

Abstract
The holD gene codes for the ψ subunit of the Escherichia coli DNA polymerase III holoenzyme, a component of the γ complex clamp loader. A holD mutant was isolated for the first time in a screen for mutations that increase the frequency of tandem repeat deletions. In contrast to tandem repeat deletions in wild-type strains, deletion events stimulated by the holD mutation require RecA. They do not require RecF, and hence do not result from the recombinational repair of gaps, arguing against uncoupling of the leading and lagging strand polymerases in the holD mutant. The holD recBC combination of mutations is lethal and holD recBts recCts strains suffer DNA double-strand breaks (DSBs) at restrictive temperature. DSBs require the presence of the Holliday junction-specific enzymes RuvABC and are prevented in the presence of RecBCD. We propose that impairment of replication due to the holD mutation causes the arrest of the entire replisome; consequently, Holliday junctions are formed by replication fork reversal, and unequal crossing over during RecA- and RecBCD-mediated re-incorporation of reversed forks causes the hyper-recombination phenotype.
Keywords: deletion/DNA polymerase/double-strand breaks/homologous recombination/repair
Introduction
Alterations in the length of repetitive sequences within or near genes can alter their expression or function. In humans, expansion of repeated sequences has been found associated with several genetic degenerative disorders (reviewed in Ashley and Warren, 1995; Djian, 1998). Two classes of pattern of tract instability have been described: small changes as little as a few nucleotides in length and rearrangements that involve long sequences. With the aim of understanding better the circumstances in which recombination between long repeated sequences is stimulated, we isolated and characterized mutants where the frequency of this type of rearrangement was increased (Bierne et al., 1997b). This revealed that different recombination processes are specifically stimulated by mutations in DNA repair or DNA replication genes.
In wild-type Escherichia coli chromosomes, the frequency of recombination between long tandem repeats is unaffected by the inactivation of RecA, the main homologous recombination protein, and is also independent of proteins involved in the pre-synaptic (RecBCD, RecFOR) and post-synaptic (RuvABC) steps of homologous recombination (Lovett et al., 1993; Bierne et al., 1997b). This suggests that such deletions can occur independently of the classical homologous recombination pathways. A systematic determination of recombination frequencies in mutants affected for DNA replication showed that mutations in most of the E.coli genes required for chromosomal replication affect tandem repeat recombination (Saveson and Lovett, 1997). The mechanism involved was studied in a dnaBts mutant, impaired for the main replicative helicase. Stimulation of deletions by dnaBts mutation was dependent on RecA and also on RecBCD, the complex that specifically initiates recombinational repair of DNA double-strand breaks (DSBs) (Saveson and Lovett, 1999). The RecBCD complex binds to double-stranded ends and promotes DNA degradation by its helicase and exonuclease activities (reviewed in Kowalczykowski, 1994; Kuzminov, 1999). Upon the encounter of a specific sequence, CHI, the activity of the complex is modified to generate a single-stranded 3′ tail on which RecBCD facilitates the loading of RecA and thus promotes the invasion of a homologous sequence (Anderson and Kowalczykowski, 1997; Churchill et al., 1999). The RecBCD dependence of tandem repeat deletions in dnaBts strains is in agreement with the observation that double-strand ends are produced in the bacterial chromosome upon replication arrest by inactivation of this helicase (Michel et al., 1997; Seigneur et al., 1998).
Using random mutagenesis, we screened for mutations that stimulate tandem repeat recombination in E.coli. Two such mutations were previously shown to map in uvrD and dnaE (Bierne et al., 1997a,b). Hyper-recombination in uvrD mutants, defective for the repair of mismatches and for the excision repair of UV lesions, was dependent on RecA and RecFOR. The RecFOR proteins are thought to catalyze recombination at single-stranded gaps by facilitating the action of RecA on gapped DNA (Umezu et al., 1993; Webb et al., 1997; reviewed in Kuzminov, 1999). We proposed that the absence of UvrD resulted in the formation of gaps and that unequal exchanges during gap repair by RecFOR-mediated recombination caused the hyper-recombination phenotype (Bierne et al., 1997a,b). DnaE is the catalytic subunit (α) of the E.coli DNA polymerase III holoenzyme (Pol III HE), the main E.coli polymerase. In contrast to uvrD and dnaBts mutations, mutations in the dnaE gene led to a similar increase in the frequency of tandem repeat deletions in the presence and absence of RecA (Bierne et al., 1997a,b). Consequently, deletions were proposed to occur by replication slippage, a RecA-independent mechanism. The dnaE mutation would cause a pause in the replication progression, leading to dissociation of the 3′ end of the growing strand from the template. Re-association on the second homologous repeat and continuation of replication would lead to the loss of one of the repeats in the newly synthesized genome.
In the present work, we describe the characterization of a third hyper-recombination mutation isolated by random mutagenesis. This mutation affects one of the E.coli Pol III HE subunits, ψ, encoded by the holD gene. Escherichia coli Pol III HE is composed of 10 different subunits, assembled in two catalytic cores, two sliding clamps and a clamp loader (reviewed in Kelman and O’Donnell, 1995). The polymerase core contains the catalytic subunit α (dnaE), the proofreading activity ε (dnaQ) and a polypeptide of unknown function, θ (holE). The two cores are held together by a dimer of the τ subunit (dnaX), which binds the clamp loader and DnaB, allowing the two cores to function in a coordinated fashion, one on the leading strand and the other on the lagging strand. Each core polymerase is tethered to the DNA by a β clamp (dnaN). The clamp loader (γ complex) contains five different polypeptides: γ (dnaX), δ (holA), δ′ (holB), χ (holC) and ψ (holD). The function of the γ complex during replication has been studied extensively in vitro. This complex was proposed to be required for loading and unloading β after completion of each Okazaki fragment, thus allowing rapid cycling of the lagging strand polymerase (reviewed in Kelman and O’Donnell, 1995; Naktinis et al., 1996; Turner et al., 1999). ψ is a small protein of 15.2 kDa that acts as a bridge between γ and χ (Kelman and O’Donnell, 1995; Glover and McHenry, 1998, 2000; Kelman et al., 1998).
In contrast to extensive studies of replication in vitro, little is known about the individual role of each Pol III HE subunit in vivo. dnaE, dnaX, holA and holB are essential genes. Knockout of the dnaQ or holC genes, although viable, confers a severe growth defect. Only deletion of holE has no noticeable phenotype (reviewed in Kelman and O’Donnell, 1995). The holD mutant isolated in the present work is the first holD mutant described. The mutation increases the frequency of recombination between tandemly repeated sequences ∼20-fold in a RecA-dependent way. In the holD mutant, recBC inactivation is lethal and causes the accumulation of DSBs. We propose a model to account for these observations that implies the arrest of the entire replisome when HolD fails to function.
Results
Isolation of a hyper-recombination mutant that maps in the holD gene
To isolate mutants in which the frequency of recombination between tandemly repeated sequences is increased, an E.coli strain that carries a tandem repeat of 624 bp in the lacZ gene was used (Bierne et al., 1997b). The duplication inactivates lacZ and deletion of one of the repeats restores a Lac+ phenotype. One clone in which deletion frequency was increased ∼20-fold was studied (JJC616; Tables I and II). The mutation was mapped by Hfr conjugation and P1 transduction at min 99 on the genetic map of E.coli and P1 transduced in the wild-type isogenic strain JJC520, using the linked mdoB or thr markers and screening for the hyper-recombination phenotype (JJC867, JJC947 and JJC1303; Tables I and II). Among genes that map at min 99, we tested holD, which encodes the ψ subunit of the Pol III HE. Introduction of the wild-type holD gene, carried on the vector pAM34, in the hyper-recombination mutant decreased the level of tandem repeat deletion to that observed in the wild-type strain, whereas the vector had no significant effect (Table II). We conclude that the mutation responsible for the hyper-recombination phenotype of this strain is located in the holD gene.
