

Abstract
Given the importance of the Rho GTPase family member Rac1 and the Rac1/Cdc42 effector PAK1 in T-cell activation, we investigated the requirements for their activation by the T-cell receptor (TCR). Rac1 and PAK1 activation required the tyrosine kinases ZAP-70 and Syk, but not the cytoplasmic adaptor Slp-76. Surprisingly, PAK1 was activated in the absence of the transmembrane adaptor LAT while Rac1 was not. However, efficient PAK1 activation required its binding sites for Rho GTPases and for PIX, a guanine nucleotide exchange factor for Rho GTPases. The overexpression of βPIX that either cannot bind PAK1 or lacks GEF function blocked PAK1 activation. These data suggest that a PAK1–PIX complex is recruited to appropriate sites for activation and that PIX is required for Rho family GTPase activation upstream of PAK1. Furthermore, we detected a stable trimolecular complex of PAK1, PIX and the paxillin kinase linker p95PKL. Taken together, these data show that PAK1 contained in this trimolecular complex is activated by a novel LAT- and Slp-76-independent pathway following TCR stimulation.
Keywords: PAK/PIX/PKL/signal transduction/TCR
Introduction
Specific T-cell recognition of antigen triggers a cascade of diverse intracellular signaling events that includes protein phosphorylation, lipid hydrolysis, intracellular calcium flux, lipid phosphorylation, small GTPase activation, cytoskeletal rearrangements and many others. This complex series of events is required for productive T-cell activation as defined by either T-cell proliferation, differentiation or activation of effector functions (reviewed by van Leeuwen and Samelson, 1999; Kane et al., 2000). Among the earliest events in T-cell receptor (TCR) signal transduction is the activation of the Src tyrosine kinase family members Lck and FynT, which directly phosphorylate immunoreceptor tyrosine-based activation motifs (ITAMs) in the ζ and CD3γ, δ and ε chains. The phosphorylated ITAMs recruit the Syk family tyrosine kinases ZAP-70 and Syk via tandem SH2 domains in their N-termini. The TCR-associated Src and Syk kinases are then able to effect, directly or indirectly, the tyrosine phosphorylation of multiple substrates.
One critical substrate of the Src and Syk kinases is the transmembrane adaptor molecule LAT. The palmitoylation of LAT targets it to cholesterol-rich lipid rafts and is required for its function in the TCR signaling pathway (Zhang et al., 1998b; Lin et al., 1999). Tyrosine-phosphorylated LAT recruits additional proteins to the membrane such as Grb2 and PLCγ1, which are critical for the activation of the Ras and phosphatidylinositol signaling pathways (Zhang et al., 1998a). Indeed, LAT-deficient Jurkat T cells are defective in the activation of both the Ras signaling pathway and the phosphatidylinositol signaling pathway (Finco et al., 1998; Zhang et al., 1999).
Yet another target of the Src and Syk kinases is the cytoplasmic adaptor Slp-76. Slp-76 can interact with multiple proteins including PLCγ1 (D.Yablonski, manuscript in preparation), Grb2 (Jackman et al., 1995), Gads, LAT (via Gads) (Asada et al., 1999; Liu et al., 1999), Vav1 (Wu et al., 1996), Lck (Sanzenbacher et al., 1999) and Nck (Bubeck Wardenburg et al., 1998). Amidst this array of interactions, the precise mechanism of Slp-76 function in the context of TCR-mediated signaling remains unclear. However, it is clear from the study of a Slp-76-deficient Jurkat T cell that Slp-76, like LAT, is required for the optimal activation of the Ras and phosphatidylinositol signaling pathways (Yablonski et al., 1998b).
While both Ras activation and calcium flux are necessary for T-cell activation, Rho family GTPases (Rac1, Cdc42 and Rho) also appear to play an important role. The expression of a dominant-negative Rac1 allele inhibits TCR-induced interleukin-2 (IL-2) promoter activation (Genot et al., 1996) and dominant-negative Cdc42 blocks T-cell polarization towards antigen presenting cells (Stowers et al., 1995). Furthermore, Vav1, a guanine nucleotide exchange factor (GEF) for Rac1 and Cdc42, is required for efficient positive and negative selection of T cells. In mature T cells, Vav1 is important for TCR-induced calcium flux and perhaps also Ras pathway activation (Turner et al., 1997; Fischer et al., 1998; Holsinger et al., 1998; Costello et al., 1999).
The study of PAK1, an effector of Rac1/Cdc42, further demonstrates the importance of the Rho family of GTPases in T-cell activation. After TCR engagement, PAK1 associates with the cytoplasmic adaptor Nck and becomes catalytically activated (Bubeck Wardenburg et al., 1998; Yablonski et al., 1998a). Furthermore, a dominant-negative allele of PAK1 blocks the TCR-induced activation of Erk, a downstream effector of the Ras pathway, and of the nuclear factor of activated T cells (NFAT), a critical transcriptional element in the IL-2 promoter. The activation of Rac1/Cdc42 and PAK1 is believed to require the association of Vav1, Slp-76, Nck and PAK1 (Bubeck Wardenburg et al., 1998). In a model to explain these observations, Slp-76 recruits Vav1 to a complex containing Nck and PAK1. Vav1 catalyzes GTP loading of Cdc42 or Rac1, which, in turn, activates the Nck-associated PAK1.
The study of PAK1 activation in other cell types has revealed a possible role for the PAK-interacting exchange factor or PIX (also known as Cool) (Bagrodia et al., 1998; Manser et al., 1998; Daniels et al., 1999; Yoshii et al., 1999). Cloned as a PAK-interacting protein, PIX is a GEF for Rac1 and possibly for Cdc42. Thus far, α and β genes have been cloned and each appears to have multiple splice forms. Interestingly, αPIX, but not βPIX, has an N-terminal calponin homology domain similar to that of Vav1. However, distinct from Vav1, both αPIX and βPIX contain an SH3 domain that binds with unusually high affinity to a polyproline stretch in PAK1. Finally, PAK1 activation in fibroblasts and neuronal cells may involve the p95PKL family of PIX-interacting proteins (p95PKL, Cat and GIT), which link PAK1 to the integrin-associated adaptor paxillin (Bagrodia et al., 1999; Turner et al., 1999; Zhao et al., 2000).
In the present study, we examined the requirements for PAK1 and Rac1 activation by TCR signaling. PAK1 activation required ZAP-70, but not LAT, Slp-76 or Nck. Similar to PAK1, Rac1 activation also required ZAP-70 but not Slp-76. However, unlike PAK1, Rac1 was not activated in the absence of LAT. Finally, we showed that PIX and p95PKL are critical for the activation of PAK1 by the TCR. These data demonstrate that PAK1 and Rac1 are activated by novel mechanisms that are distinct from the TCR-mediated activation of the Ras and phosphatidylinositol signaling pathways.
