

Abstract
Human cytomegalovirus (HCMV) encodes several genes that disrupt the major histocompatibility complex (MHC) class I antigen presentation pathway. We recently described the HCMV-encoded US6 gene product, a 23 kDa endoplasmic reticulum (ER)-resident type I integral membrane protein that binds to the transporter associated with antigen processing (TAP), inhibits peptide translocation and prevents MHC class I assembly. The functional consequence of this inhibition is to prevent the cell surface expression of class I bound viral peptides and their recognition by HCMV-specific cytotoxic T cells. Here we describe a novel mechanism of action for US6. We demonstrate that US6 inhibits the binding of ATP by TAP1. This is a conformational effect, as the ER lumenal domain of US6 is sufficient to inhibit ATP binding by the cytosolic nucleotide binding domain of TAP1. US6 also stabilizes TAP at 37°C and prevents conformational rearrangements induced by peptide binding. Our findings suggest that the association of US6 with TAP stabilizes a conformation in TAP1 that prevents ATP binding and subsequent peptide translocation.
Keywords: ABC transporter/antigen presentation/human cytomegalovirus/TAP/US6
Introduction
Major histocompatibility complex (MHC) class I molecules are expressed on the plasma membrane of all nucleated cells and present peptides derived from cytoplasmic proteins. Cytotoxic T lymphocytes monitor these MHC class I molecules for any antigenic peptides derived from viral and malignant proteins, and eliminate infected or transformed cells. The peptides that are presented on MHC class I molecules are predominantly generated by proteolytic processing of proteins in the cytoplasm by the multi-subunit proteasome complex reviewed by Pamer and Cresswell (1998). The transporter associated with antigen processing (TAP) is localized to the endoplasmic reticulum (ER) membrane and translocates these peptides from the cytoplasm into the ER lumen, which is the site of the assembly of MHC class I molecules (Abele and Tampe, 1999). The translocation of peptides into the ER by TAP represents a key stage in the assembly of the MHC class I molecule, since only peptide-loaded class I molecules can leave the ER for transport to the cell surface.
TAP is a member of the ATP binding cassette (ABC) transporter superfamily whose members utilize energy from ATP hydrolysis to translocate a variety of substrates across membranes (Schneider and Hunke, 1998; Holland and Blight, 1999). TAP is unusual for a mammalian ABC transporter, because it is not encoded in a single protein but consists of two separate polypeptides. The two subunits, TAP1 and TAP2, each have an N-terminal polytopic membrane domain and a C-terminal cytoplasmic nucleotide binding domain (NBD), and form a heterodimer (Kelly et al., 1992). The highly conserved NBDs of ABC transporters act as molecular motors, coupling the hydrolysis of ATP to translocation of the substrate by the membrane domains. In contrast to the NBDs, the membrane domains of TAP share little homology with the other ABC transporters and are specialized for the binding and translocation of peptides. Sequential C-terminal truncations suggest that the N-terminus of TAP1 is cytoplasmic and the membrane domain spans the membrane seven times, whereas the N-terminus of TAP2 is lumenal and the membrane domain spans the membrane eight times (Vos et al., 1999). The NBDs of TAP1 and TAP2 can associate with each other (Lapinski et al., 2000), and the TAP1 and TAP2 subunits are thought to align in a tail/tail to head/head orientation (Vos et al., 2000).
The peptide-binding site is shared between TAP1 and TAP2 and has been mapped to cytoplasmic loops in the membrane domains of TAP1 and TAP2 (Androlewicz and Cresswell, 1994; Androlewicz et al., 1994; Nijenhuis and Hammerling, 1996; Nijenhuis et al., 1996). TAP can bind and translocate a range of different sized peptides, although peptides with 8–12 residues in length are optimal for transport (Koopmann et al., 1996). These are the same size or slightly longer than those that bind MHC class I molecules, which bind peptides of 8–10 amino acids in length. Compared with MHC class I, TAP has a relatively broad sequence specificity, although there are some amino acid preferences at the N- and C-terminus of the peptide (Abele and Tampe, 1999). In contrast to the peptide binding specificity of TAP, little is known about the mechanism of peptide translocation across the ER membrane. The NBDs play a pivotal role, as a mutant TAP that is unable to bind nucleotide due to mutations in the Walker A motifs of both TAP1 and TAP2 is completely inactive and, surprisingly, even unable to bind peptide (Knittler et al., 1999). Translocation requires ATP hydrolysis, as non-hydrolysable ATP analogues allow peptide binding but not translocation (Neefjes et al., 1993; Meyer et al., 1994). However, at present there is no information on how the energy derived from the hydrolysis of ATP is harnessed for peptide translocation.