Table I. Strains and plasmids.
| Strain | Genotype | Origin or reference |
|---|---|---|
| CAG12079 | fuc::Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG12135 | recD1901::Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG18430 | mdoB::Tn10 (zjj202::Tn10) | Singer et al. (1989); Nichols et al. (1998) |
| CAG18442 | thr-34::Tn10 | Singer et al. (1989); Nichols et al. (1998) |
| CAG18619 | mdoB::Tn10kan (zjj3188::Tn10kan) | Singer et al. (1989); Nichols et al. (1998) |
| GY9701 | ΔrecA938::cm (p-RecA) | R.Devoret; Michel et al. (1997) |
| LN2926 | Hfr PK191 rpsL Δ(recBC)Ap | Cornet et al. (1994) |
| JJC330 (SK129) | recB270 recC271 | S.Kushner; Michel et al. (1997) |
| WA576 | recF400::Tn5 | W.Wackernagel |
| JJC40 | AB1157 hsdR | laboratory collection |
| JJC99 | wild type | Bierne et al. (1997b) |
| JJC443 | lexAind3 mal::Tn10 | Bierne et al. (1997a) |
| JJC754 | ΔruvABC::cm | Seigneur et al. (1998) |
| JJC942 | trpA9605am his-29am | laboratory collection |
| JJC1144 | holDQ10am mdoB::Tn10kan | P1 JJC867 * JJC99 selection KanR, screening for LBS |
| JJC1159 | holDQ10am mdoB::Tn10kan [pAM-holD] | JJC1144 transformed by pAM-holD |
| JJC1220 | recB270 recC271 fuc::Tn10 | P1 CAG12079 * JJC330, selection TetR, screening for exo–, UVS |
| JJC1288 | holDQ10am mdoB::Tn10kan [pAM-holD] ΔruvABC::cm | P1 JJC754 * JJC1159, selection CmR |
| JJC1291 | holDQ10am mdoB::Tn10kan [pAM-holD] ΔruvABC::cm Δ(recA-srl)::Tn10 | P1 JJC557 * JJC1288, selection TetR |
| JJC1294 | holDQ10am mdoB::Tn10kan ΔruvABC::cm Δ(recA-srl)::Tn10 | JJC1291 cured of pAM-holD |
| JJC1298 | Hfr PK191 rpsL recD1901::Tn10 | P1 CAG12135 * LN2926, selection TetR |
| JJC1299 | Hfr PK191 rpsL recD1901::Tn10 ΔrecA938::cm | P1 GY9701 * JJC1298, selection CmR |
| JJC1366 | ΔruvABC::cm | P1 JJC754 * JJC99, selection CmR |
| JJC1367 | ΔruvABC::cm Δ(recA-srl)::Tn10 | P1 JJC557 * JJC1366, selection TetR |
| JJC520 derivatives. All JJC520 derivatives carry a 624 bp duplication in lacZ (lacZ624dup) adjacent to a CmR marker | ||
| JJC520 | lacZ624dup CmR | Bierne et al. (1997b) |
| JJC557 | Δ(recA-srl)::Tn10 | Bierne et al. (1997b) |
| JJC561 | recF400::Tn5 | Bierne et al. (1997b) |
| JJC616 | holDQ10am | ethylmethane sulfonate mutagenesis of JJC520 |
| JJC617 | holDQ10am mdoB::Tn10kan | P1 CAG18619 * JJC616, selection KanR, screening for LBS |
| JJC867 | holDQ10am mdoB::Tn10kan | P1 JJC617 * JJC520 selection KanR |
| JJC868 | holDQ10am mdoB::Tn10kan Δ(recA-srl)::Tn10 | P1 JJC557 * JJC867 selection TetR |
| JJC947 | holDQ10am mdoB::Tn10 | P1 JJC616-mdoB::Tn10 * JJC520, selection TetR, screening for LBS |
| JJC1096 | holDQ10am mdoB::Tn10kan [pAM-holD] | JJC867 transformed by pAM-holD |
| JJC1097 | lexAind3 mal::Tn10 | P1 JJC443 * JJC867, selection TetR, screening for UVS |
| JJC1157 | holDQ10am mdoB::Tn10 [pAM-holD] | JJC947 transformed by pAM-holD |
| JJC1163 | holDQ10am mdoB::Tn10 [pAM-holD] recF400::Tn5 | P1 WA576 * JJC1157, selection KanR |
| JJC1164 | holDQ10am mdoB::Tn10 recF400::Tn5 | JJC1163 cured of pAM-holD |
| JJC1218 | holDQ10am mdoB::Tn10kan [pAM-holD] recB270 recC271 fuc::Tn10 | P1 JJC1220 * JJC1096 selection TetR |
| JJC1219 | holDQ10am mdoB::Tn10kan recB270 recC271 fuc::Tn10 | JJC1218 cured of pAM-holD |
| JJC1297 | thr-34::Tn10 | P1 CAG18442 * JJC520, selection TetR |
| JJC1300 | holDQ10am mdoB::Tn10kan [pAM-holD] recD1901::Tn10 ΔrecA938::cm | conjugation from JJC1299 to JJC1096, selection TetR CmR; screening for UVS |
| JJC1303 | holDQ10am | P1 JJC867 * JJC1297, selection Thr+, screening LBS |
| JJC1364 | holDQ10am recD1901::Tn10 | conjugation from JJC1298 to JJC1303, selection TetR CmR |
Table II. Mutation in the holD gene stimulates deletion of tandem repeats.
| Strain | Genotype | Recombinant proportion × 104 | Relative values | C.f.u. LB/MM |
|---|---|---|---|---|
| JJC520 | wild type | 1.9 ± 0.7 (21) | 1 | 0.9 ± 0.2 (12) |
| JJC616 | holD | 25 ± 13 (7) | 13 | ND |
| JJC867a | holD | 50 ± 4 (33) | 26 | 0.02 ± 0.013 (15) |
| JJC867 [pAM-holD]a | holD HolD+ | 2.9 ± 0.3 (3) | 1.5 | 0.97 ± 0.4 (7) |
| JJC867 [pAM34]a | holD | 32 ± 6 (3) | 17 | 0.0025 ± 0.0016 (5) |
aSimilar results were obtained in JJC947 and JJC1303 in which the hyper-recombination phenotype was co-transduced with mdoB::Tn10 and Thr+, respectively. In these mutants, the presence of the holD-carrying plasmid also restored a wild-type level of recombination and LB resistance (not shown).
Figures in parentheses indicate the number of determinations.
The holD gene of the hyper-recombination strain carries an amber mutation, suppressed by an unknown suppressor
The nucleotide sequence of the holD gene was determined in JJC520 (wild type) and JJC867 (holD). The only change in JJC867 compared with the wild-type strain was a C to T mutation at position 28, which changes the tenth amino acid, a glutamine, to an amber nonsense codon. The presence of an amber suppressor mutation in JJC520 was shown previously with the use of a plasmid carrying an amber mutant reporter gene (Bierne et al., 1997b) and was confirmed here by the observation that JJC520 supported the growth of λam phages (not shown). In spite of our efforts, we could not identify the suppressor mutation carried in this strain (see Materials and methods). Nevertheless, the presence of an amber suppressor in the holDam strain suggests that a mutated full-size protein is synthesized.
Phenotype of the holD mutant
The phenotype of the holD mutant strain was analyzed. The holD mutation conferred sensitivity to rich medium since holD liquid cultures or holD colonies grown on minimal medium had a reduced plating efficiency on LB (Table II). The ∼1% of bacteria that could form colonies on LB plates had lost the hyper-recombination phenotype and may have lost the holD mutation or acquired an additional amber suppressor. Sensitivity to rich medium is a property shared by several replication mutants in E.coli and is generally attributed to the difficulty in coping with an increased number of replication forks during propagation in rich medium. The sensitivity to rich medium was fully suppressed by introduction of the holD wild-type allele on a plasmid (Table II). In addition, the holD mutation conferred a mild sensitivity to high doses of UV irradiation (Figure 1) and a slight mutator phenotype, as the proportion of RifR cells in liquid cultures was increased nearly 7-fold by the holD mutation (3.3 × 10–7 in holD versus 4.8 × 10–8 in wild-type cells).