Results
Activation of PAK1 requires the tyrosine kinase ZAP-70
We have shown previously that PAK1 is activated by TCR stimulation and that this activation requires the Src kinase Lck (Yablonski et al., 1998a). In order to identify additional requirements for PAK1 activation by TCR stimulation, we measured the activation of endogenous PAK1 in the P116 Jurkat mutant cell line, which does not express ZAP-70 or Syk (Williams et al., 1998). Parental Jurkat cells, the P116 cells or P116 cells stably transfected with a wild-type ZAP-70 cDNA were stimulated with an anti-TCR antibody for 3 min or a buffer control. Endogenous PAK1 was immunoprecipitated and in vitro kinase activity, using histone H4 as an exogenous substrate, was measured, as has been described previously (Yablonski et al., 1998a). Fold activation of PAK1 kinase activity after TCR stimulation was normalized to that of wild-type Jurkat cells in the same experiment. The typical fold activation of wild PAK1 kinase activity in wild-type Jurkat cells ranged between 6- and 8-fold. Baseline kinase activity was the same in all cells and with all mutant PAK1 constructs (data not shown). As shown in Figure 1, P116 cells failed to induce PAK1 kinase activity following TCR stimulation. However, P116 cells stably reconstituted with a wild-type ZAP-70 cDNA induced PAK1 kinase activity nearly as well as parental Jurkat cells after TCR stimulation. Therefore, TCR-induced PAK1 activation requires ZAP-70.

Fig. 1. PAK1 activation requires ZAP-70 but not LAT. The cells indicated were stimulated for 3 min with C305 (+) or a buffer control (–) at 37°C. Endogenous PAK1 was immunoprecipitated with an anti-PAK1 antibody and subjected to an in vitro kinase assay as described in Materials and methods. The fold increase in H4 phosphorylation after TCR stimulation was normalized to the fold activation of parental Jurkat cells in the same experiment. This percentage of wild-type Jurkat PAK1 activation was averaged over n experiments and plotted above. Error bars indicate the range or standard error over n experiments. P116 (n = 2) is a ZAP-70-deficient Jurkat cell. P116 C39 (n = 2) is a ZAP-70 reconstituted P116 cell. J.CaM2 (n = 11) is a LAT-deficient cell. J.CaM2 LAT3 (n = 3) and J.CaM2 LAT9 (n = 2) are two J.CaM2 cells independently reconstituted with LAT.
PAK1 activation occurs in the absence of LAT
One important downstream target of ZAP-70 is the lipid raft-anchored adaptor LAT. LAT is a critical link between the TCR-associated Src and Syk tyrosine kinases and the activation of the Ras and phosphatidylinositol pathways (Finco et al., 1998; Zhang et al., 1999). Since PAK1 activation was strictly dependent on ZAP-70 and Lck, we asked whether LAT is also required for PAK1 activation by measuring PAK1 kinase activity in the LAT-deficient Jurkat cell line, J.CaM2. As shown in Figure 1, J.CaM2 cells reproducibly induced PAK1 kinase activity after 3 min of TCR stimulation. At time points of up to 15 min of TCR stimulation, the activation of PAK1 in J.CaM2 was similar to that in wild-type Jurkat cells (data not shown). The induction of PAK1 kinase activity in two stable clones of J.CaM2 reconstituted with a wild-type LAT cDNA was similar to that of J.CaM2 and Jurkat. These data show that PAK1 can be activated independently of LAT.
TCR-mediated PAK1 activation is independent of Slp-76 and Nck
That PAK1 could be activated independently of LAT demonstrated a fundamental difference between the activation of the Ras and phosphatidylinositol pathways and the activation of PAK1. To confirm this difference, we asked whether the cytoplasmic adaptor Slp-76, which functions in concert with LAT to activate the Ras and phosphatidylinositol pathways, was also dispensable for PAK1 activation by TCR stimulation. We measured the TCR induction of PAK1 kinase activity in a Jurkat mutant line, J14, which does not express Slp-76 (Yablonski et al., 1998b). The TCR-inducible activation of endogenous PAK1 immunoprecipitated from J14 was essentially identical to that of wild-type Jurkat T cells (Figure 2A), showing that PAK1 can be activated by the TCR in the absence of Slp-76.

Fig. 2. TCR-mediated PAK1 activation is independent of Slp-76 and Nck. (A) Parental Jurkat cells and the Slp-76-deficient Jurkat subline, J14, were tested for PAK1 kinase activity as in Figure 1 (n = 4). (B) Jurkat T cells were transfected with either 5 µg of pEF-HA-PAK1 (WT), 12 µg pEF-HA-PAK1P13A (P13A) or 5 µg pEFBos (vector) to achieve equal expression levels. Fourteen hours later, the cells were stimulated for 3 min with C305 (+) or a buffer control (–) at 37°C. Anti-Nck immunoprecipitates were analyzed by 7.5% SDS–PAGE and blotted with an anti-HA antibody (top panel) or an anti-Nck antibody (middle panel). Whole cell lysates were blotted with an anti-HA antibody (bottom panel) showing equivalent expression of HA-PAK1. The experiment shown is representative of two independent experiments. (C) As in (B), but anti-HA immunoprecipitates were tested for kinase activity (n = 4). The increased mean fold activation and large range of PAK1 P13A was due to a single experiment out of four in which PAK1 P13A was uncharacteristically activated 3.5-fold above wild-type PAK1.
Previously, a complex of Nck/Slp-76/Vav1 was proposed to be necessary for PAK1 activation by the TCR (Bubeck Wardenburg et al., 1998). Although Slp-76-independent PAK1 activation was inconsistent with such a model, we asked whether Nck might still be required for PAK1 activation. We generated a mutant of PAK1 (P13A) described previously that is unable to interact with Nck in vitro and in fibroblasts (Bokoch et al., 1996). We first confirmed that in Jurkat cells, P13A PAK1 failed to associate inducibly with Nck. Either hemagglutinin (HA)-tagged wild-type PAK1 or HA-tagged P13A PAK1 was transfected into Jurkat cells. Nck was immunoprecipitated with an anti-Nck antiserum and its association with PAK1 was detected with an anti-HA antibody. Whereas wild-type PAK1 associated inducibly with Nck as previously reported (Yablonski et al., 1998a), the mutant P13A PAK1 failed to do so (Figure 2B, lanes 3 and 4). The inducible interaction of the polyproline stretch in PAK1 and the second SH3 domain of Nck is likely to be regulated by conformational changes in one or both proteins. The kinase activity of HA-tagged wild-type or P13A PAK1 was then measured by an in vitro kinase assay of anti-HA immune complexes. In spite of its failure to bind Nck, P13A PAK1 kinase activity was induced by TCR stimulation to a level comparable to that of wild-type PAK1 (Figure 2C). These data, taken together, demonstrate that PAK1 activation requires neither Slp-76 nor direct binding to Nck.
Rac1 activation by the TCR requires LAT and ZAP-70 but not Slp-76
Since we found that PAK1 activation does not require LAT or Slp-76, we asked whether the activation of Rac1, a potential activator of PAK1, had similar requirements. Endogenous, GTP-bound Rac1 was specifically precipitated using glutathione S-transferase (GST) fused to the G-protein binding domain (GBD) of PAK1 (Manser et al., 1998) and then detected with a Rac1-specific antibody (Figure 3A, top panels). TCR stimulation alone was sufficient to activate Rac1 in Jurkat cells as described previously (Kuhne et al., 2000). A specific increase in the amount of GTP-bound Rac1 was detected after TCR stimulation in the Slp-76-deficient J14 cell line but not the LAT-deficient J.CaM2 or ZAP-70-deficient P116 cell lines. Although J14 cells did not activate Rac1 as robustly as Jurkat cells, the J14 cell line, generated by random mutagenesis, is likely to have additional deficiencies that could explain this partial defect. Indeed, in multiple experiments, the reconstitution of the J14 cell line with Slp-76 (J14 76-11) did not markedly enhance the activation of Rac1, demonstrating that the presence of Slp-76 does not have an effect on Rac1 activation. However, reconstitution of the J.CaM2 cell line with a wild-type LAT cDNA (J.CaM2 LAT3) restored Rac1 activation. Two independently reconstituted J.CaM2 cells showed similar results (data not shown). Reconstitution of P116 cells with a wild-type ZAP-70 cDNA (P116 C39) also restored Rac1 activation by the TCR. Similar trends in the mutant cell lines were observed at later time points as Rac1 activation declined (data not shown). These data show that Rac1 activation can occur in the absence of Slp-76 but not in the absence of ZAP-70 or LAT.
