binding site (has been discussed above), allosteric effects of DNA binding (to be discussed below), and RPA phosphorylation (will not be discussed here).
General model of RPA–ssDNA interaction
Upon ssDNA binding, RPA changes from a globular to an elongated conformation. We hypothesize that this conformational change is associated with the flexibility of the interdomain linker between DBD-A and DBD-B (linker 290–300), with additional contribution provided by linkers connecting the RPA70-NTD with DBD-A (linker 110–180) and DBD-B with DBD-C (linker 420–432). In the absence of DNA, interdomain and intersubunit interactions stabilize the globular conformation (Figure 5A). In this conformation, all three linkers are susceptible to a proteolytic cleavage.
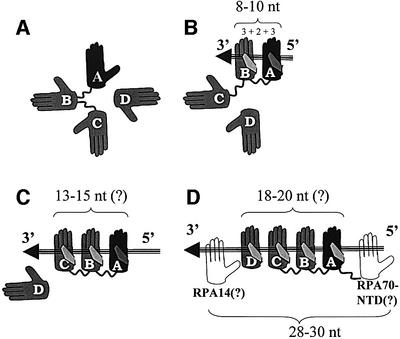
Fig. 5. Suggested multistep ssDNA-binding pathway. (A) Free RPA in globular conformation. (B) 8 nt binding by DBD-A and DBD-B (8–10 nt binding mode). (C) 13–15 nt binding by DBD-A, DBD-B and DBD-C. (D) 18–20 nt binding by DBD-A, -B, -C and -D. The four DBDs together with RPA70-NTD and RPA14 occlude 28–30 nt (30 nt binding mode). DBDs are represented by palms and labeled as A, B, C and D, respectively. DBD-A is colored in black. DNA is represented by an arrow; 5′- and 3′-ends of DNA are indicated. See text for more details.
We propose that DNA binding rearranges the domains and orients them in tandem on DNA via a multistep pathway. In agreement with the existing model, RPA initially binds DNA in the 8–10 nt binding mode, which is associated with DNA binding by DBD-A and DBD-B (Figure 1C), and with the globular shape of the molecule (Figure 5B). Being a prerequisite for higher order binding modes, this binding orients and stabilizes RPA on DNA and makes the linker between DBD-A and DBD-B (residues 290–300) resistant to proteolysis (Gomes et al., 1996).
In the next step, the DBD-A–DBD-B duet is joined by DBD-C (Figure 5C). Indicative of this binding is a protection of linker 420–432 from proteolytic cleavage (Gomes et al., 1996). Three factors taken together indicate that DBD-C cannot make contacts to the 5′-protruding end of ssDNA in front of DBD-A: (i) the limited size of linker 420–432; (ii) the location of the point it initiates from (amino acid 420) >50 Å away from the 5′-end, but close to the 3′-end of ssDNA; and (iii) directionality of the protein backbone at this point toward the 3′-end (Figure 1). Therefore, DBD-C should contact the 3′-end, in tandem after DBD-B. If, as postulated by Brill and Bastin-Shanower (1998), DBD-C contains an OB-fold, which binds DNA through a similar mechanism, DBD-C would be predicted to contact 3 nt, and a further 2 nt would be required to bridge the space between DBD-B and DBD-C. Thus, 13–15 nt are predicted to be contacted by the trio of DBD-A, DBD-B and DBD-C, the number that exactly coincides with the 13 nt binding mode reported by Lavrik et al. (1999). In this mode, again in agreement with results of Lavrik, RPA contacts ssDNA exclusively through the RPA70 subunit. We associate this mode with the elongated contracted conformation.
Finally, the central OB-fold of RPA32 (DBD-D) joins the trio in tandem after DBD-C (Figure 5D). Based on similar assumptions, one should predict 18–20 nt as a site directly contacted by the quartet. This correlates with the finding that RPA demonstrates different affinities to the 5′- and 3′-protruding arm of ssDNA, but only if their length is 19 nt or shorter; affinities to 23 nt arms are similar, if not equal (de Laat et al., 1998). Indeed, within our model, 20 nt and longer fragments of ssDNA protruding in either direction provide enough room to accommodate all four DBDs of RPA. In contrast, shorter fragments might restrict some domains from binding; this restriction could be different depending on the directionality of DNA.