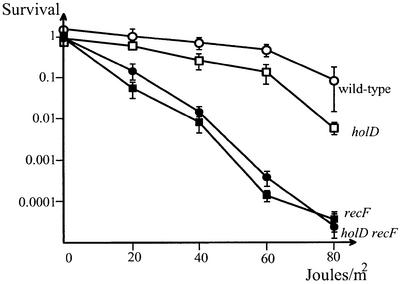
Fig. 1. UV sensitivity of recF mutants is not significantly affected by the holD mutation. Exponential cultures of cells grown to OD 0.5 were plated at appropriate dilutions on minimal medium plates that were irradiated at 2 J/m2 for various times and incubated for 48 h at 37°C. Ratios of c.f.u. (colony-forming units) on irradiated versus non-irradiated plates are shown. Open circles, JJC520 (wild type); closed circles, JJC561 (recF400::Tn5); open squares, JJC947 (holD); closed squares, JJC1164 (holD recF400::Tn5). Results are the average of 3–6 independent determinations.
Hyper-recombination in holD cells depends on the homologous recombination function of RecA
Tandem repeat deletions can be increased in E.coli replication mutants by RecA-dependent or -independent mechanisms (Bierne et al., 1997b; Saveson and Lovett, 1999). A ΔrecA::Tn10 mutation was introduced into the holD strain to determine whether RecA is required for the hyper-recombination phenotype. holD recA clones were obtained at the expected frequency and accordingly the strain presented no growth defect at 37°C compared with the holD single mutant. The holD recA cells were sensitive to rich medium (JJC868; Table III). The frequency of recombination was similar in the holD recA strain to that of wild-type cells (Table III). Introduction of the wild-type recA allele on a plasmid restored the hyper-recombination phenotype (JJC868 [pGB-RecA]; Table III). This shows that the hyper-recombination phenotype of the holD mutant requires RecA.
Table III. Tandem repeat deletions in holD mutants are dependent on RecA and independent of SOS induction, RecF and RecD.
| Strain | Genotype | Recombinant proportion × 104 | Relative values | C.f.u. LB/MM |
|---|---|---|---|---|
| JJC520 | wild type | 1.9 ± 0.7 (21) | 1 | 0.9 ± 0.2 (12) |
| JJC867 | holD | 50 ± 4 (33) | 26 | 0.02 ± 0.013 (15) |
| JJC868 | holD recA | 4.8 ± 3 (15) | 2.5 | 0.00014 ± 0.00006 (3) |
| JJC868 [pGB-recA] | holD recA RecA+ | 45 ± 20 (4) | 23 | 0.0015 ± 0.0009 (5) |
| JJC1097 | holD lexAind3 | 22 ± 10 (10) | 11 | 0.0077 ± 0.0044 (6) |
| JJC1164 | holD recF | 30 ± 15 (8) | 16 | 0.0023 ± 0.002 (7) |
| JJC1164 [pAM-holD] | holD recF HolD+ | 3.1 ± 1 (2) | 1.6 | 0.9 ± 0.1 (4) |
| JJC1364 | holD recD | 44 ± 21 (8) | 23 | ND |
As reported previously (Bierne et al., 1997a,b), recombination frequency was not significantly modified by the following mutations: ΔrecA (1.8 × 10–4), lexAind3 (0.9 × 10–4), recF400::Tn5 (1.1 × 10–4), recD1901::Tn10 (2.9 × 10–4).
Figures in parentheses indicate the number of determinations.
RecA is essential in E.coli for homologous recombination and for the induction of the SOS response, a set of repair genes induced by DNA damage (reviewed in Walker, 1996). In order to determine whether SOS induction is required for the hyper-recombination phenotype of the holD mutant, a lexAind3 mutation was used. This mutation prevents cleavage of the LexA repressor, which precludes SOS induction without affecting homologous recombination. The frequency of recombination was not modified significantly by the presence of the lexAind3 allele, which indicates that the hyper-recombination phenotype conferred by the holD allele does not require SOS induction (Table III). These results suggest that the stimulation of recombination between tandemly repeated sequences by the holD mutation requires the homologous recombination function of RecA. We therefore tested the role of proteins involved in the pre-synaptic steps of either of the two recombination pathways, RecFOR and RecBCD.
Hyper-recombination in holD cells is independent of RecF
The holD mutation affects a subunit of the γ complex of DNA Pol III HE responsible in vitro for the recycling of the lagging strand polymerase (reviewed in Kelman and O’Donnell, 1995). Impairment of the onset of Okasaki fragment synthesis could result in the formation of gaps on the lagging strand during replication. Since gap repair is mediated by the RecFOR pathway of recombination, if the hyper-recombination phenotype resulted from gap repair, it would require the presence of functional RecF protein. A recF null mutation did not modify the frequency of recombination in the holD mutant (Table III). Introduction of a plasmid carrying the wild-type holD allele in the holD recF strain decreased the frequency of recombination to the level observed in wild-type or recF strains, confirming that the hyper-recombination phenotype of the holD recF mutant is due to the holD mutation (Table III). These results indicate that the RecF protein is not required for the recombination events induced by the holD mutation. To test further whether the holD mutation caused the formation of gaps in the bacterial chromosome, sensitivity to UV irradiation was compared in recF and holD recF strains. Effectively, UV lesions on gapped DNA cannot be repaired by nucleotide excision repair (reviewed in Lin and Sancar, 1992) and therefore are mainly repaired by RecF-mediated recombination. The holD mutation did not increase the UV sensitivity of a recF strain (Figure 1) significantly, indicating that the holD mutation did not result in the accumulation of gaps.
The holD mutant requires RecBC for viability
Since the hyper-recombination phenotype of the holD mutant is dependent on RecA and independent of RecF, it presumably occurs by RecBCD-mediated recombination. However, this hypothesis could not be tested directly since attempts to introduce the null recB::Tn10 allele in the holD strain failed, suggesting that the holD recBC strain might not be viable. A conditional mutant was constructed, which carries recBts recCts mutations. The holD recBts recCts strain (JJC1219; Table I) was viable at low temperature and did not grow at high temperature, hence in conditions where RecBC was inactive. This result shows that the inactivation of RecBC is lethal in a holD strain, indicating the formation of double-strand ends in this strain.
The RecBCD complex is both a recombinase and an exonuclease. Whereas mutations in recC or recB inactivate all functions, mutations in recD inactivate only the exonuclease V activity while preserving the capacity to perform homologous recombination (Amundsen et al., 2000 and references therein). A recD::Tn10 null mutation could be introduced in the holD mutant, indicating that the exonuclease action of the RecBCD enzyme is not essential in this mutant. Recombination between tandemly repeated sequences was as high in the recD holD strains as in the holD single mutant, indicating that exonuclease V is not required for the hyper-recombination phenotype of the holD mutant (Table III).
The holD recA recD strain is not viable
holD recA and holD recD strains, in which either homologous recombination or exonuclease V action is inactivated, are viable whereas holD recBC strains, which lack both activities, are not viable. The non-viability of the holD recBC strain could result from the simultaneous inactivation of both exonuclease V and homologous recombination in this strain. To examine this possibility, we tested the viability of a holD recA recD strain. The pAM-holD plasmid was used. This plasmid carries a conditional replication origin, which functions only in the presence of isopropyl-β-d-thiogalactopyranoside (IPTG). In the absence of IPTG, the plasmid does not replicate and is lost during bacterial growth. Upon propagation of holD strains carrying the pAM-holD plasmid in the absence of IPTG, the plasmid was lost as expected and the holD mutant was recovered. A holD recA recD [pAM-holD] strain was constructed (JJC1300; Table I) and cells were propagated in the absence of IPTG to cure the plasmid (see Materials and methods). No holD recA recD cells could be isolated, indicating that this strain is not viable. Consequently, (i) the holD recA strain requires the exonuclease V activity of RecBCD for viability and (ii) the holD recD strain depends on the homologous recombination functions of RecA and RecBC for viability.