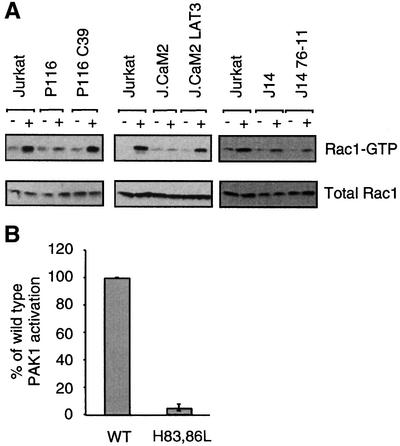
Fig. 3. Rac1 activation by the TCR requires ZAP-70 and LAT, but not Slp-76. (A) The cells indicated were stimulated for 2 min with C305 (+) or a buffer control (–) and then lysed. Rac1-GTP was specifically precipitated by incubation with GST–PAK1–CRIB domain for 15 min followed by a rapid wash with lysis buffer. Bound Rac1 was detected by SDS–PAGE and a Rac1-specific antibody (top panel) and compared with total Rac1 (bottom panel). These results are representative of at least three independent experiments. The cells tested were: the ZAP-70-deficient P116 cell, or the ZAP-70 reconstituted P116 C39, the LAT-deficient J.CaM2 cell, a LAT reconstituted J.CaM2 LAT3, the Slp-76-deficient J14 cell and a Slp-76 reconstituted J14 76-11. (B) Jurkat T cells were transfected with either 5 µg of pEF-HA-PAK1 (WT) or 5 µg of pEF-HA-PAK1H83L,H86L (H83,86L) to achieve equal expression. Kinase activity of the HA-tagged PAK1 was measured as in Figure 2C (n = 3).
Because PAK1 was activated in the absence of Rac1 activation in the LAT-deficient J.CaM2 cell, we asked whether Rho family GTPase binding is dispensable for PAK1 activation in response to TCR stimulation. To do so, we generated the H83,86L mutant of PAK1 described previously, which does not bind to Rac1 or Cdc42 (Manser et al., 1997). We transfected the wild type or the H83,86L mutant of HA-epitope-tagged PAK1 into Jurkat cells and assayed their catalytic activation following TCR stimulation. In contrast to the inducible activation of the P13A mutant PAK1, the H83,86L PAK1 was not activated by TCR stimulation (Figure 3B). These data demonstrate that an intact GTPase binding site is required for PAK1 activation in response to TCR stimulation.
Association with PIX is required for PAK1 activation
Previous studies from our laboratory and others had suggested that PAK1 might function downstream of the GEF Vav1, which was thought to require interaction with Slp-76 and LAT for function (Bubeck Wardenburg et al., 1998; Yablonski et al., 1998a; van Leeuwen and Samelson, 1999). In view of the LAT and Slp-76 independence of PAK1 activation, we asked whether another GEF, PIX, is important for PAK1 activation by the TCR. First, we confirmed that PIX could interact with the N-terminus of PAK1 in T cells, as has been described in fibroblasts (Manser et al., 1998). Immunoprecipitates from Jurkat cells of transfected wild-type PAK1 contained multiple isoforms of α and β PIX as detected by an anti-PIX antibody (Figure 4A). Similar results were seen with the immunoprecipitation of endogenous PAK1 (Figure 6B). The interaction between PAK1 and PIX was not enhanced by TCR stimulation although a portion of the PAK1-associated αPIX shifted to a lower-mobility form after stimulation. This mobility shift might indicate phosphorylation or other post-translational modification. Although there appears to be a reduction in PAK1-associated αPIX after TCR stimulation, this was not reproducible in multiple experiments. The interaction between PAK1 and PIX is mediated by the SH3 domain of PIX and a polyproline region in the N-terminus of PAK1. In agreement with previous results (Manser et al., 1998), mutation of residues P192 and R193 in PAK1 to glycine and alanine, respectively, abrogated the co-immunoprecipitation of the multiple PIX isoforms with PAK1 in Jurkat cells (Figure 4A). We then tested the ability of the TCR to activate this P192G,R193A PAK1. As shown in Figure 4B, this allele of PAK1 was poorly activated by TCR stimulation in Jurkat cells.

Fig. 4. PIX binding is required for PAK1 activation. (A) Jurkat T cells were transfected with either 5 µg of pEF-HA-PAK1 (WT) or 3 µg of pEF-HA-PAK1P192G,R193A (P192G, R193A) to achieve equal expression. The cells indicated were stimulated for 2 min with C305 (+) or a buffer control (–) and then lysed. Anti-HA immunoprecipitates were analyzed for PIX content by 7.5% SDS–PAGE and western blotting with an anti-PIX antibody (top panel) or an anti-HA antibody (bottom panel). Results are representative of three independent experiments. (B) PAK1 kinase assays were performed on anti-HA immunoprecipitates prepared as in (A) (n = 3).
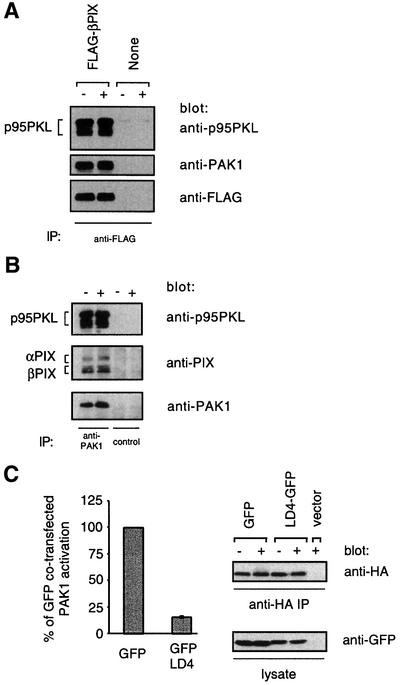
Fig. 6. p95PKL–PIX–PAK1 is required for efficient PAK1 activation by the TCR. (A) Either Jurkat cells stably expressing wild-type FLAG-βPIX or untransfected Jurkat cells were stimulated for 2 min with C305 (+) or a buffer control (–) and then lysed. Anti-FLAG immunoprecipitates were analyzed by 7.5% SDS–PAGE and blotting with an anti-p95PKL antibody (top panel) or an anti-PAK1 antibody (middle panel). The blot was then stripped and reprobed with an anti-FLAG antibody (bottom panel). These data are representative of two independent experiments. (B) Jurkat cells were stimulated for 2 min with C305 (+) or a buffer control (–) and then lysed. Endogenous PAK1 was immunoprecipitated from Jurkat cells with a C-terminally directed anti-PAK1 antibody (lanes 1 and 2) or the same antibody pre-incubated with a PAK1-blocking peptide (lanes 3 and 4). The immunoprecipitates were analyzed by 7.5% SDS–PAGE and western blotting with an anti-p95PKL antibody (top panel) and anti-PAK1 antibody (bottom panel). The top panel was stripped and reprobed with an anti-PIX antibody (middle panel), which explains the relatively weaker PIX signal. (C) Jurkat T cells were co-transfected with 15 µg of the GFP parental vector and 5 µg of pEF-HA-PAK1 or with 15 µg of LD4–GFP and 20 µg of pEF-HA-PAK1 to equalize expression of PAK1. Six hours after transfection, the cells were stimulated for 2 min with C305 or a buffer control at 37°C and PAK1 kinase activity was measured (left panel, n = 3). Equivalent immunoprecipitation of HA-PAK1 was confirmed by an anti-HA western blotting (right top panel) and expression of GFP and LD4–GFP was confirmed by an anti-GFP western blot from whole cell lysate (right bottom panel).