The 18–20 nt binding mode is associated with the elongated extended conformation and 30 nt occluded binding site (30 nt binding mode). There is no conflict between the 18–20 nt contacted site and the 30 nt mode. 28–30 nt was defined to be the occluded size per RPA trimer. The remaining 10–12 nt can fill the space between trimers. Although there are no data supporting direct interaction of the remaining two RPA OB-folds, RPA70-NTD and RPA14, with DNA, the two might just occupy this space. Interestingly, if we formally count 5 nt (3 nt to contact an OB-fold and 2 nt to bridge the space) per two remaining OB-folds, RPA70-NTD and RPA14, the total comes to 28–30.
The binding of DBD-A and DBD-B to DNA should, in its turn, be a multistep process. Most likely, it is initiated by DBD-A (Figure 5; black palm), as the domain with higher activity and flexibility, which binds the 5′-end of ssDNA, and then supported by DBD-B. This scenario is in agreement with the experimental observations that RPA binds to DNA with a 5′ to 3′ polarity (de Laat et al., 1998; Iftode and Borowiec, 2000).
A comparison of the structure of RPA70181–432 with that of the RPA14/32 core complex suggested that RPA might switch from a globular to an elongated conformation by means of ‘helical shuffling’. The RPA14/32 core dimer is stabilized by contacts between two C-terminal helices provided by the RPA14 and RPA32 core. In the RPA14/32 core crystals, two independent dimers contributed their two helical interfaces to form the four-helix bundle, which stabilized the dimer-of-dimers (Bochkarev et al., 1999). Two helices from the second dimer were proposed to compensate for a helical environment that is normally provided by RPA70, the subunit not included in the dimeric complex. The C-terminal domain of RPA70 has been suggested to contribute a helix or two to the trimerization interface. DBD-B also contains a C-terminal helix. Like the crystals of the RPA14/32 core, the crystals of RPA70181–432 are stabilized by a hydrophobic two-helix packing arrangement (Figure 6). Molecular contacts suggest that, in the context of intact RPA, DBD-B might contribute the C-terminal helix to interact with the multi-helical trimerization interface. Unlike stable helical interaction within the trimerization core, an interaction of the helix provided by DBD-B might be reversible (here helical shuffling), depending on the DNA-binding mode; the association of this helix with the multi-helical interface could stabilize the globular conformation, whereas DNA binding could induce its dissociation.
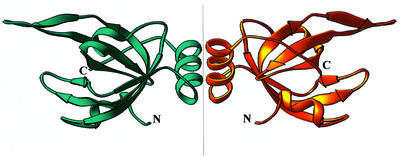
Fig. 6. Hydrophobic helix–helix interaction between two independent DBD-Bs stabilizes the crystals. The non-crystallographic axis is represented with a dashed line.
Possible allosteric regulation of RPA–protein interactions
A reorientation of DBDs induced by DNA binding may play a regulatory role for the RPA–protein interaction. Proteins such as p53, XPA, UNG and many others interact with RPA via a ‘two-point interaction’, in which two distinct areas of the interacting protein contact two distinct areas of RPA. In all known cases of the two-point interaction where detailed information is available, one contact is originated from the N-terminal half of RPA70 and the other from either RPA70-CTD or RPA32, which is adjacent to RPA70-CTD. We speculate that, in the globular conformation, the two contacting areas are juxtaposed. In this conformation, the two-point interaction with RPA is enabled for a group of proteins associating with free RPA. After DNA binding, the N- and C-termini of RPA70 move apart, as do the two binding areas. This reorientation allosterically switches the two-point interaction, making it unfavorable for proteins associating with free RPA in favor of those having higher affinity to the DNA-bound forms of RPA.