The holD mutant suffers DSBs in the absence of RecBC or in recA cells deficient for exonuclease V
The RecBCD complex is specifically required for the repair of DSBs since it initiates homologous recombination at double-stranded DNA ends. In order to determine whether the non-viability of the holD recBC strain results from DSB formation in this strain, the presence of linear DNA in vivo was directly tested by pulsed field gel electrophoresis (PFGE). The circular chromosome of E.coli does not enter gels in PFGE, in contrast to linear molecules. Therefore, the proportion of DNA that enters pulsed field gels reflects the extent of chromosome linearization. In vivo tritium labeling allows a quantitative determination of the proportion of DNA that migrates in gels, and gentle cell lysis in plugs ensures that the linear molecules detected are formed in vivo (Michel et al., 1997). Very little linear DNA can be detected in RecBCD+ cells in which linear DNA is either degraded or repaired by homologous recombination (wild-type cells, Table IV; Michel et al., 1997; Seigneur et al., 1998). The proportion of linear DNA was compared in recBts recCts and holD recBts recCts strains at low (RecBC mainly active) and high (RecBC inactive) temperature. The presence of the holD mutation led to a strong increase in the proportion of linear DNA upon inactivation of RecBC (compare JJC330 to JJC1219 at 42°C; Table IV). An effect of the holD mutation could also be observed at 30°C, which probably results from a saturation of the residual RecBCD activity in the holD recBts recCts mutant at low temperature. These results show that the holD mutation leads to the accumulation of linear DNA in a recBC context, hence to the occurrence of DSBs.
Table IV. The holD mutation causes the formation of RuvABC-dependent DSBs.
| Strain | Genotype | Percentage of linear DNA |
N | |
|---|---|---|---|---|
| 30°C | 42°C | |||
| JJC40 | wild type | 4.7 ± 0.4 | 4.4 ± 1.2 | 2/3 |
| JJC330 | recBts recCts | 6.1 ± 0.6 | 23.1 ± 2 | 2/2 |
| JJC1219 | holD recBts recCts | 32.8 ± 6.4 | 75.8 ± 6.9 | 4/4 |
| JJC557 [p-Gam] | recA Gam+ | 18.8 ± 3.5 | 33.2 ± 3.5 | 2/3 |
| JJC868 [p-Gam] | holD recA Gam+ | 13.7 ± 4.9 | 53.3 ± 3.7 | 4/6 |
| JJC1367 [p-Gam] | recA ruvABC Gam+ | 18.2 ± 0.6 | 33.6 ± 0.8 | 2/3 |
| JJC1294 [p-Gam] | holD recA ruvABC Gam+ | 15.5 ± 0.2 | 28 ± 3.9 | 2/5 |
| JJC1294 [p-Gam] [pRuvABC] | holD recA ruvABC Gam+ RuvABC+ | 20.2 ± 7.2 | 47.3 ± 3.3 | 4/8 |
N is the number of determinations at 30°C/42°C. Results in JJC40 were published previously (Seigneur et al., 1998). Results in JJC330 are confirmatory of previous results (Seigneur et al., 1998).
Since the holD recA recD mutant is not viable, we tested whether simultaneous inactivation of exonuclease V and RecA in the holD mutant also leads to an increased level of linear DNA. We used the λ Gam protein to inhibit the exonuclease V action of RecBCD. Gam was carried on a plasmid, under the control of a promoter active only at high temperature (pSF117; Murphy, 1991; called p-Gam here). In holD recA cells carrying this plasmid, exonuclease V is active at low temperature, which allows propagation of the strain, whereas at high temperature exonuclease V is inactivated by the induction of Gam. PFGE analysis showed an increase in the level of linear DNA due to the holD mutation in recA [p-Gam] cells at 42°C (compare JJC557[p-Gam] and JJC868[p-Gam]; Table IV). These results confirm that holD mutants devoid of homologous recombination and exonuclease V suffer DSBs due to the holD mutation.
DSB formation is RuvABC dependent in holD strains
The DSBs caused by the holD mutation in recBC and recA Gam+ cells may result from pauses of the replication forks when the γ complex fails to promote the lagging strand synthesis. Replication fork arrest caused by the inhibition of a replicative helicase was previously shown to cause chromosomal DSBs that result from the action of a Holliday junction specific enzyme: the RuvABC complex (Michel et al., 1997; Seigneur et al., 1998; reviewed in Michel, 2000). The RuvAB complex binds specifically to Holliday junctions and promotes branch migration of recombination intermediates. The RuvC endonuclease binds to the RuvAB–DNA complex and catalyzes Holliday junction resolution (reviewed in West, 1997). We tested whether RuvABC plays a role in DSB formation in holD mutants. For unknown reasons, we could not introduce the ruvABC mutation into the holD recBts recCts strain. Therefore, we tested the effects of the ruvABC mutation in the holD recA Gam+ cells. Inactivation of RuvABC led to a significant decrease in the level of DSB formation from 53 to 28% (compare JJC868[pGam] and JJC1294[pGam]; Table IV). Consequently, the holD mutation did not significantly modify the level of DSBs in a ruvABC context (compare JJC1294[pGam] and JJC1367[pGam]; Table IV). The amount of linear DNA was almost fully restored by the introduction of ruvABC wild-type genes on a plasmid (JJC1294[pGam][pRuvABC]; Table IV), whereas the presence of a plasmid carrying inactive ruvABC genes did not significantly modify the level of breakage (26% on average; data not shown). We conclude from these experiments that the occurrence of DSBs caused by the holD mutation requires functional RuvABC proteins.
The holD mutation does not cause DNA degradation in recA cells
Theoretically, RuvABC-mediated DSBs could occur either by direct breakage of the chromosome (Figure 2A), or after formation of a Holliday junction (Figure 2B). These models can be distinguished in recA RecBCD+ cells (Figure 2). Direct breakage of the E.coli chromosome leads to the formation of a long linear chromosomal arm, which is a substrate for exonuclease V action of RecBCD in cells defective for homologous recombination, leading to extensive DNA degradation (reviewed in Kuzminov, 1995; Figure 2A). In contrast, replication fork reversal leads to the formation of a small double-stranded tail, therefore to a non-detectable amount of DNA degradation in recA RecBCD+ cells (Seigneur et al., 1998; Figure 2B). To distinguish between these two models, DNA degradation was compared in holD+ and holD mutants in either recA+ or ΔrecA contexts (Figure 3). As reported previously, no DNA degradation was detected in wild-type cells whereas 30–40% of the chromosomal DNA was degraded in 3 h in the recA single mutant (Figure 3; Skarstad and Boye, 1993). The holD mutation caused a modest level of DNA degradation in recA+ cells and no detectable DNA degradation in the recA strain (Figure 3). The absence of holD-induced DNA degradation in the recA context is not compatible with the hypothesis of a direct breakage of chromosomes (Figure 2A) and rather supports the hypothesis of replication fork reversal (Figure 2B).
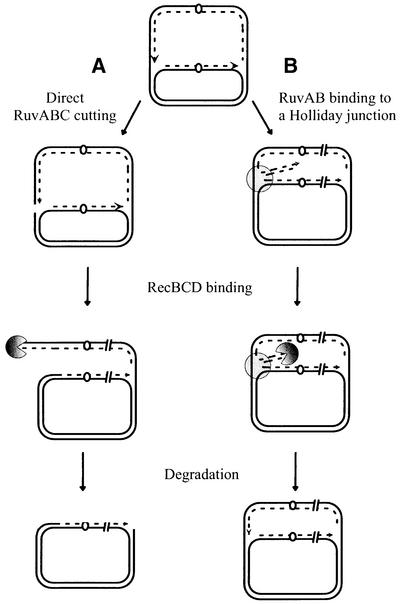
Fig. 2. In recA strains, extensive DNA degradation is expected if blocked replication forks are directly broken, not if they form a Holliday junction. (A) The fork is directly broken. (B) A Holliday junction is formed.
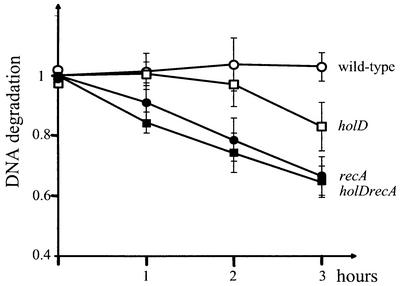
Fig. 3. DNA degradation in recA strains is not significantly affected by the holD mutation. DNA degradation was measured at different times as described previously (see Materials and methods; Seigneur et al., 1998). Open circles, JJC520 (wild type); closed circles, JJC557 (recA); open squares, JJC867 (holD); closed squares, JJC868 (holD recA). Results are the average of 3–5 independent determinations.