Herpes viruses have evolved several gene products that interfere with MHC class I antigen presentation and help infected cells to evade viral recognition by CTL (Krmpotic et al., 1999; Tortorella et al., 2000). TAP represents an important target for viral inhibition as, apart from some signal sequence-derived peptides, most class I binding peptides require transport from the cytosol to the ER. Two viral inhibitors of human TAP have been characterized, ICP47 and US6. The herpes simplex virus (HSV)-derived cytosolic protein ICP47 acts as a high affinity substrate that blocks TAP by binding to its peptide-binding site, thereby preventing the binding of antigenic peptides (Fruh et al., 1995; Hill et al., 1995; Ahn et al., 1996; Tomazin et al., 1996). A second viral TAP inhibitor, US6, is encoded by the human cytomegalovirus (HCMV) and differs from ICP47. US6 is an ER-localized type I integral membrane protein that associates with TAP and inhibits peptide translocation without affecting peptide binding (Ahn et al., 1997; Hengel et al., 1997; Lehner et al., 1997), a function encoded by the lumenal domain of US6 (Ahn et al., 1997). In this study we examine the inhibition of TAP by US6. We demonstrate that US6 has a unique mechanism of action. US6 inhibits the binding of ATP by TAP1 and also prevents the conformational rearrangement of TAP induced by peptide binding. Thus, by preventing TAP from utilizing its energy source, ATP, US6 effectively inhibits peptide translocation. Our data provide insights into both viral inhibition of TAP and peptide translocation by TAP.
Results
US6 inhibits ATP binding by TAP
US6 associates with TAP and inhibits peptide translocation into the lumen of the ER, but the molecular basis of this inhibition is unknown. Since ATP binding and hydrolysis are essential for the function of not only TAP but all ABC transporters, we wanted to determine whether US6 affected the binding of ATP by TAP. We therefore compared the ATP-binding capacity of TAP in cell lysates prepared from the Pala cell line, with Pala cells transfected with the US6 gene (Pala-US6) (Lehner et al., 1997). ATP binding was assayed with ATP derivatives that can be cross-linked directly to TAP by photoactivation. Cell lysates were prepared in the detergent digitonin, which maintains the interaction between US6 and TAP. These were incubated with 8-azido 5′-[α-32P]adenosine triphosphate ([α-32P]8N3ATP), and cross-linking via the azido (N3) group was induced by short-wave UV irradiation at 254 nm. The TAP heterodimer was immunoprecipitated with the anti-TAP1 antibody 148.3 and labelled proteins visualized with a phosphoimager. [α-32P]8N3ATP labelling of Pala cell lysates revealed a 70 kDa product consistent with either TAP1 or TAP2 (Figure 1A). Labelling of the 70 kDa band required UV cross-linking and could be competed out by cold ATP. Furthermore, no similar sized product was observed in TAP-negative T2 cell lysates. As the immunoprecipitation was performed under native conditions, the labelled species could correspond to either TAP1 or TAP2. In contrast, no labelling was seen of any protein of the same size in cell lysates prepared from Pala-US6 in the concentration range 2.5–10 µM [α-32P]8N3ATP (Figure 1B). This was not the consequence of lower TAP expression in Pala-US6 cells, as equivalent amounts of TAP1 were recovered in parallel immunoprecipitations, as shown by immunoblotting (Figure 1C). Therefore, these data demonstrate a direct inhibition of ATP binding to TAP by US6.