Materials and methods
The construct pET15b-RPA70181–432 was described previously (Pfuetzner et al., 1997). The protein was expressed in Escherichia coli BL21 (DE3) cells (Stratagene) as a fusion to an N-terminal His6 tag, purified according to original protocol (Novagen) using HiTrap Chelating and HiTrap Heparin columns (Pharmacia Biotech), concentrated to 8–10 mg/ml, and stored frozen in 15 mM HEPES pH 7.0, 350 mM NaCl and 5 mM dithiothreitol. To incorporate selenomethionine (Se-Met) in the protein, BL21 (DE3) cells were grown in a methionine-depleted medium as previously described (Van Duyne et al., 1993; Bochkarev et al., 1999). The Se-Met-containing protein was purified using the same protocol. Crystals suitable for data collection were grown by the method of sitting drop vapor diffusion against a reservoir solution containing 0.1 M K,NaH2PO4, 0.1 M Tris pH 8.5 and 3.8 M NaCl. The sitting drops were formed by mixing 2 µl of reservoir solution and 2 µl of the protein. The space group was P212121 and the unit cell parameters 63.5 × 84.9 × 119.1 Å. There were two independent molecules per asymmetric unit. MAD data at four-wavelength (2.7 Å) and high resolution data set (2.5 Å) were collected from a single Se-Met-modified RPA70181–432 crystal on the 19ID beamline at the Structural Biology Center, Argonne National Laboratory. Diffraction was anisotropic and extended to 2.3 Å in one direction and 2.7 Å in another. Data were integrated and scaled with Denzo/Scalepack (Otwinowski and Minor, 1997). Coordinates of eight Se atoms were located and initially refined in PHASES (Furey and Swaminathan, 1997) and then in SHARP (de La Fortelle and Bricogne, 1997). In the experimental map, continuous density was observed for both crystallographically independent molecules in the interval 181–426, except for a fragment 290–297 in the second molecule, which corresponds to the interdomain linker (Figure 2). The model was built with O (Jones et al., 1991) and refined against 2.5 Å resolution data using the CNS program (Brunger et al., 1998). The final model contains 3795 non-hydrogen atoms and 32 ordered water molecules, has a working R-factor of 21.5% and free R-factor of 28.2% for 19 729 reflections (86.2%), with F > 1.5σ between 20.0 and 2.5 Å. Eighty-three percent of amino acids are in most favorable areas with one outlier, Asn239. Characteristically, the same amino acid was in disallowed conformation in the RPA70181–422–ssDNA structure. Data collection and refinement statistics are summarized in Table I. The atomic coordinates have been deposited at the Protein Data Bank (ID code 1FGU).
Table I. Data collection and refinement statistics.
| Data processing (DENZO/SCALEPACK) |
|
|
|
|
|
| Experiment |
Se-Met MAD |
|
|
|
High resolution |
| Data set | 1 (peak) | 2 (inflection) | 3 (high energy) | 4 (low energy) | (low energy) |
| Wavelength (Å)/energy (KeV) | 0.9793/12.660 | 0.9795/12.658 | 0.961/12.9 | 1.033/12.0 | 1.033/12.0 |
| Anomalous or isomorphousa | ano | ano | ano | ano | iso |
| Resolution (Å) | 2.7 | 2.7 | 2.7 | 2.7 | 2.5 |
| No. of reflections measured | 161 599 | 162 599 | 162 452 | 162 769 | 247 836 |
| No. of independent reflections | 33 748 | 33 910 | 33 905 | 33 948 | 22 963 |
| R-factor (%) (overall/outer shell) | 4.9/38.3 | 4.2/36.9 | 4.2/40.6 | 3.7/30.8 | 5.2/19.0 |
| I/σ (overall/outer shell) | 25.7/2.7 | 26.4/2.8 | 25.5/2.4 | 29.3/3.4 | 37.1/4.6 |
| Completeness (%) |
98.8/98.4 |
99.2/98.7 |
99.1/98.5 |
99.2/98.2 |
98.6/96.0 |
| Experimental phase calculation (SHARP) |
|
|
Structure refinement (CNS) |
|
|
| Resolution (Å) | 20–2.7 | resolution (Å) | 20–2.5 | ||
| No. of Se sites | 8 | No. of reflections with F > 1.5σ | 19 729 (86.2%) | ||
| No. of phased reflections | 15 874 | R/R-free (%) | 21.5/28.1 | ||
| FOM (acentric) | 0.59 | No. of protein atoms | 3795 | ||
| FOM (centric) | 0.50 | water molecules | 32 | ||
| model r.m.s.d. from ideality bonds/angles (Å/°) | 0.019/2.2 | ||||
The parameters are defined as in SCALEPACK, SHARP and CNS.
aI(+) and I(–) are processed as independent (ano) or equivalent (iso).
Acknowledgments
Acknowledgements
We thank Aled Edwards and Joan Conaway for critical reading of the manuscript, and members of the Structural Biology Center (SBC) 19ID beamline at the Advanced Photon Source (APS), Argonne National Laboratory (ANL) for support. This work was supported by NIH grant GM61192-01 (to A.B.). Use of the ANL SBC beamlines at APS was supported by the US Department of Energy, Office of Biological and Environmental Research, under contract W-31-109-ENG-38.