Discussion
In this work, we characterized the properties of the first holD mutant isolated. We show that this mutation causes lethality in combination with recBC mutation and stimulates homologous recombination between tandemly repeated sequences in a RecA-dependent way. The holD mutation induces the formation of RuvABC-dependent DSBs in the bacterial chromosome of recA strains lacking exonuclease V activity. Our results point to the arrest of the entire replication machinery upon HolD defect, causing replication fork reversal and, in turn, RecA- and RecBCD-mediated homologous recombination.
Simultaneous arrest of leading and lagging strand polymerases in holD mutants
The HolD subunit of Pol III HE, also called ψ, is part of the γ complex, thought to catalyze the recycling of the lagging strand polymerase at each Okazaki fragment (reviewed in Kelman and O’Donnell, 1995). The current model of γ complex action, based on in vitro experiments, is as follows. Loading of the sliding clamp β at the 3′ end of the RNA primer, which allows the binding of the lagging strand core polymerase, is catalyzed by the δ subunit (Turner et al., 1999). This loading requires the action of χ, which clears the 3′ end of the RNA primer by displacing the primase and perhaps SSB (Glover and McHenry, 1998; Kelman et al., 1998; Yuzhakov et al., 1999). The γ polypeptide binds and hydrolyzes ATP, inducing a conformational change in the γ complex that exposes δ (Turner et al., 1999; Ason et al., 2000 and references therein). ψ, encoded by holD, acts as a bridge between the ATPase γ and the protein χ (Xiao et al., 1993; Glover and McHenry, 2000). Although the precise role of ψ has remained elusive, it may contribute to the correct positioning of the γ and χ polypeptides for the energy-driven loading of the β clamp at the onset of Okazaki fragment synthesis. In vitro, the absence of χ decreases the processivity of Pol III HE, which indicates that χ is involved in efficient chain elongation (Kelman et al., 1998). In vivo, a mutation that affects the onset of Okasaki fragment synthesis could theoretically cause the formation of gaps in the lagging strand if lagging strand synthesis restarts after interruption, or the formation of a large single-stranded DNA region on the lagging strand at the fork if the synthesis on this strand does not restart while the leading strand synthesis goes on. The presence of numerous and/or large gaps is expected (i) to increase the UV sensitivity of the strain, since the nucleotide excision repair proteins do not act on single-stranded DNA, (ii) to increase the UV sensitivity of recF mutants, since lesions on gapped DNA are repaired mainly by RecF-mediated homologous recombination and (iii) to stimulate RecF-mediated homologous recombination. In contrast, our results show that (i) the holD mutant is only mildly sensitive to UV irradiation, (ii) the holD mutation does not increase the sensitivity to UV irradiation of a recF strain significantly and (iii) recombination between tandemly repeated sequences is not affected by a recF mutation. We conclude that the holD mutation characterized in this work does not lead to the accumulation in vivo of single-strand DNA on the chromosome, hence to the uncoupling of leading and lagging strand synthesis. This suggests that, in vivo, impairment of the recycling of the lagging strand polymerase by a mutation in the γ complex may lead to the arrest of the entire replication machinery.
A model for RuvABC-dependent DSBs in a holD recBC context
In contrast to the lack of effect of the recF mutation in the holD mutant, the RecBC complex is essential for viability in this strain. Therefore, for viability the strain requires a protein that acts at double-stranded DNA ends. However, the low level of DNA degradation in the holD single mutant and the observation that DNA degradation is not increased by the holD mutation in the recA strain argue against the formation of a high amount of linear DNA in holD or holD recA strains. Furthermore, previous studies have shown that to survive the occurrence of DSBs in the duplicated part of its chromosome, E.coli requires a functional RecA protein (Cromie et al., 2000; reviewed in Kuzminov, 1995). In contrast, we found that the holD recA strain is viable. Altogether, these observations suggest that most of the linear chromosomes detected in the holD recBts recCts strain do not form in holD single mutants, hence that RecBCD prevents breakage.
The DSBs caused by the holD mutation depend on the presence of the RuvABC proteins. These proteins have been shown previously to act at arrested replication forks blocked by the inactivation of a replicative helicase (Seigneur et al., 1998; reviewed in Michel, 2000). Based on genetic analysis of the rep mutant, defective for a helicase that participates in chromosome replication, a model was proposed for RuvABC-dependent DSB formation upon replication fork arrest (Figure 4). In a first step, a Holliday junction forms at blocked forks by annealing of neo-synthesized strands (Figure 4, A); then, binding of the RuvAB complex extends a double-stranded tail on which RecBCD can act (Figure 4, B); finally, RecBC- and RecA-mediated homologous recombination leads to re-integration of the tail into the chromosome, which, in turn, allows replication restart (Figure 4, C1–C2). Resolution of the Holliday junction by RuvC restores a viable chromosome (Figure 4, C3). Alternatively, RecBCD-mediated DNA degradation and removal of the RuvABC complex can lead to the restoration of a replication fork (Figure 4, D). In recBC or recA recD mutants, both the homologous recombination (Figure 4, C1–C3) and the degradation pathways (Figure 4, D) are inactive, and resolution of the Holliday junction by RuvC leads to a DSB (Figure 4, E). This model can remarkably explain our observations in the holD strain. It accounts for the lethality of the holD recBC and holD recA recD mutants, and for the formation of RuvABC-dependent DSBs (Figure 4, E). It can also explain the paradoxical observation that the holD recA mutant relies on exonuclease V activity for viability, whereas no DNA degradation caused by the holD mutation was detected in this strain. Indeed, degradation of a short DNA double-stranded tail would be sufficient for viability and would not be detectable (Figure 4, D). We could not test directly whether DSBs in the holD recBts recCts strain result from RuvABC action; however, the weak amount of DNA degradation in the holD mutant suggests that most of the linear DNA detected by PFGE may not be formed in the presence of RecBC. Therefore, we propose that most of the DSBs detected in the holD recBts recCts strain also result from the action of RuvABC on reversed replication forks. The similarity of the properties of helicase and holD mutants reinforces the idea that in holD cells the two DNA polymerases are arrested in concert when the mutated HolD protein fails to function.
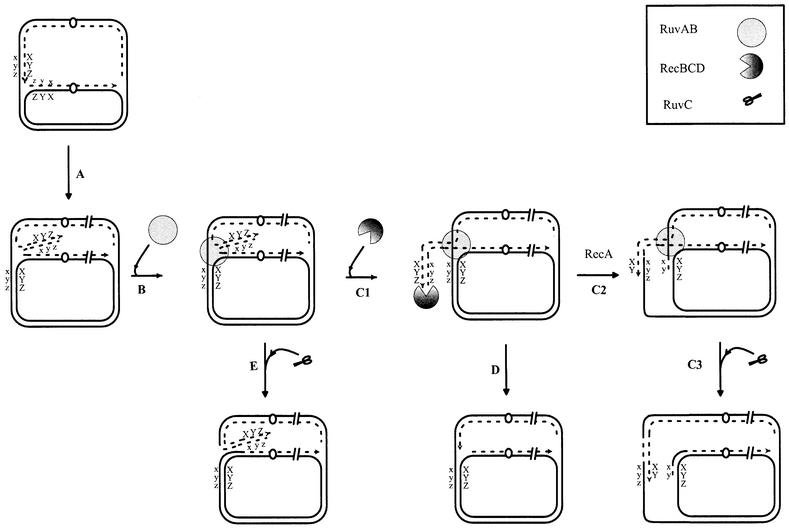
Fig. 4. Model for processing of arrested replication forks by recombination proteins in the holD mutants (adapted from Seigneur et al., 1998). In step A, the replication fork is blocked due to a defect in the lagging strand polymerase in the holD mutant. The two newly synthesized strands anneal, forming a Holliday junction that is stabilized by RuvAB binding (step B). Pathway C: RecBCD binds to the double-stranded tail (C1) and initiates a genetic exchange mediated by RecA (C2), RuvC resolves the first Holliday junction bound by RuvAB (C3). Pathway D: RecBCD-mediated degradation of the tail progresses up to the RuvAB-bound Holliday junction. Replication can restart when RecBCD has displaced the RuvAB complex. Pathway E: RuvC resolves the RuvAB-bound Holliday junction. Breakage at both forks results in the linear DNA detected by PFGE. Continuous and discontinuous lines represent the template and the newly synthesized strands of the chromosome, respectively; the arrow indicates the 3′ end of the growing strand.