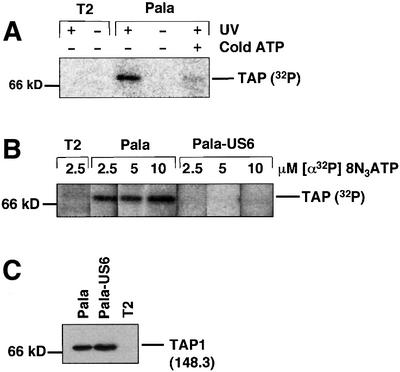
Fig. 1. Photoaffinity labelling of TAP with [α-32P]8N3ATP is inhibited by US6. (A) [α-32P]8N3ATP labelling of TAP. Digitonin cell lysates from T2 and Pala cells (106 per reaction) were labelled with [α-32P]8N3ATP (as described in Materials and methods) in the presence (+) or absence (–) of UV light and cold ATP (1 mM). Following a TAP1 (148.3) immunoprecipitation, eluted proteins were separated on a 10% SDS–polyacrylamide gel prior to exposure to a phosphoimager screen. (B and C) US6 inhibits TAP labelling by [α-32P]8N3ATP. Lysates from T2-, Pala- and US6-transfected Pala cells were photolabelled with 2.5, 5 and 10 µM [α-32P]8N3ATP, immunoprecipitated as described above and separated on a 10% SDS–polyacrylamide gel prior to exposure to a phosphoimager screen (B). A parallel immunoprecipitation with a TAP1 (148.3) mAb from unlabelled cell lysates was performed, and probed with TAP1 (R.RING4C) antisera (C).
In addition to labelling TAP with [α-32P]8N3ATP, we made use of a novel ATP-photoaffinity analogue, 8-azido 5′-γ-biotin adenosine triphosphate (8N3ATPγbiotin), in which the biotin group is attached to the terminal γ-phosphoryl of ATP, rendering this group non-hydrolysable (Figure 2A). 8N3ATPγbiotin labelling of Pala cell lysates was followed by TAP immunoprecipitation, SDS–PAGE and transfer to membranes, which revealed a 70 kDa band on streptavidin-HRP chemiluminescence (Figure 2B). Labelling of the 70 kDa TAP band was specific as it was not seen in the absence of UV cross-linking, was competed out by cold ATP and was not seen in the TAP-negative T2 cell line (Figure 2B). Labelling was concentration dependent, with maximal labelling observed in this experiment at 34 µM 8N3ATPγbiotin (Figure 2C). The 70 kDa species was not labelled in Pala-US6 cell lysates in reactions with either 8.5 or 17 µM 8N3ATPγbiotin (Figure 2C), even though equivalent amounts of TAP were recovered in parallel immunoprecipitations from Pala and Pala-US6 cells (Figure 2D). However, at the highest concentration used (34 µM 8N3ATPγbiotin), labelling of this species was observed in Pala-US6 cell lysates (Figure 2C). Therefore, US6 does not abolish ATP binding by TAP, as labelling was observed at the highest concentration of 8N3ATPγbiotin, but instead appears to reduce the overall affinity of ATP binding by TAP.
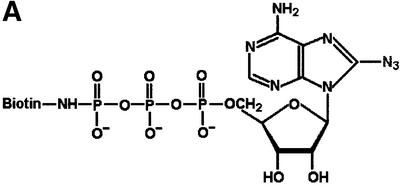
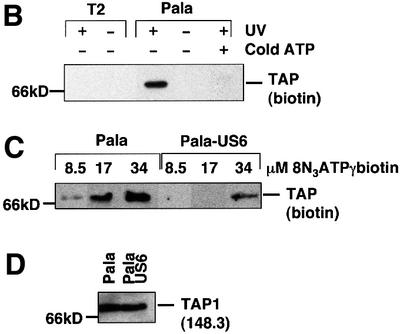
Fig. 2. Photoaffinity labelling of TAP with 8N3ATPγbiotin is inhibited by US6. (A) Chemical structure of 8N3ATPγbiotin. (B) 8N3ATPγbiotin specifically labels TAP. Digitonin lysates of T2 and Pala cells (106 per reaction) were labelled with 34 µM 8N3ATPγbiotin (as described in Materials and methods) in the presence (+) or absence (–) of UV light and cold ATP (136 µM). Following a TAP1 (148.3) mAb immunoprecipitation, eluted proteins were separated on a 10% SDS–polyacrylamide gel, transferred to PVDF membranes and probed with Extravidin-HRP conjugate (Sigma). Reactive bands were detected by chemiluminescence. (C and D) US6 inhibits TAP photolabelling by 8N3ATPγbiotin. Digitonin lysates of Pala- and US6-transfected Pala cells (106 per reaction) were photolabelled with 8.5, 17 and 34 µM 8N3ATPγbiotin, immunoprecipitated with a TAP1 (148.3) mAb (as described above), and eluted proteins were separated on a 10% SDS–polyacrylamide gel, transferred to PVDF membranes and visualized with Extravidin-HRP (C). A parallel immunoprecipitation with a TAP1 (148.3) mAb from unlabelled cell lysates was performed, and probed with TAP1 (R.RING4C) antisera (D).