References
- Blackwell L.J. and Borowiec,J.A. (1994) Human replication protein A binds single-stranded DNA in two distinct complexes. Mol. Cell. Biol., 14, 3993–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell L.J., Borowiec,J.A. and Masrangelo,I.A. (1996) Single-stranded-DNA binding alters human replication protein A structure and facilitates interaction with DNA-dependent protein kinase. Mol. Cell. Biol., 16, 4798–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochkarev A., Pfuetzner,R.A., Edwards,A.M. and Frappier,L. (1997) Structure of the single-stranded-DNA-binding domain of replication protein A bound to DNA. Nature, 385, 176–181. [DOI] [PubMed] [Google Scholar]
- Bochkarev A., Bochkareva,E., Frappier,L. and Edwards,A.M. (1999) The crystal structure of the complex of replication protein A subunits RPA32 and RPA14 reveals a mechanism for single-stranded DNA binding. EMBO J., 18, 4498–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochkareva E., Frappier,L., Edwards,A.M. and Bochkarev,A. (1998) The RPA32 subunit of human replication protein A contains a single-stranded DNA-binding domain. J. Biol. Chem., 273, 3932–3936. [DOI] [PubMed] [Google Scholar]
- Bochkareva E., Korolev,S. and Bochkarev,A. (2000) The role for zinc in replication protein A. J. Biol. Chem., 275, 27332–27338. [DOI] [PubMed] [Google Scholar]
- Braun K.A., Lao,Y., He,Z., Ingles,C.J. and Wold,M.S. (1997) Role of protein–protein interactions in the function of replication protein A (RPA): RPA modulates the activity of DNA polymerase α by multiple mechanisms. Biochemistry, 36, 8443–8454. [DOI] [PubMed] [Google Scholar]
- Brill S.J. and Bastin-Shanower,S. (1998) Identification and characterization of the fourth single-stranded-DNA binding domain of replication protein A. Mol. Cell. Biol., 18, 7225–7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger A.T. et al. (1998) Crystallography and NMR system (CNS): A new software system for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Carson M. (1997) Ribbons. Methods Enzymol., 276, 493–505. [PubMed] [Google Scholar]
- de Laat W.L., Appeldoorn,E., Sugasawa,K., Weterings,E., Jaspers,N.G. and Hoeijmakers,J.H. (1998) DNA-binding polarity of human replication protein A positions nucleases in nucleotide excision repair. Genes Dev., 12, 2598–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de La Fortelle E. and Bricogne,G. (1997) Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol., 276, 472–494. [DOI] [PubMed] [Google Scholar]
- Furey W. and Swaminathan,S. (1997) PHASES-95: A program package for the processing and analysis of diffraction data from macromolecules. Methods Enzymol., 277, 590–620. [DOI] [PubMed] [Google Scholar]
- Golub E.I., Gupta,R.C., Haaf,T., Wold,M.S. and Radding,C.M. (1998) Interaction of human Rad51 recombination protein with single-stranded DNA binding protein, RPA. Nucleic Acids Res., 26, 5388–5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes X.V. and Wold,M.S. (1996) Functional domains of the 70-kilodalton subunit of human replication protein A. Biochemistry, 35, 10558–10568. [DOI] [PubMed] [Google Scholar]
- Gomes X.V., Henricksen,L.A. and Wold,M.S. (1996) Proteolytic mapping of human replication protein A: evidence for multiple structural domains and a conformational change upon interaction with single-stranded DNA. Biochemistry, 35, 5586–5595. [DOI] [PubMed] [Google Scholar]
- Hays S.L., Firmenich,A.A., Massey,P., Banerjee,R. and Berg,P. (1998) Studies of the interaction between Rad52 protein and the yeast single-stranded DNA binding protein RPA. Mol. Cell. Biol., 18, 4400–4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath M.P., Schweiker,V.L., Bevilacqua,J.M., Ruggles,J.A. and Schultz,S.C. (1998) Crystal structure of the Oxytricha nova telomere end binding protein complexed with single strand DNA. Cell, 95, 963–974. [DOI] [PubMed] [Google Scholar]
- Iftode C. and Borowiec,J.A. (2000) 5′→ 3′ molecular polarity of human replication protein A (hRPA) binding to pseudo-origin DNA substrates. Biochemistry, 39, 11970–11981. [DOI] [PubMed] [Google Scholar]