In order to confirm the requirement for PIX binding for PAK1 activation by the TCR, we stably transfected epitope-tagged wild-type (WT), Dbl homology mutant (DH*) or SH3 mutant (SH3*) βPIX into Jurkat cells. Several stable clones expressing each βPIX allele were isolated and screened for approximately equivalent overexpression of βPIX. Stable clones expressed at least 5-fold more βPIX than non-transfected parental cells as detected by an anti-PIX antibody that recognizes the SH3 domain of PIX (Figure 5A, top panel). The disrupted SH3 domain of SH3* βPIX is not detected by the anti-PIX antibody and equivalent expression was confirmed using the FLAG epitope tag (Figure 5A, bottom panel). Endogenous PAK1 activation was measured from these βPIX-overexpressing clones. While clones overexpressing WT βPIX activated PAK1 as well as the Jurkat parental line, clones expressing DH* or SH3* mutant βPIX were significantly impaired in their ability to activate PAK1 (Figure 5B). In contrast to the impaired PAK1 activation, the overall pattern of tyrosine phosphorylation following TCR stimulation in all clones was similar (data not shown). Taken together, these data show that the PAK1–PIX interaction is critical for TCR-mediated PAK1 activation.
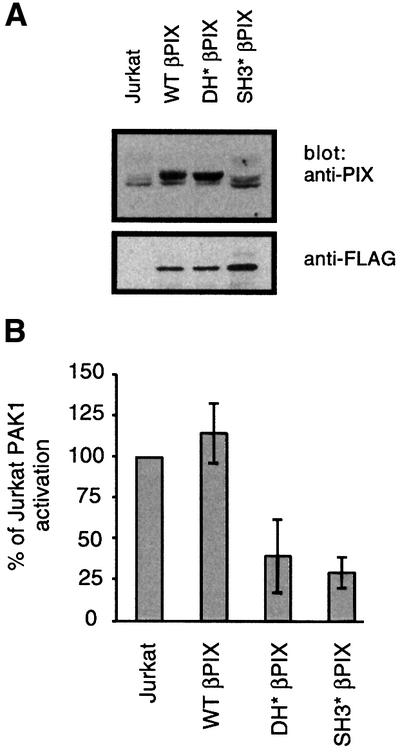
Fig. 5. Overexpression of βPIX mutants blocks PAK1 activation. (A) Anti-PIX immunoblot of 1 × 106 cells from parental Jurkat, representative clones overexpressing either wild-type βPIX (WT βPIX), L238R, L239S βPIX (DH* βPIX) or W34P, W44G βPIX (SH3* βPIX). (B) Endogenous PAK1 kinase assays performed as in Figure 1B on representative clones expressing the indicated βPIX allele (n = 3). Results are representative of least three independent clones expressing each PIX allele (data not shown).
A PKL–PIX–PAK1 complex is important for PAK1 activation by the TCR
The ability of the SH3* mutant of βPIX to block PAK1 activation suggested that a βPIX–PAK1 complex might be recruited to upstream components for PAK1 activation. Thus, the overexpressed SH3* mutant βPIX may compete with wild-type PIX for binding to these upstream components, thereby preventing endogenous PAK1 from being recruited and activated. One possible candidate that might link PIX to the TCR was the recently cloned p95PKL, which interacts with PIX and paxillin (Turner et al., 1999). Indeed, anti-FLAG immunoprecipitates from Jurkat cells stably expressing FLAG-tagged wild-type βPIX contained multiple isoforms of p95PKL as identified by western blotting with an anti-p95PKL antibody (Figure 6A). Like the interaction between PAK1 and PIX, the association between PIX and p95PKL was not induced by TCR stimulation. As expected, endogenous PAK1 was also present in these anti-PIX immune complexes. Importantly, anti-PAK1 immunoprecipitates from untransfected Jurkat cells contained both p95PKL and PIX (Figure 6B). These data taken together demonstrate that p95PKL, PIX and PAK1 form a trimolecular complex constitutively in vivo.
In a previous study, p95PKL was shown to link PIX to the integrin-associated adaptor paxillin by binding directly to both proteins (Turner et al., 1999). Having demonstrated that p95PKL interacts with βPIX and PAK1 in T cells, we asked whether p95PKL was functionally important for PAK1 activation. To interfere with p95PKL function, we fused 20 amino acids spanning the LD4 region of paxillin to the N-terminus of green fluorescent protein (GFP), analogous to an approach used in a previous study (Turner et al., 1999). By binding directly to the paxillin binding site in p95PKL, the LD4–GFP competitively inhibits interactions between p95PKL and paxillin. LD4–GFP or GFP alone was co-transfected into Jurkat T cells with HA-tagged wild-type PAK1. The kinase activity of the HA-tagged PAK1 was measured with an in vitro kinase assay on anti-HA immune complexes. Relative to the expression of GFP alone, expression of LD4–GFP potently inhibited PAK1 activation induced by TCR stimulation (Figure 6C). These data suggest that p95PKL may be important in PAK1 activation by the TCR and may function by recruiting PIX–PAK1 to upstream components of the PAK1 activation pathway.
Discussion
We have further elucidated the TCR signaling pathways that activate PAK1 and Rac1. A previous study implicated Vav1, Nck and Slp-76 in TCR-mediated PAK1 and Rac1 activation. In this model, Slp-76 served as a bridge between an Nck–PAK1 complex and Vav1. Mutant forms of these molecules were demonstrated to block PAK1 activation by the TCR (Bubeck Wardenburg et al., 1998). We have taken advantage of the known P13A mutation in PAK1 and the Slp-76-deficient Jurkat cell to test this model without overexpressing dominant-negative proteins. Surprisingly, we show that Nck binding to PAK1 is dispensable for PAK1 activation by the TCR. Moreover, PAK1 is activated in a Slp-76-deficient Jurkat cell. While a Nck–Slp-76–Vav1 complex may play some role in the activation of PAK1, our data suggest that this complex is not necessary for PAK1 activation following TCR stimulation.
In order to characterize this Slp-76/Nck-independent pathway, we measured the activation of PAK1 in the LAT-deficient J.CaM2 cell. Surprisingly, we observed reproducible activation of endogenous PAK1 in the absence of LAT. The activation of PAK1 in J.CaM2 contrasts with the almost complete absence of Erk1/2 activation and calcium flux in these cells (Finco et al., 1998). These results are consistent with our previous data that PAK1 activation is independent of Ras (Yablonski et al., 1998a). Taken together, they demonstrate a fundamental difference between the requirements for PAK1 activation and the requirements for the activation of the Ras and phosphatidylinositol pathways. Furthermore, we believe that these are among the first descriptions of a LAT- and Slp-76-independent pathway. Our finding that Lck and ZAP-70 are absolutely required for PAK1 activation demonstrates that this novel LAT- and Slp-76-independent pathway diverges at the level of the Src and Syk kinases (Figure 7).To confirm that PAK1 is activated by a LAT- and Slp-76-independent pathway, we asked whether Rac1, a Rho GTPase that activates PAK1 in many systems, is activated by a similar mechanism. Like PAK1, Rac1 activation by the TCR did not require Slp-76, but did require ZAP-70. However, unlike PAK1, Rac1 was absolutely dependent on LAT for its activation by the TCR. The surprising Slp-76 independence but LAT dependence of Rac1 activation shows that Rac1 is activated by a mechanism that is distinct from both the PAK1 pathway and Ras/phosphatidylinositol pathways. Consistent with this conclusion, we have observed that doses of phorbol 12-myristate 13-acetate and ionomycin that strongly activate the Ras pathway and the NFAT transcriptional element are poor activators of Rac1 (data not shown). One possible caveat to these studies is the constitutive activation of the PI-3 kinase pathway in Jurkat cells caused by deficiency of PTEN, the tumor suppressor lipid and protein phosphatase (Shan et al., 2000). This constitutive activation may bypass requirements for PAK1 and Rac1 activation.