US6 inhibits ATP binding to TAP1, and promotes ATP binding to TAP2
To determine which of the TAP subunits are affected by US6, cell lysates were labelled with 68 µM 8N3ATPγbiotin to allow TAP labelling of both Pala and Pala-US6 cell lysates (Figure 3). At this concentration, similar TAP labelling was seen in cell lysates from Pala and Pala-US6 cells (Figure 3A, compare lanes 1 and 3). In order to determine which subunits were labelled, TAP1 and TAP2 were dissociated by heating at 37°C with 0.5% SDS, and the individual subunits were immunoprecipitated with either TAP1- or TAP2-specific antibodies. Dissociation of the TAP heterodimer in SDS and subsequent TAP1 immunoprecipitation revealed significant labelling of TAP1 in Pala (wild-type) cell lysates (Figure 3A, lane 2). In marked contrast, no detectable 8N3ATPγbiotin-labelled TAP1 was present in immunoprecipitates of the TAP1 subunit from Pala-US6 cell lysates (Figure 3A, compare lanes 2 and 4). This was despite equivalent recovery of TAP1 in parallel immunoprecipitations as demonstrated by immunoblotting (Figure 3B). This indicates that US6 inhibits ATP binding to TAP1 and that another site binds to ATP in the presence of US6, probably TAP2. To confirm the identity of this second site, the SDS-dissociated TAP heterodimer was precipitated with a TAP2 antibody. Only limited labelling of TAP2 was observed in cell lysates prepared from Pala cells (Figure 3C, lane 2); however, TAP2 was more efficiently labelled in Pala-US6 cell lysates (Figure 3C, compare lanes 2 and 4). Again there was an equivalent recovery of TAP2 in parallel immunoprecipitations of SDS-dissociated TAP as shown by immunoblotting (Figure 3D, lanes 2 and 4). Therefore, in wild-type cell lysates TAP binds to ATP predominantly on the TAP1 subunit. In contrast, labelling reactions with lysates from US6-transfected cells reveal that US6 prevents ATP binding to TAP1; US6 instead allows ATP to bind to TAP2. Labelling of TAP2 requires high concentrations of the ATP analogue. This is consistent with TAP2 from US6-transfected cells having a lower affinity for ATP than TAP1 from wild-type cell lysates.
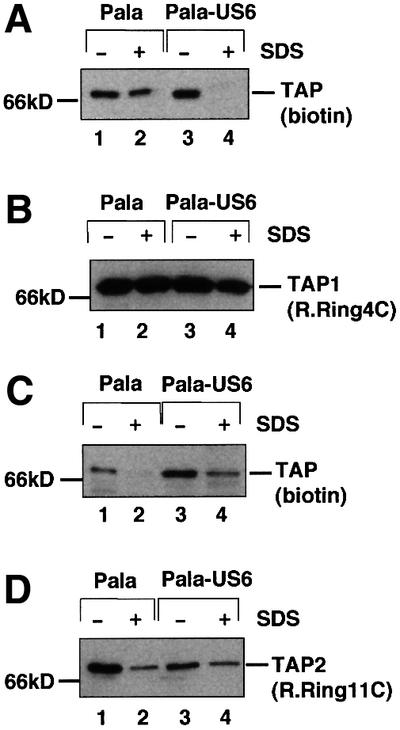
Fig. 3. US6 inhibits 8N3ATPγbiotin photolabelling of TAP1, and promotes ATP labelling of TAP2. (A–D) Digitonin extracts from Pala- and US6-transfected Pala cells (2 × 106 per reaction) were photolabelled with 68 µM 8N3ATPγbiotin and each lysate was divided into four. In two samples (lanes 2 and 4), the TAP heterodimer was dissociated by incubation at 37°C for 15 min in 0.5% SDS (+); the other two samples (lanes 1 and 3) were not heated with SDS prior to immunoprecipitation (–). (A and C) TAP was immunoprecipitated with either the TAP1 mAb (148.3) (A) or the TAP2 mAb (435.3) (C). Immunoprecipitates were separated on 10% SDS–polyacrylamide gels and labelled products detected with Extravidin-HRP. Parallel immunoprecipitations with the TAP1 (148.3) mAb (B) or the TAP2 (435.3) mAb (D) were performed, and separated proteins transferred onto PVDF membranes and probed with TAP1 (R.RING4C) antisera (B) or TAP2 (R.RING11C) antisera (D).