Alternative pathways for tandem repeat recombination
Stimulation of recombination between tandemly repeated sequences by mutations in the holoenzyme Pol III has been reported previously (Bierne et al., 1997b; Saveson and Lovett, 1997). In dnaEts strains, affected in the catalytic polymerase subunit α, stimulation of deletion was independent of recombination proteins and was therefore proposed to occur by replication slippage (Bierne et al., 1997b). In the holD strain described here, RecBCD-mediated recombination is taking place, since RecBC inactivation is lethal. We propose that RecBCD acts at reversed replication forks to restore a viable chromosome by re-incorporation of the double-stranded tail generated by fork reversal (Figure 4, C). Unequal crossing over during the fork repair process would cause the increased recombination between tandemly repeated sequences. Similarly to the holD mutation, dnaBts mutations increased tandem repeat deletion (Saveson and Lovett, 1997, 1999) and caused RuvABC-dependent DSBs (Michel et al., 1997; Seigneur et al., 1998). The hyper-recombination phenotype of dnaBts mutants was dependent on RecA and RecBCD, and could also result from the re-incorporation of a double-stranded tail formed by replication fork reversal. Our results show that impairment of different components of the replisome can stimulate recombination at arrested replication fork by different mechanisms. They support a new model of recombination between tandemly repeated sequences, which implies a replication arrest due to a DNA polymerase mutation, and erroneous repair of reversed replication forks.
Replication fork reversal
Recent studies suggest that the reversal of replication forks can occur by several different pathways in E.coli, depending on the cause of replication arrest. RecA is required for the formation of the RuvABC substrate in the dnaBts strain, whereas it is not required in a rep mutant (Seigneur et al., 2000). In the holD strain, a high level of RuvABC-catalyzed DSBs is observed in recA mutants (Table IV), indicating that RecA is not essential for their formation. The observation that RuvABC inactivation does not restore the viability of the holD recBts recCts strain at high temperature, whereas it renders the rep recBts recCts strain thermoresistant, may be due to different mechanisms of replication fork reversal in rep and holD mutants. The RecG protein, which can bind to forked DNA and to Holliday junctions in vitro, was proposed to catalyze replication fork reversal at forks blocked by UV lesions (McGlynn and Lloyd, 2000). In contrast, RecG does not seem to act at forks blocked by helicase inactivation, since recG mutation has no effect on RuvABC-dependent DSBs in a rep mutant and only a minor effect in dnaBts strains (M.Seigneur and B.Michel, unpublished results). A putative role of RecG in replication fork reversal in holD strains remains to be tested.
We can imagine three pathways for the re-establishment of a functional replication fork after reversal: (i) re-integration by homologous recombination (Figure 4, C); (ii) degradation of the double-stranded tail by an exonuclease (Figure 4, D); and (iii) migration of the Holliday junction back to the original configuration by a branch migration enzyme (in Figure 4, step A is a reversible reaction). In E.coli rep and holD mutants, the latter pathway is either rare or does not lead to the formation of a viable chromosome since RecBCD is essential for viability. Homologous recombination may be favored because it facilitates the binding of the primosomal proteins, a set of proteins that assemble at recombination intermediates to load a new replisome (Kuzminov and Stahl, 1999; Motamedi et al., 1999; reviewed in Kogoma, 1997; Marians, 2000; Sandler and Marians, 2000). The distribution of the recombination hotspot CHI is not random on the E.coli chromosome (Blattner et al., 1997). The bias in the orientation of CHI sites is such that RecBCD progressing on the double-stranded tail formed by replication fork reversal encounters CHI sites in the active orientation very frequently (one every 5 kb on average; Blattner et al., 1997), which stimulates homologous recombination. Consequently, re-integration of a reversed fork by homologous recombination may be the major pathway in Rec+ cells, causing the hyper-recombination phenotype of the holD strain. Nevertheless, this is not the only pathway since holD recA and rep recA strains (Seigneur et al., 1998) are viable. The lethality of rep recD recA and holD recD recA combinations indicates that DNA degradation is the alternative pathway. The mechanisms of re-establishment of a replication fork may depend on the enzymes that have catalyzed the formation of the Holliday junction. For example, RecG was proposed to promote both the formation of reversed forks and their branch migration back to the original configuration during UV repair (McGlynn and Lloyd, 2000).
In eukaryotes, reversed forks could presumably be reset either by homologous recombination or by backward branch migration. The former model is supported by the observation that replication can be initiated from a recombination intermediate formed by DSB repair (Chen et al., 1998; Holmes and Haber, 1999; reviewed in Haber, 2000). The latter model is supported by the properties of eukaryotic enzymes belonging to the RecQ subfamily of helicases, which have been proposed to catalyze such a reaction. Sgs1 in Saccharomyces cerevisiae, Rqh in Schizosaccharomyces pombe and the Bloom syndrome protein (BLM) in human were shown to act at Holliday junctions, and were proposed to participate in genome replication by disrupting Holliday junctions formed upon replication arrest (Doe et al., 2000; Gangloff et al., 2000; Karow et al., 2000).
Hyper-recombination associated with a mutation in a DNA polymerase gene or with the encounter of replication forks with a replication barrier was reported in yeast (Zou and Rothstein, 1997; Kobayashi et al., 1998; Defossez et al., 1999; reviewed in Rothstein et al., 2000). Furthermore, the existence of links between replication impairment and recombination was also described in higher eukaryotes (Sonoda et al., 1998; Karow et al., 2000). The present work is a new illustration of a role for DNA recombination proteins during replication progression. It extends the replication fork reversal model to cells affected in a DNA polymerase subunit involved in the synthesis of the lagging strand.
Materials and methods
Bacterial strains and plasmids
The bacterial strains used in this study are shown in Table I. The construction of strain JJC520, which carries a 624 bp duplication in the lacZ gene adjacent to a CmR gene, the mutagenesis of JJC520 to isolate hyper-recombination clones and the mapping of the mutation by Hfr crossing have been described previously (Bierne et al., 1997a,b). All E.coli strains used in this study were constructed by classical techniques of P1 transduction or Hfr conjugation, and grown on standard media, Luria broth supplemented with 25 mg/ml thymine and minimal medium M63 (Miller, 1992). The UV-sensitive phenotype of lexAind and recA, recBC, recF mutants and the exonuclease-deficient phenotype conferred by the recBCD mutation were verified as described previously (Seigneur et al., 1998). The sensitivity to rich medium of holD strains was systematically verified for each experiment. To measure deletion frequencies, isolated colonies were picked from minimal medium plates and resuspended in 1 ml of minimal medium. Dilutions of these bacterial suspensions were plated on several minimal medium plates containing X-gal. LacZ+ and lacZ colonies were counted; the average of the proportion of LacZ+ among lacZ clones in several colonies was calculated.
pGBara-recA and pSF117 (pBR322-Gam+, called p-Gam here) were described previously (Michel et al., 1997). pAM-holD was constructed by cloning in the pAM34 vector (Gil and Bouché, 1991) a 937 bp PCR fragment carrying the holD gene. This plasmid carries an inducible replication origin that functions only in the presence of IPTG. Segregation of the plasmid was performed in minimal medium. Overnight cultures grown in the presence of 50 µg/ml IPTG and 40 µg/ml ampicillin were diluted 1000-fold in minimal medium devoid of IPTG and of ampicillin. These overnight cultures were diluted again 1000-fold in the same medium. The second cultures propagated in the absence of IPTG either did not grow, indicating that cured cells were not viable (e.g. holD recA recD strain), or contained >99% of cured bacteria that could be isolated by plating and tested (e.g. holD and holD recA strains). holD recF and holD recBts recCts strains were constructed by the introduction of recF or recBts recCts mutations in the holD [pAM-holD] strain, respectively, and curing of the plasmid by propagation in the absence of IPTG as described above.