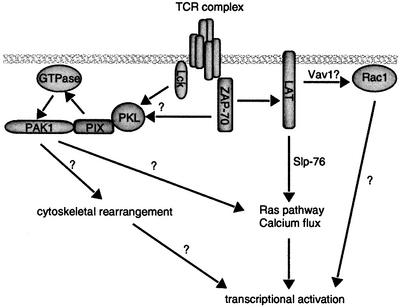
Fig. 7. Proposed model for PAK1 and Rac1 activation by the TCR. PAK1 activation requires Lck and ZAP-70, but occurs independently of LAT and Slp-76. Rac1 activation requires ZAP-70 and LAT, but is independent of Slp-76. These contrast the requirement of ZAP-70, LAT and Slp-76 for Ras and phosphatidylinositol pathway activation. Each of these divergent pathways is required to activate the transcriptional responses of T-cell activation. PAK1 activity most likely contributes to cytoskeletal reorganization as well. Undoubtedly, cytoskeletal rearrangements require contributions from many other components besides PAK1, but these have been omitted for clarity.
Previous studies had implicated Vav1 as a GEF upstream of PAK1 by binding a complex of Nck/Slp-76/Gads/LAT (Bubeck Wardenburg et al., 1998; van Leeuwen and Samelson, 1999). Our observation that PAK1 activation was independent of LAT, Slp-76 and Nck suggests that Vav1 may not play an important role in PAK1 activation by the TCR. In the light of this surprising result, we asked whether another GEF, PIX, is important in TCR-mediated PAK1 activation. We first showed that PAK1 constitutively interacts with multiple isoforms of PIX in Jurkat cells. Furthermore, a mutant PAK1 that cannot bind PIX (P192G,R193A) is significantly impaired in its activation by the TCR. These data suggest two simple models of PIX function: (i) PIX might be important for the recruitment of PAK1 to the membrane for activation or (ii) PIX might be important as a GEF to allow GTPase-mediated PAK1 activation.
We used the overexpression of mutants of βPIX to address these possibilities. Overexpression of a βPIX mutant that does not have GEF function (DH*) inhibited PAK1 activation by the TCR. This is consistent with the second model, which suggested that βPIX is required for the localized activation of a Rho GTPase, which then activates PAK1 (Figure 7). Indeed, this model is also consistent with our finding that PAK1 requires Rho GTPase binding for activation by TCR stimulation. However, this GTPase is probably not Rac1 in view of Rac1’s dependence on LAT for activation. In support of this hypothesis, Rac1 activation was not affected by the overexpression of wild-type, DH* or SH3* βPIX (data not shown). Cdc42 is an excellent candidate GTPase for PAK1 activation since we have shown previously that a dominant-negative allele of Cdc42 inhibits PAK1 activation by the TCR (Yablonski et al., 1998a). However, we have been unable to reproducibly observe endogenous Cdc42 activation in Jurkat cells.
That the SH3* βPIX could block PAK1 activation is consistent with our first model, which suggested that PIX is responsible for recruiting PAK1 to appropriate sites for activation by binding to additional upstream components of this cascade. One candidate for such an upstream component is p95PKL, recently identified in fibroblasts as a 95 kDa, tyrosine-phosphorylated protein that can interact with PIX and the integrin-associated adaptor paxillin (Bagrodia et al., 1999; Turner et al., 1999). In Jurkat cells, we showed that p95PKL, PIX and PAK1 form a trimolecular complex in vivo. We asked whether p95PKL is important for TCR-mediated PAK1 activation in T cells by overexpressing the paxillin LD4 repeat, which binds to p95PKL, as a GFP fusion protein. Indeed, compared with overexpression of GFP alone, LD4–GFP potently blocked PAK1 activation by the TCR. These data, to our knowledge, are the first demonstration of a role for p95PKL and PIX in a receptor-mediated activation of PAK1.
While the platelet-derived growth factor and EphB2 receptor tyrosine kinases appear to utilize PI-3 kinase and PIX to activate PAK1 (Yoshii et al., 1999), the mechanism by which the TCR and its associated tyrosine kinases, Lck and ZAP-70, activates p95PKL/PIX/PAK1 is unclear. The interactions between PKL, PIX and PAK1 described here appear to be unaffected by TCR stimulation and we have not observed obvious tyrosine phosphorylation of any of the three proteins. Based on work in fibroblasts, we might predict a complex containing paxillin/p95PKL/PIX/PAK1 in T cells. However, we have been unable to detect paxillin in complex with p95PKL or PIX and vice versa in T cells (data not shown). Nonetheless, interactions between a paxillin superfamily member and p95PKL are intriguing in view of reported observations that paxillin and Lck inducibly associate in T cells (Ostergaard et al., 1998). This inducible interaction could bring a paxillin–p95PKL–PIX–PAK1 complex to the TCR complex where PAK1 could be activated. Another possible mechanism for p95PKL–PIX–PAK1 recruitment to the TCR complex may be its association with FAK as suggested by a GIT1–FAK interaction detected in fibroblasts (Zhao et al., 2000). Since others have reported that FAK or the FAK-like Pyk2 can interact with Lck (Berg and Ostergaard, 1997), Fyn (Ganju et al., 1997; Qian et al., 1997) and ZAP-70 (Katagiri et al., 2000), this interaction might also link the p95PKL–PIX–PAK1 complex to the TCR. These possibilities are currently being investigated.
The relevant downstream targets of PAK1 in TCR signaling are not yet known. We have shown previously that a dominant-negative allele of PAK1 blocks TCR-mediated activation of NFAT. Surprisingly, this block could not be overcome by either pharmacological activation of the Ras pathway or calcium mobilization alone, suggesting that PAK1 acts upstream of Ras pathway activation and calcium flux (Yablonski et al., 1998a). One possible mechanism for these observations may be PAK1’s activation of Raf1 (Chaudhary et al., 2000; Sun et al., 2000), although this alone does not explain PAK1’s effect on the calcium-dependent pathway. Another possible mechanism may be PAK1’s effects on the cytoskeleton (reviewed by Bagrodia and Cerione, 1999). The regulation of actin polymerization specifically may explain PAK1’s influence on NFAT activation since cytochalasin D, an actin depolymerizing agent, can block TCR-induced NFAT and IL-2 reporter activation (Holsinger et al., 1998). However, it should be emphasized that many molecular events besides PAK1 activation may be required to orchestrate the cytoskeletal rearrangements of T-cell activation.