US6 inhibits ATP binding by the TAP–MHC class I loading complex
Labelling with 8N3ATP compounds clearly demonstrates that US6 inhibits ATP binding by TAP. However, because of the low affinity of 8N3ATP compounds for the NBDs, only a small proportion of TAP molecules will be photolabelled. Therefore, to determine whether US6 inhibits ATP binding by the entire population of TAP molecules, the ATP-binding capacity of TAP was assayed by binding to ATP-agarose beads (Knittler et al., 1999). Proteins that bound to the ATP-agarose (pellet) were eluted with EDTA and compared with the proteins in the unbound fraction (supernatant) by SDS–PAGE and immunoblotting (Figure 4A). Both TAP1 and TAP2 bound to ATP-agarose in cell lysates prepared in the detergent digitonin from untransfected Pala cells. In contrast, in Pala-US6 cell lysates very little TAP1 and TAP2 were present in the ATP-agarose fraction. Furthermore, in Pala-US6 cell lysates no US6 was observed in the ATP-agarose fraction, but was all present in the unbound supernatant fraction. Therefore, these data are consistent with US6 expression causing a substantial reduction in the ATP-binding capacity of TAP, and are consistent with our previous results in which we show an inhibition of labelling with 8N3ATP analogues (Figures 1 and 2).
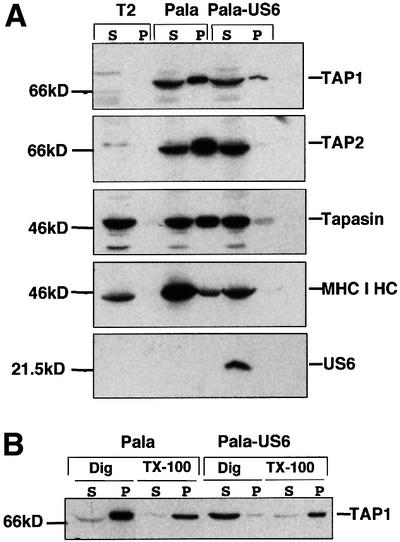
Fig. 4. US6 inhibits the binding of TAP to ATP-agarose. (A) US6 inhibits ATP binding by TAP and TAP-associated proteins. Digitonin extractions of T2, Pala and Pala-US6 cells were incubated with ATP-agarose for 1 h. The final ATP concentration was 13 µM. Agarose bound (P) and unbound (S) proteins separated on either 10 or 12% SDS–polyacrylamide gels. Immunoblots of the ATP-agarose fractions were probed with TAP1- (148.3), TAP2- (435.3), MHC class I heavy chain (HC10)-, tapasin (R.gp48N)- and US6 (R.US6N)-specific antibodies. (B) US6 inhibition of ATP binding by TAP is lost following TAP–US6 dissociation in Triton X-100. Pala- and US6-transfected Pala cells were extracted in 1% digitonin (Dig) and, after removal of the nuclear fraction, each lysate was divided into two aliquots. Triton X-100 (TX-100) (1% final) was added to one aliquot and samples were incubated with ATP-agarose for 0.5 h. Equal cell equivalents of the ATP-agarose bound pellet (P) and unbound supernatant (S) fractions were separated on a 10% SDS–polyacrylamide gel, transferred onto PVDF membranes and probed with TAP1 (148.3) antibodies.