Genetic tests to ascertain the presence of an amber suppressor mutation in JJC520 were a plasmid carrying an α-amylase amber mutant gene [in contrast to a previous mis-interpretation (Bierne et al., 1997b), this test does not allow identification of the amino acid incorporated; Declerck et al., 1990] and λam phages. The λam phages allowed it to be determined that the suppressor could not be supG, therefore the lysT gene was not sequenced.
Sequence analysis and research of a suppressor mutation
DNA preparation and sequence analysis were performed as described (Seigneur et al., 1998). The holD locus was amplified by PCR from JJC520 (holD520+) and JJC867 (holD867) colonies. The sequence of two independent PCR products from each strain was determined entirely on one strand with the use of three primers: GTGGTGGAATTCCTGGGTATG, TACAGCAACTGGGCATTACCCA and GAAGCACGGAATTCCAGCCATA.
To identify the suppressor mutation carried in JJC520, known amber suppressor tRNA genes were sequenced. glnUW, glnXV, leuX, serU, tyrT, tyrU, trpT were amplified by PCR. The sequence was determined entirely on both strands with the use of two primers. Sequences were compared with the DNA sequences in GenBank (Blattner et al., 1997) and found to be identical, showing the absence of any mutation in these tRNA genes. We then attempted to clone the suppressor gene from JJC520. Chromosomal DNA purified from JJC520 and partially digested with either Sau3A, EcoRI, HindIII or both EcoRI and HindIII was cloned in pBR322 or pGB2 vectors. These banks were used to transform an indicator strain, JJC942, that carried a his and a trp amber mutation, selecting for His+ Trp+ clones. No gene that conferred a His+ Trp+ phenotype could be isolated, whereas, as expected, the his and trp wild-type alleles were cloned. We conclude that the strain may carry a non-clonable or multigenic amber suppressor.
Measurement of DSBs by PFGE
Measurement of DSBs was performed as described previously (Seigneur et al., 1998). Briefly, for chromosome labeling, cells were grown in minimal medium in the presence of tritiated thymidine and deoxyadenosine. Cells were collected, washed and embedded in agarose plugs. Gentle lysis was performed in plugs. Plugs were used for PFGE and the proportion of DNA migrating was determined by cutting each lane into slices and counting the tritium present in the wells and in the gel slices (Seigneur et al., 1998). To avoid DNA damage during PFGE, the apparatus was routinely washed with 0.1% SDS.
Measurement of DNA degradation
Measurement of DNA degradation was as described previously (Seigneur et al., 1998). Briefly, cells were grown in the presence of tritiated thymidine and deoxyadenosine at 37°C until OD 0.4. Cells were then collected, washed, suspended at OD 0.15 in cold medium containing 40 µg/ml cold thymidine and incubated at 37°C. The amount of trichloroacetic acid (TCA)-precipitated tritium was determined at time 0 (transfer to cold medium) and every hour for 3 h. TCA-precipitated DNA was bound to Millipore filters, washed on a Millipore multi-filtration unit and counted in a Beckman counter.
Acknowledgments
Acknowledgements
We thank all laboratories that sent us strains and plasmids, and G.Maenhaut-Michel for λam phages. We thank M.F.Bredèche, A.Vivrel and G.Grompone for performing some of the experiments, and N.Galleron for her help in sequence analysis. We are very grateful to V.Bidnenko, C.Bruand, D.Canceill, P.Noirot and E.Viguera for helpful reading of the manuscript. M.-J.F. is supported by a TMR Marie Curie Research Training Grant from the European Community. This work is supported in part by the Programme de Recherche Fondamentale en Microbiologie, Maladies Infectieuses et Parasitaires.
References
- Amundsen S.K., Taylor,A.F. and Smith,G.R. (2000) The RecD subunit of the Escherichia coli RecBCD enzyme inhibits RecA loading, homologous recombination and DNA repair. Proc. Natl Acad. Sci. USA, 97, 7399–7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson D.G. and Kowalczykowski,S.C. (1997) The translocating RecBCD enzyme stimulates recombination by directing RecA protein onto ssDNA in a χ-regulated manner. Cell, 90, 77–86. [DOI] [PubMed] [Google Scholar]
- Ashley C.T.J. and Warren,S.T. (1995) Trinucleotide repeat expansion and human disease. Annu. Rev. Genet., 29, 703–728. [DOI] [PubMed] [Google Scholar]
- Ason B., Bertram,J.G., Hingorani,M.M., Beechem,J.M., O’Donnell,M., Goodman,M.F. and Bloom,L.B. (2000) A model for Escherichia coli DNA polymerase III holoenzyme assembly at primer/template ends—DNA triggers a change in binding specificity of the γ complex clamp loader. J. Biol. Chem., 275, 3006–3015. [DOI] [PubMed] [Google Scholar]
- Bierne H., Seigneur,M., Ehrlich,S.D. and Michel,B. (1997a) uvrD mutations enhance tandem repeat deletion in the Escherichia coli chromosome via SOS induction of the RecF recombination pathway. Mol. Microbiol., 26, 557–567. [DOI] [PubMed] [Google Scholar]
- Bierne H., Vilette,D., Ehrlich,S.D. and Michel,B. (1997b) Isolation of a dnaE mutation which enhances RecA-independent homologous recombination in the Escherichia coli chromosome. Mol. Microbiol., 24, 1225–1234. [DOI] [PubMed] [Google Scholar]
- Blattner F.R. et al. (1997) The complete genome sequence of Escherichia coli K-12. Science, 277, 1453–1474. [DOI] [PubMed] [Google Scholar]
- Chen C., Umezu,K. and Kolodner,R.D. (1998) Chromosomal rearrangements occur in S.cerevisiae rfa1 mutator mutants due to mutagenic lesions processed by double-strand-break repair. Mol. Cell, 2, 9–22. [DOI] [PubMed] [Google Scholar]
- Churchill J.J., Anderson,D.G. and Kowalczykowski,S.C. (1999) The RecBC enzyme loads RecA protein onto ssDNA asymmetrically and independently of χ, resulting in constitutive recombination activation. Genes Dev., 13, 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromie G.A., Millar,C.B., Schmidt,K.H. and Leach,D.R.F. (2000) Palindromes as substrates for multiple pathways of recombination in Escherichia coli. Genetics, 154, 513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Declerck N., Joyet,P., Gaillardin,C. and Masson,J.M. (1990) Use of amber suppressors to investigate the thermostability of Bacillus licheniformisα-amylase. Amino acid replacements at 6 histidine residues reveal a critical position at His-133. J. Biol. Chem., 265, 15481–15488. [PubMed] [Google Scholar]
- Defossez P.A., Prusty,R., Kaeberlein,M., Lin,S.J., Ferrigno,P., Silver,P.A., Keil,R.L. and Guarente,L. (1999) Elimination of replication block protein Fob1 extends the life span of yeast mother cells. Mol. Cell, 3, 447–455. [DOI] [PubMed] [Google Scholar]
- Djian P. (1998) Evolution of simple repeats in DNA and their relation to human disease. Cell, 94, 155–160. [DOI] [PubMed] [Google Scholar]
- Doe C.L., Dixon,J., Osman,F. and Whitby,M.C. (2000) Partial suppression of the fission yeast rqh1(–) phenotype by expression of a bacterial Holliday junction resolvase. EMBO J., 19, 2751–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S., Soustelle,C. and Fabre,F. (2000) Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nature Genet., 25, 192–194. [DOI] [PubMed] [Google Scholar]
- Gil D. and Bouché,J.P. (1991) ColE1-type vectors with fully repressible replication. Gene, 105, 17–22. [DOI] [PubMed] [Google Scholar]
- Glover B.P. and McHenry,C.S. (1998) The χΨ subunits of DNA polymerase III holoenzyme bind to single-stranded DNA-binding protein (SSB) and facilitate replication of an SSB-coated template. J. Biol. Chem., 273, 23476–23484. [DOI] [PubMed] [Google Scholar]
- Glover B.P. and McHenry,C.S. (2000) The DnaX-binding subunits δ′ and Ψ are bound to γ and not τ in the DNA polymerase III holoenzyme. J. Biol. Chem., 275, 3017–3020. [DOI] [PubMed] [Google Scholar]