The role of Vav1 in PAK1 and Rac1 activation remains unclear. The data presented here do not exclude a role for Vav1 in Rac1 or PAK1 activation by the TCR. However, our findings do contradict the simple model that Rac1 and PAK1 are activated by a Vav1–Slp-76–Gads–LAT complex. Consistent with this, others have shown that the Slp-76–Vav1 interaction is not required for their synergistic activation of the IL-2 promoter and that Slp-76 and Vav1 can function in distinct pathways (Raab et al., 1997; Fang and Koretzky, 1999). Nevertheless, Vav1 may yet be responsible for the activation of Rac1 downstream of LAT but independent of Slp-76. Although Vav1 is hypophosphorylated in the LAT-deficient J.CaM2 cell line (Finco et al., 1998), recent studies demonstrate that both negative and positive regulatory sites of tyrosine phosphorylation appear to be present in Vav1 (Kuhne et al., 2000; Lopez-Lago et al., 2000). These recent observations and our data showing Slp-76-independent Rac1 activation suggest that the link between LAT, Vav1 and Rac1 requires additional investigation.
In this study, we have defined requirements for the activation of PAK1 by the TCR. Surprisingly, PAK1 activation does not require the adaptors LAT or Slp-76. For optimal activation by the TCR, PAK1 requires its binding site for Rho GTPases and its binding site for a PIX–p95PKL complex. These data imply that molecules originally identified as focal adhesion signaling molecules are important for TCR signaling. This overlap between elements of focal adhesions and elements of the TCR complex as well as recent descriptions of the highly ordered and dynamic T cell–APC interface (van der Merwe et al., 2000) suggest that the TCR organizes a complex structure that resembles, in part, a focal adhesion at the T cell–APC interface. We propose that the relatively upstream activation of the PAK1 pathway helps orchestrate the cytoskeletal rearrangements that assist in the formation and maintenance of this dynamic signaling machine.
Materials and methods
Reagents
The cell lines used were the Jurkat T cell line J.E6-1 (Weiss et al., 1984), the Slp-76-deficient Jurkat subline J14, the Slp-76 reconstituted J14 76-11 (Yablonski et al., 1998b), the LAT-deficient Jurkat subline J.CaM2 (Goldsmith et al., 1988), the LAT reconstituted J.CaM2 LAT3 and J.CaM2 LAT9 cell lines (Finco et al., 1998), the ZAP-70-deficient P116 Jurkat subline and the ZAP-70 reconstituted P116 C39 (Williams et al., 1998). Cells were grown in RPMI-1640 with 5% fetal calf serum (FCS) supplemented with penicillin, streptomycin and glutamine at 37°C in humidified 5% CO2. TCR stimulations were performed with the anti-Jurkat TCR Vβ C305 mAb (Weiss and Stobo, 1984). Prior to any stimulation, CD3 surface expression was confirmed by fluorescence-activated cell sorter (FACS) analysis. The M2 anti-FLAG antibody was obtained from Sigma. The anti-PAK1 antibodies, sc882 and sc881, were obtained from Santa Cruz Biotechnology. The 12CA5 anti-HA antibody was obtained from Boehringer Mannheim. The polyclonal anti-Nck antibody was provided by Joseph Schlessinger (New York University). The anti-Rac1 antibody and the anti-p95PKL antibody were both obtained from Transduction Laboratories. The anti-GFP antibody was obtained from Clontech. The anti-PIX antibody was described previously (Manser et al., 1998).
Transfections
Jurkat T cells (2 × 107) in 0.4 ml of serum-free RPMI-1640 were transiently transfected by electroporation using the Gene Pulser (Bio-Rad) at 250 V and 960 µF. Following transfection, cells were incubated in RPMI containing 10% FCS. Stable transfectants were isolated by performing the above protocol, but 48 h after transfection, cells were plated under limiting dilution conditions in 2 mg/ml G418 (Calbiochem). Approximately 4 weeks later, clonal cell populations were isolated, screened for equivalent surface expression of CD3 by FACS analysis and equivalent expression of the FLAG epitope-tagged βPIX by western blotting.
Constructs
PAK1 was mutated by overlapping PCR to generate a proline to alanine change at codon 13, histidine to leucine change at codons 83 and 86, a proline to glycine change at codon 192 and an arginine to alanine change at 193. Briefly, sense and antisense oligos were synthesized that contained the desired mutation and flanking sequence. PCR was performed with an oligo in the polylinker of pcDNA hPAK1 and an antisense oligo containing the desired mutation. A second PCR was performed with the sense oligo containing the mutation and an oligo 3′ to the ClaI site internal to the hPAK1 cDNA. These overlapping pieces were assembled with a final PCR to generate the mutated PAK1 N-terminus and then placed into the full-length pEF-hPAK1 vector (Yablonski et al., 1998a) using the polylinker Asp718 site and the internal ClaI site. All PAK1-containing cDNAs were grown at 30°C to minimize spontaneous mutation of the kinase. All mutants were transfected into 293 HEK cells and PAK1 kinase assays were performed to confirm kinase activity. The LD4–GFP construct was constructed by ligation of annealed oligos encoding the human paxillin LD4 sequence (underlined) MATRELDELMASLSDFKFMAQGGG preceded by a 5′ Kozak ACC sequence into the pEGFP-N1 vector (Clontech) between the EcoRI and PinAI sites. Bacterially expressed GST–CRIB was prepared as described previously (Manser et al., 1998). Wild-type βPIX, DH domain mutant βPIX (L238R, L239S), SH3 domain mutant βPIX (W34P, W44G) (Manser et al., 1998) were inserted into the pcdef3 vector for efficient expression in Jurkat T cells. All constructs were confirmed by sequencing.
Cell stimulation, immunoprecipitation and western blotting
Cells were harvested and washed in phosphate-buffered saline (PBS) and resuspended at 108 cells/ml of PBS and incubated at 37°C for 10 min. Cells were stimulated with a 1:500 dilution of C305 for 2–3 min followed by rapid centrifugation and resuspension of the pellet by vigorous vortexing in a lysis buffer appropriate for the experiment. For immunoprecipitations, the lysis buffer was identical to that used for the PAK1 kinase assays. After clarification of the lysate by centrifugation in a microfuge at 13 000 r.p.m. for 10 min at 4°C, the supernatant was spun at 55K for 15 min in a tabletop ultracentrifuge. The supernatant was tumbled with 30 µl of protein A-expressing pansorbin bacteria (Calbiochem) for 20 min at 4°C. The supernatant was then tumbled with the relevant antibody prebound to the relevant beads for 1.5 h at 4°C. The beads were washed four times with lysis buffer and resuspended in reducing sample buffer for SDS–PAGE. Gels were transferred to Immobilon P (Millipore Corporation) and probed with primary antibodies as described in the text and the appropriate secondary antibodies coupled to horseradish peroxidase. Detection was performed with enhanced chemiluminescence (Amersham).
Rac1 activation assay
Specific isolation of Rac1-GTP was performed as described previously (Manser et al., 1998; Kuhne et al., 2000). Briefly, cells were stimulated as described above and lysed in lysis buffer containing 0.5% Triton X-100, 25 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM EGTA, 20 mM β-glycerophosphate, 4% glycerol, 10 mM NaF, 2 mM sodium orthovanadate, 5 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 5 µg/ml leupeptin, 5 µg/ml pepstatin. After 10 min at 4°C, the lysates were clarified in a microfuge for 10 min at 13 000 r.p.m. The supernatant was incubated with 20 µg of GST–CRIB for 15 min at 4°C with rotation. The pellets were washed once with 500 µl of lysis buffer and resuspended in 40 µl of sample buffer. GST–CRIB-associated Rac1 was identified by SDS–PAGE and western blotting with a Rac1-specific antiserum.