In addition to translocating peptides into the ER lumen, TAP acts as a molecular scaffold for the final stage of MHC class I folding, namely the binding of peptide (Lehner and Trowsdale, 1998). This macromolecular assembly, in which nascent MHC class I molecules associate with TAP via tapasin (Sadasivan et al., 1996; Ortmann et al., 1997), is thought to represent the physiologically relevant form of TAP. We therefore determined whether the interaction of the other components of this TAP–MHC class I loading complex with ATP was also affected by US6 (Figure 4A). In Pala cell lysates, tapasin and MHC class I heavy chain were detected in the ATP-agarose fraction as part of the TAP–MHC class I loading complex. Control lysates prepared from T2 cells (which express neither TAP1 nor TAP2) showed no tapasin or MHC class I heavy chain in the ATP-agarose fraction. In contrast to wild-type Pala, in lysates prepared from Pala-US6 cells, little tapasin and no MHC class I heavy chain were detected in the ATP-agarose fraction, presumably due to the reduced binding of TAP to ATP. These data are, therefore, consistent with US6 expression causing a substantial reduction in the ATP-binding capacity of the TAP–MHC class I loading complex.
The US6 inhibition of ATP binding by TAP may represent an irreversible disruption of TAP’s NBDs or represent a more subtle conformational effect on the NBDs. To discriminate between these possibilities we took advantage of the observation that solubilization of cells in the detergent digitonin preserves the TAP–US6 association, whereas this interaction is disrupted by addition of Triton X-100 such that US6 and TAP are dissociated (data not shown). Cells were lysed in either digitonin alone, or digitonin followed by 1% Triton X-100 and lysates incubated with ATP-agarose (Figure 4B). TAP bound ATP-agarose efficiently in Pala cell lysates in both digitonin and Triton X-100. In contrast, in Pala-US6 digitonin lysates the majority of TAP did not bind ATP-agarose, but the addition of Triton X-100 restored TAP’s ability to bind to the ATP-agarose beads (Figure 4B). This seems to indicate that the inhibition of ATP binding by TAP is dependent upon US6 association with TAP, and is reversible following US6 dissociation from TAP.
US6 does not associate with single chain TAP1
The data discussed above are consistent with the inhibition of ATP binding to TAP, and more specifically to the TAP1 subunit, by US6. To gain a fuller understanding of the mechanism of inhibition of TAP by US6 we determined whether US6 inhibits ATP binding by single chain TAP1 expressed in the absence of TAP2. The C-terminus of US6 was tagged with the FLAG epitope (US6FLAG) and transfected into T2-TAP1 cells (Figure 5A). In this cell line, only the single chain TAP1 subunit is expressed (J.T.Karttunen, P.J.Lehner, S.Sen Gupta, E.W.Hewitt and P.Cresswell, in preparation) and a substantial proportion of it binds to ATP-agarose (Figure 5B), suggesting that TAP1, in the absence of TAP2, folds into a stable conformation. However, in T2-TAP1 cells transfected with US6FLAG, no inhibition of ATP-agarose binding by TAP1 was observed (Figure 5B). The most obvious explanation for the failure of US6 to inhibit ATP binding by TAP1 was that these two molecules do not interact. We therefore probed the association between TAP1 and US6 by immunoprecipitation with an antibody directed against the FLAG epitope tag (Figure 5C). In control HeLa-M cells (which express both TAP subunits) transfected with US6FLAG, TAP1 co-immunoprecipitated with US6FLAG (Figure 5C). However, we were unable to detect any TAP1 in parallel immunoprecipitations from T2-TAP1 cells transfected with US6-FLAG (Figure 5C). This was despite comparable levels of expression of both TAP1 and US6FLAG in the transfected HeLa-M and T2-TAP1 cell lines (Figure 5A). We conclude that US6 cannot associate with single chain TAP1 alone. Although single chain TAP1 may not fold correctly and, therefore, be unable to adopt the conformation recognized by US6, a significant proportion of single chain TAP1 does bind to ATP-agarose. US6 may therefore interact with either TAP2 alone or the TAP1/2 heterodimer. This hypothesis could not be tested as TAP2 appears to be extremely unstable when expressed in isolation and we were unable to generate a stable single chain TAP2 expressing cell line. This is in keeping with observations from patients with bare lymphocyte syndrome who have mutations that abolish TAP1 expression, and TAP2 is no longer detected despite expressing a wild-type TAP2 gene (Furukawa et al., 1999).