- Haber J.E. (2000) Recombination: a frank view of exchanges and vice versa. Curr. Opin. Cell Biol., 12, 286–292. [DOI] [PubMed] [Google Scholar]
- Holmes A.M. and Haber,J.E. (1999) Double-strand break repair in yeast requires both leading and lagging strand DNA polymerases. Cell, 96, 415–424. [DOI] [PubMed] [Google Scholar]
- Karow J.K., Constantinou,A., Li,J.L., West,S.C. and Hickson,I.D. (2000) The Bloom’s syndrome gene product promotes branch migration of Holliday junctions. Proc. Natl Acad. Sci. USA, 97, 6504–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelman Z. and O’Donnell,M. (1995) DNA polymerase III holoenzyme: structure and function of a chromosomal replicating machine. Annu. Rev. Biochem., 64, 171–200. [DOI] [PubMed] [Google Scholar]
- Kelman Z., Yuzhakov,A., Andjelkovic,J. and O’Donnell,M. (1998) Devoted to the lagging strand—the χ subunit of DNA polymerase III holoenzyme contacts SSB to promote processive elongation and sliding clamp assembly. EMBO J., 17, 2436–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T., Heck,D.J., Nomura,M. and Horiuchi,T. (1998) Expansion and contraction of ribosomal DNA repeats in Saccharomyces cerevisiae: requirement of replication fork blocking (Fob1) protein and the role of RNA polymerase I. Genes Dev., 12, 3821–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogoma T. (1997) Stable DNA replication: interplay between DNA replication, homologous recombination and transcription. Microbiol. Mol. Biol. Rev., 61, 212–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczykowski S.C., DixonA.K., Eggleston,S.D., Lauder,W. and Rehauer,M. (1994) Biochemistry of homologous recombination in Escherichia coli. Microbiol. Rev., 58, 401–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzminov A. (1995) Collapse and repair of replication forks in Escherichia coli.Mol. Microbiol., 16, 373–384. [DOI] [PubMed] [Google Scholar]
- Kuzminov A. (1999) Recombinational repair of DNA damage in Escherichia coli and bacteriophage λ. Microbiol. Mol. Biol. Rev., 63, 751–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzminov A. and Stahl,F.W. (1999) Double-strand end repair via the RecBC pathway in Escherichia coli primes DNA replication. Genes Dev., 13, 345–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.J. and Sancar,A. (1992) (A)BC excinuclease—the Escherichia coli nucleotide excision repair enzyme. Mol. Microbiol., 6, 2219–2224. [DOI] [PubMed] [Google Scholar]
- Lovett S.T., Drapkin,P.T., Sutera,V.A. and Gluckman-Peskind,T.J. (1993) A sister-strand exchange mechanism for recA-independent deletion of repeated DNA sequences in Escherichia coli. Genetics, 135, 631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marians K.J. (2000) PriA-directed replication fork restart in Escherichia coli. Trends Biochem. Sci., 25, 185–189. [DOI] [PubMed] [Google Scholar]
- McGlynn P. and Lloyd,R.G. (2000) Modulation of RNA polymerase by (P)ppGpp reveals a RecG-dependent mechanism for replication fork progression. Cell, 101, 35–45. [DOI] [PubMed] [Google Scholar]
- Michel B. (2000) Replication fork arrest and DNA recombination. Trends Biochem. Sci., 25, 173–178. [DOI] [PubMed] [Google Scholar]
- Michel B., Ehrlich,S.D. and Uzest,M. (1997) DNA double strand breaks caused by replication arrest. EMBO J., 16, 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J.H. (1992) A Short Course in Bacterial Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Motamedi M.R., Szigety,S.K. and Rosenberg,S.M. (1999) Double-strand-break repair recombination in Escherichia coli: physical evidence for a DNA replication mechanism in vivo. Genes Dev., 13, 2889–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy K.C. (1991) λ-Gam protein inhibits the helicase and χ-stimulated recombination activities of Escherichia coli RecBCD enzyme. J. Bacteriol., 173, 5808–5821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naktinis V., Turner,J. and O’Donnell,M. (1996) A molecular switch in a replication machine defined by an internal competition for protein rings. Cell, 84, 137–145. [DOI] [PubMed] [Google Scholar]
- Nichols B.P., Shafiq,O. and Meiners,V. (1998) Sequence analysis of Tn10 insertion sites in a collection of Escherichia coli strains used for genetic mapping and strain construction. J. Bacteriol., 180, 6408–6411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein R., Michel,B. and Gangloff,S. (2000) Replication fork pausing and recombination or ‘gimme a break’. Genes Dev., 14, 1–10. [PubMed] [Google Scholar]
- Sandler S.J. and Marians,K.J. (2000) Role of PriA in replication fork reactivation in Escherichia coli. J. Bacteriol., 182, 9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saveson C.J. and Lovett,S.T. (1997) Enhanced deletion formation by aberrant DNA replication in Escherichia coli. Genetics, 146, 457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saveson C.J. and Lovett,S.T. (1999) Tandem repeat recombination induced by replication fork defects in Escherichia coli requires a novel factor, RadC. Genetics, 152, 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seigneur M., Bidnenko,V., Ehrlich,S.D. and Michel,B. (1998) RuvAB acts at arrested replication forks. Cell, 95, 419–430. [DOI] [PubMed] [Google Scholar]
- Seigneur M., Ehrlich,D. and Michel,B. (2000) Resolution of Holliday junctions by RuvABC prevents dimer formation in rep mutants and UV-irradiated cells. Mol. Microbiol., 37, 180–191. [DOI] [PubMed] [Google Scholar]
- Singer M. et al. (1989) A collection of strains containing genetically linked alternating antibiotic resistance elements for genetic mapping of Escherichia coli. Microbiol. Rev., 53, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarstad K. and Boye,E. (1993) Degradation of individual chromosomes in RecA mutants of Escherichia coli. J. Bacteriol., 175, 5505–5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda E., Sasaki,M.S., Buerstedde,J.M., Bezzubova,O., Shinohara,A., Ogawa,H., Takata,M., YamaguchiIwai,Y. and Takeda,S. (1998) Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J., 17, 598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner J., Hingorani,M.M., Kelman,Z. and O’Donnell,M. (1999) The internal workings of a DNA polymerase clamp-loading machine. EMBO J., 18, 771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umezu K., Chi,N.-W. and Kolodner,R. (1993) Biochemical interaction of the Escherichia coli RecF, RecO and RecR proteins with RecA protein and single-stranded DNA binding protein. Proc. Natl Acad. Sci. USA, 90, 3875–3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker G.C. (1996) The SOS response of Escherichia coli. In Neidhardt,F.C. et al. (eds), Escherichia coli and Salmonella typhimurium, Cellular and Molecular Biology, 2nd edn. American Society for Microbiology, Washington, DC, pp. 1400–1417.
- Webb B.L., Cox,M.M. and Inman,R.B. (1997) Recombinational DNA repair: the RecF and RecR proteins limit the extension of RecA filaments beyond single-strand DNA gaps. Cell, 91, 347–356. [DOI] [PubMed] [Google Scholar]
- West S.C. (1997) Processing of recombination intermediates by the RuvABC proteins. Annu. Rev. Genet., 31, 213–244. [DOI] [PubMed] [Google Scholar]
- Xiao H., Crombie,R., Dong,Z., Onrust,R. and O’Donnell,M. (1993) DNA polymerase III accessory proteins. III. holC and holD encoding χ and Ψ. J. Biol. Chem., 268, 11773–11778. [PubMed] [Google Scholar]
- Yuzhakov A., Kelman,Z. and O’Donnell,M. (1999) Trading places on DNA—a three-point switch underlies primer handoff from primase to the replicative DNA polymerase. Cell, 96, 153–163. [DOI] [PubMed] [Google Scholar]
- Zou H. and Rothstein,R. (1997) Holliday junctions accumulate in replication mutants via a RecA homolog-independent mechanism. Cell, 90, 87–96. [DOI] [PubMed] [Google Scholar]