PAK1 kinase assay
PAK1 kinase assays were performed as described in Yablonski et al. (1998a). In all experiments equal amounts of immunoprecipitated kinase were confirmed by western blotting (data not shown). Quantitation of histone H4 phosphorylation was performed by Fuji Multimager analysis of 32P-labeled histone H4 identified after SDS–PAGE or by Cherenkov counting of 40 µl of the 50 µl kinase reaction spotted onto Whatman 3 mm paper after extensive washing in buffer containing 10% trichloroacetic acid and 10 mM sodium pyrophosphate. Background measurements were taken either by immunoprecipitation in the presence of the sc882 blocking peptide or by anti-HA immunoprecipitations on untransfected cells. Typical activation of PAK1, either endogenous or transfected, in Jurkat was 6- to 8-fold. To allow averaging of kinase assays performed on separate days, fold activation for each cell line or PAK1 allele was normalized to the activation of wild-type PAK1 measured in the same experiment. This percentage was averaged over n experiments as reported in the figure legend. Error bars show the standard error of the mean when n > 2 or the range when n = 2. In all experiments, the background PAK1 activity was similar.
Acknowledgments
Acknowledgements
We thank all members of the Weiss laboratory for useful discussion and L.Kane and M.Tomlinson for careful reading of the manuscript. This work was supported in part by National Institutes of Health grant CA72531. G.M.K. is supported by a National Institutes of Health medical scientist training grant. A.W. is an Investigator of the Howard Hughes Medical Institute.
References
- Asada H. et al. (1999) Grf40, a novel Grb2 family member, is involved in T cell signaling through interaction with SLP-76 and LAT. J. Exp. Med., 189, 1383–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagrodia S. and Cerione,R.A. (1999) Pak to the future. Trends Cell Biol., 9, 350–355. [DOI] [PubMed] [Google Scholar]
- Bagrodia S., Taylor,S.J., Jordon,K.A., Van Aelst,L. and Cerione,R.A. (1998) A novel regulator of p21-activated kinases. J. Biol. Chem., 273, 23633–23636. [DOI] [PubMed] [Google Scholar]
- Bagrodia S., Bailey,D., Lenard,Z., Hart,M., Guan,J.L., Premont,R.T., Taylor,S.J. and Cerione,R.A. (1999) A tyrosine-phosphorylated protein that binds to an important regulatory region on the cool family of p21-activated kinase-binding proteins. J. Biol. Chem., 274, 22393–22400. [DOI] [PubMed] [Google Scholar]
- Berg N.N. and Ostergaard,H.L. (1997) T cell receptor engagement induces tyrosine phosphorylation of FAK and Pyk2 and their association with Lck. J. Immunol., 159, 1753–1757. [PubMed] [Google Scholar]
- Bokoch G.M., Wang,Y., Bohl,B.P., Sells,M.A., Quilliam,L.A. and Knaus,U.G. (1996) Interaction of the Nck adapter protein with p21-activated kinase (PAK1). J. Biol. Chem., 271, 25746–25749. [DOI] [PubMed] [Google Scholar]
- Bubeck Wardenburg J., Pappu,R., Bu,J.Y., Mayer,B., Chernoff,J., Straus,D. and Chan,A.C. (1998) Regulation of PAK activation and the T cell cytoskeleton by the linker protein SLP-76. Immunity, 9, 607–616. [DOI] [PubMed] [Google Scholar]
- Chaudhary A., King,W.G., Mattaliano,M.D., Frost,J.A., Diaz,B., Morrison,D.K., Cobb,M.H., Marshall,M.S. and Brugge,J.S. (2000) Phosphatidylinositol 3-kinase regulates raf1 through pak phosphorylation of serine 338. Curr. Biol., 10, 551–554. [DOI] [PubMed] [Google Scholar]
- Costello P.S., Walters,A.E., Mee,P.J., Turner,M., Reynolds,L.F., Prisco,A., Sarner,N., Zamoyska,R. and Tybulewicz,V.L. (1999) The Rho-family GTP exchange factor Vav is a critical transducer of T cell receptor signals to the calcium, ERK and NF-κB pathways. Proc. Natl Acad. Sci. USA, 96, 3035–3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels R.H., Zenke,F.T. and Bokoch,G.M. (1999) αPix stimulates p21-activated kinase activity through exchange factor-dependent and -independent mechanisms [published erratum appears in J. Biol. Chem., 274, 15292]. J. Biol. Chem., 274, 6047–6050. [DOI] [PubMed] [Google Scholar]
- Fang N. and Koretzky,G.A. (1999) SLP-76 and Vav function in separate, but overlapping pathways to augment interleukin-2 promoter activity. J. Biol. Chem., 274, 16206–16212. [DOI] [PubMed] [Google Scholar]
- Finco T.S., Kadlecek,T., Zhang,W., Samelson,L.E. and Weiss,A. (1998) LAT is required for TCR-mediated activation of PLCγ1 and the Ras pathway. Immunity, 9, 617–626. [DOI] [PubMed] [Google Scholar]
- Fischer K.D. et al. (1998) Vav is a regulator of cytoskeletal reorganization mediated by the T-cell receptor. Curr. Biol., 8, 554–562. [DOI] [PubMed] [Google Scholar]
- Ganju R.K., Hatch,W.C., Avraham,H., Ona,M.A., Druker,B., Avraham,S. and Groopman,J.E. (1997) RAFTK, a novel member of the focal adhesion kinase family, is phosphorylated and associates with signaling molecules upon activation of mature T lymphocytes. J. Exp. Med., 185, 1055–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genot E., Cleverley,S., Henning,S. and Cantrell,D. (1996) Multiple p21ras effector pathways regulate nuclear factor of activated T cells. EMBO J., 15, 3923–3933. [PMC free article] [PubMed] [Google Scholar]
- Goldsmith M.A., Dazin,P.F. and Weiss,A. (1988) At least two non-antigen-binding molecules are required for signal transduction by the T-cell antigen receptor. Proc. Natl Acad. Sci. USA, 85, 8613–8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsinger L.J. et al. (1998) Defects in actin-cap formation in Vav-deficient mice implicate an actin requirement for lymphocyte signal transduction. Curr. Biol., 8, 563–572. [DOI] [PubMed] [Google Scholar]
- Jackman J.K., Motto,D.G., Sun,Q., Tanemoto,M., Turck,C.W., Peltz,G.A., Koretzky,G.A. and Findell,P.R. (1995) Molecular cloning of SLP-76, a 76-kDa tyrosine phosphoprotein associated with Grb2 in T cells. J. Biol. Chem., 270, 7029–7032. [DOI] [PubMed] [Google Scholar]
- Kane L.P., Lin,J. and Weiss,A. (2000) Signal transduction by the TCR for antigen. Curr. Opin. Immunol., 12, 242–249. [DOI] [PubMed] [Google Scholar]
- Katagiri T., Takahashi,T., Sasaki,T., Nakamura,S. and Hattori,S. (2000) Protein-tyrosine kinase Pyk2 is involved in interleukin-2 production by Jurkat T cells via its tyrosine 402. J. Biol. Chem., 275, 19645–19652. [DOI] [PubMed] [Google Scholar]
- Kuhne M.R., Ku,G. and Weiss,A. (2000) A guanine nucleotide exchange factor-independent function of Vav1 in transcriptional activation. J. Biol. Chem., 275, 2185–2190. [DOI] [PubMed] [Google Scholar]