- Iftode C., Daniely,Y. and Borowiec,J.A. (1999) Replication protein A (RPA): the eukaryotic SSB. Crit. Rev. Biochem. Mol. Biol., 34, 141–180. [DOI] [PubMed] [Google Scholar]
- Ikegami T., Kuraoka,I., Saijo,M., Kodo,N., Kyogoku,Y., Morikawa,K., Tanaka,K. and Shirakawa,M. (1998) Solution structure of the DNA- and RPA-binding domain of the human repair factor XPA. Nature Struct. Biol., 5, 701–706. [DOI] [PubMed] [Google Scholar]
- Jacobs D.M., Lipton,A.S., Isern,N.G., Daughdrill,G.W., Lowry,D.F., Gomes,X. and Wold,M.S. (1999) Human replication protein A: global fold of the N-terminal RPA-70 domain reveals a basic cleft and flexible C-terminal linker. J. Biomol. NMR, 14, 321–331. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kim C., Snyder,R.O. and Wold,M.S. (1992) Binding properties of replication protein A from human and yeast cells. Mol. Cell. Biol., 12, 3050–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavrik O.I., Kolpashchikov,D.M., Weisshart,K., Nasheuer,H.P., Khodyreva,S.N. and Favre,A. (1999) RPA subunit arrangement near the 3′-end of the primer is modulated by the length of the template strand and cooperative protein interactions. Nucleic Acids Res., 27, 4235–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.H. and Kim,D.K. (1995) The role of the 34-kDa subunit of human replication protein A in simian virus 40 DNA replication in vitro. J. Biol. Chem., 270, 12801–12807. [DOI] [PubMed] [Google Scholar]
- Lin Y.L., Chen,C., Keshav,K.F., Winchester,E. and Dutta,A. (1996) Dissection of functional domains of the human DNA replication protein complex replication protein A. J. Biol. Chem., 271, 17190–17198. [DOI] [PubMed] [Google Scholar]
- Miller S.D., Moses,K., Jayaraman,L. and Prives,C. (1997) Complex formation between p53 and replication protein A inhibits the sequence-specific DNA binding of p53 and is regulated by single-stranded DNA. Mol. Cell. Biol., 17, 2194–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzin A.G. (1993) OB(oligonucleotide/oligosaccharide binding)-fold: common structural and functional solution for non-homologous sequences. EMBO J., 12, 861–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otterlei M. et al. (1999) Post-replicative base excision repair in replication foci. EMBO J., 18, 3834–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Pfuetzner R.A., Bochkarev,A., Frappier,L. and Edwards,A.M. (1997) Replication protein A. Characterization and crystallization of the DNA binding domain. J. Biol. Chem., 272, 430–434. [DOI] [PubMed] [Google Scholar]
- Philipova D., Mullen,J.R., Maniar,H.S., Lu,J., Gu,C. and Brill,S.J. (1996) A hierarchy of SSB protomers in replication protein A. Genes Dev., 10, 2222–2233. [DOI] [PubMed] [Google Scholar]
- Raghunathan S., Ricard,C.S., Lohman,T.M. and Waksman,G. (1997) Crystal structure of the homo-tetrameric DNA binding domain of Escherichia coli single-stranded DNA-binding protein determined by multiwavelength x-ray diffraction on the selenomethionyl protein at 2.9-Å resolution. Proc. Natl Acad. Sci. USA, 94, 6652–6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghunathan S., Kozlov,A.G., Lohman,T.M. and Waksman,G. (2000) Structure of the DNA binding domain of E.coli SSB bound to ssDNA. Nature Struct. Biol., 7, 648–652. [DOI] [PubMed] [Google Scholar]
- Skinner M.M. et al. (1994) Structure of the gene V protein of bacteriophage f1 determined by multiwavelength x-ray diffraction on the selenomethionyl protein. Proc. Natl Acad. Sci. USA, 91, 2071–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stigger E., Drissi,R. and Lee,S.H. (1998) Functional analysis of human replication protein A in nucleotide excision repair. J. Biol. Chem., 273, 9337–9343. [DOI] [PubMed] [Google Scholar]
- Van Duyne G.D., Standaert,R.F., Karplus,P.A., Schreiber,S.L. and Clardy,J. (1993) Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol., 229, 105–124. [DOI] [PubMed] [Google Scholar]
- Wold M.S. (1997) Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem., 66, 61–92. [DOI] [PubMed] [Google Scholar]