- Lin J., Weiss,A. and Finco,T.S. (1999) Localization of LAT in glycolipid-enriched microdomains is required for T cell activation. J. Biol. Chem., 274, 28861–28864. [DOI] [PubMed] [Google Scholar]
- Liu S.K., Fang,N., Koretzky,G.A. and McGlade,C.J. (1999) The hematopoietic-specific adaptor protein gads functions in T-cell signaling via interactions with the SLP-76 and LAT adaptors. Curr. Biol., 9, 67–75. [DOI] [PubMed] [Google Scholar]
- Lopez-Lago M., Lee,H., Cruz,C., Movilla,N. and Bustelo,X.R. (2000) Tyrosine phosphorylation mediates both activation and downmodulation of the biological activity of Vav. Mol. Cell. Biol., 20, 1678–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manser E., Huang,H.Y., Loo,T.H., Chen,X.Q., Dong,J.M., Leung,T. and Lim,L. (1997) Expression of constitutively active α-PAK reveals effects of the kinase on actin and focal complexes. Mol. Cell. Biol., 17, 1129–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manser E., Loo,T.H., Koh,C.G., Zhao,Z.S., Chen,X.Q., Tan,L., Tan,I., Leung,T. and Lim,L. (1998) PAK kinases are directly coupled to the PIX family of nucleotide exchange factors. Mol. Cell, 1, 183–192. [DOI] [PubMed] [Google Scholar]
- Ostergaard H.L., Lou,O., Arendt,C.W. and Berg,N.N. (1998) Paxillin phosphorylation and association with Lck and Pyk2 in anti-CD3- or anti-CD45-stimulated T cells. J. Biol. Chem., 273, 5692–5696. [DOI] [PubMed] [Google Scholar]
- Qian D., Lev,S., van Oers,N.S., Dikic,I., Schlessinger,J. and Weiss,A. (1997) Tyrosine phosphorylation of Pyk2 is selectively regulated by Fyn during TCR signaling. J. Exp. Med., 185, 1253–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab M., da Silva,A.J., Findell,P.R. and Rudd,C.E. (1997) Regulation of Vav-SLP-76 binding by ZAP-70 and its relevance to TCR ζ/CD3 induction of interleukin-2. Immunity, 6, 155–164. [DOI] [PubMed] [Google Scholar]
- Sanzenbacher R., Kabelitz,D. and Janssen,O. (1999) SLP-76 binding to p56lck: a role for SLP-76 in CD4-induced desensitization of the TCR/CD3 signaling complex. J. Immunol., 163, 3143–3152. [PubMed] [Google Scholar]
- Shan X., Czar,M.J., Bunnell,S.C., Liu,P., Liu,Y., Schwartzberg,P.L. and Wange,R.L. (2000) Deficiency of PTEN in Jurkat T cells causes constitutive localization of Itk to the plasma membrane and hyperresponsiveness to CD3 stimulation. Mol. Cell. Biol., 20, 6945–6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowers L., Yelon,D., Berg,L.J. and Chant,J. (1995) Regulation of the polarization of T cells toward antigen-presenting cells by Ras-related GTPase CDC42. Proc. Natl Acad. Sci. USA, 92, 5027–5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H., King,A.J., Diaz,H.B. and Marshall,M.S. (2000) Regulation of the protein kinase Raf-1 by oncogenic Ras through phosphatidylinositol 3-kinase, Cdc42/Rac and Pak. Curr. Biol., 10, 281–284. [DOI] [PubMed] [Google Scholar]
- Turner C.E., Brown,M.C., Perrotta,J.A., Riedy,M.C., Nikolopoulos,S.N., McDonald,A.R., Bagrodia,S., Thomas,S. and Leventhal,P.S. (1999) Paxillin LD4 motif binds PAK and PIX through a novel 95-kD ankyrin repeat, ARF-GAP protein: A role in cytoskeletal remodeling. J. Cell Biol., 145, 851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner M., Mee,P.J., Walters,A.E., Quinn,M.E., Mellor,A.L., Zamoyska,R. and Tybulewicz,V.L. (1997) A requirement for the Rho-family GTP exchange factor Vav in positive and negative selection of thymocytes. Immunity, 7, 451–460. [DOI] [PubMed] [Google Scholar]
- van der Merwe P.A., Davis,S.J., Shaw,A.S. and Dustin,M.L. (2000) Cytoskeletal polarization and redistribution of cell-surface molecules during T cell antigen recognition. Semin. Immunol., 12, 5–21. [DOI] [PubMed] [Google Scholar]
- van Leeuwen J.E. and Samelson,L.E. (1999) T cell antigen-receptor signal transduction. Curr. Opin. Immunol., 11, 242–248. [DOI] [PubMed] [Google Scholar]
- Weiss A. and Stobo,J.D. (1984) Requirement for the coexpression of T3 and the T cell antigen receptor on a malignant human T cell line. J. Exp. Med., 160, 1284–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss A., Wiskocil,R.L. and Stobo,J.D. (1984) The role of T3 surface molecules in the activation of human T cells: a two-stimulus requirement for IL 2 production reflects events occurring at a pre-translational level. J. Immunol., 133, 123–128. [PubMed] [Google Scholar]
- Williams B.L., Schreiber,K.L., Zhang,W., Wange,R.L., Samelson,L.E., Leibson,P.J. and Abraham,R.T. (1998) Genetic evidence for differential coupling of Syk family kinases to the T-cell receptor: reconstitution studies in a ZAP-70-deficient Jurkat T-cell line. Mol. Cell. Biol., 18, 1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Motto,D.G., Koretzky,G.A. and Weiss,A. (1996) Vav and SLP-76 interact and functionally cooperate in IL-2 gene activation. Immunity, 4, 593–602. [DOI] [PubMed] [Google Scholar]
- Yablonski D., Kane,L.P., Qian,D. and Weiss,A. (1998a) A Nck-Pak1 signaling module is required for T-cell receptor-mediated activation of NFAT, but not of JNK. EMBO J., 17, 5647–5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yablonski D., Kuhne,M.R., Kadlecek,T. and Weiss,A. (1998b) Uncoupling of nonreceptor tyrosine kinases from PLC-γ1 in an SLP-76-deficient T cell. Science, 281, 413–416. [DOI] [PubMed] [Google Scholar]
- Yoshii S. et al. (1999) αPIX nucleotide exchange factor is activated by interaction with phosphatidylinositol 3-kinase. Oncogene, 18, 5680–5690. [DOI] [PubMed] [Google Scholar]
- Zhang W., Sloan-Lancaster,J., Kitchen,J., Trible,R.P. and Samelson,L.E. (1998a) LAT: the ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell, 92, 83–92. [DOI] [PubMed] [Google Scholar]
- Zhang W., Trible,R.P. and Samelson,L.E. (1998b) LAT palmitoylation: its essential role in membrane microdomain targeting and tyrosine phosphorylation during T cell activation. Immunity, 9, 239–246. [DOI] [PubMed] [Google Scholar]
- Zhang W., Irvin,B.J., Trible,R.P., Abraham,R.T. and Samelson,L.E. (1999) Functional analysis of LAT in TCR-mediated signaling pathways using a LAT-deficient Jurkat cell line. Int. Immunol., 11, 943–950. [DOI] [PubMed] [Google Scholar]
- Zhao Z., Manser,E., Loo,T.H. and Lim,L. (2000) Coupling of PAK-interacting exchange factor PIX to GIT1 promotes focal complex disassembly. Mol. Cell. Biol., 20, 6354–6363. [DOI] [PMC free article] [PubMed] [Google Scholar]