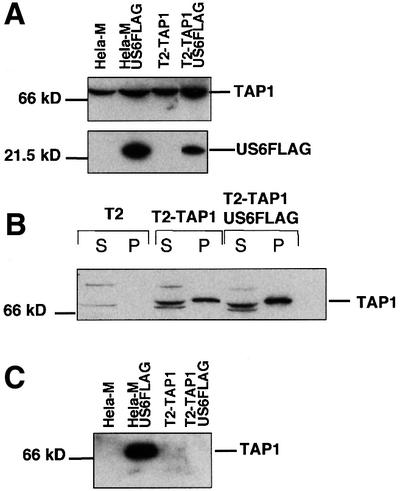
Fig. 5. US6 does not inhibit ATP binding by single chain TAP1. (A) Expression of US6FLAG in T2-TAP1 cells. Cell lysates were run on 10 and 12% SDS–polyacrylamide gels and immunoblotted with FLAG (M2) and TAP1 (148.3) antibodies. (B) US6 does not inhibit ATP binding by single chain TAP1. Digitonin extracts of T2, T2-TAP1 and T2-TAP1-US6FLAG cells were incubated with ATP-agarose for 1 h, and ATP-agarose bound (P) and unbound supernatant (S) fractions separated on a 10% SDS–polyacrylamide gel, transferred onto PVDF membranes and probed with TAP1 (148.3) antibodies. (C) US6 does not associate with single chain TAP1. Digitonin cell lysates were immunoprecipitated with the FLAG mAb M2. Eluted proteins were separated on a 10% SDS–polyacrylamide gel and immunoblotted with TAP1 (R.RING4C)-specific antisera.
US6 stabilizes TAP at 37°C in the absence of ATP
The conformational stability of TAP appears to be dependent upon bound nucleotides. At 37°C, in the absence of nucleotide, TAP undergoes a conformational change in which the reactivity of the TAP molecule to specific antibodies is reduced (van Endert, 1999). Under the same conditions TAP is partially stabilized by the addition of tri- and dinucleotides, suggesting a role for nucleotide in the conformational stability of TAP. Since our data demonstrate that US6 modulates nucleotide binding by TAP, we wondered whether US6 affects the nucleotide-dependent conformational stability of TAP. Pala and Pala-US6 membranes were incubated for 30 min at 4 or 37°C, either with 2 mM ATP or without additional nucleotide. The membranes were then solubilized in digitonin and anti-TAP1 immunoprecipitated proteins resolved on a 10% SDS–polyacrylamide gel, followed by immunoblotting with TAP1-specific antisera (Figure 6A). At 4°C, TAP appeared stable, and incubation with ATP had no effect on the level of TAP1 detected. TAP1 immunoreactivity was significantly reduced upon incubation at 37°C, but was partially stabilized with the addition of 2 mM ATP, consistent with the published observations (van Endert, 1999). In membranes isolated from Pala-US6 cells, however, US6 appeared to stabilize TAP1 at 37°C, even in the absence of ATP (Figure 6A). Thus, US6 association with TAP stabilizes the conformation recognized by anti-TAP1 antibodies at 37°C, regardless of the addition of ATP. Our results demonstrate that, in addition to inhibiting ATP binding, US6 has a parallel effect on the conformational stability of TAP.
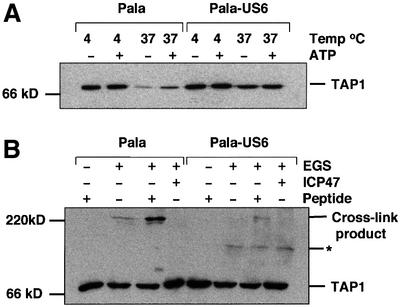
Fig. 6. US6 stabilizes TAP at 37°C but inhibits peptide-stimulated chemical cross-linking. (A) US6 stabilizes TAP at 37°C. Membranes from Pala- and US6-transfected Pala cells were incubated at either 4 or 37°C for 1 h in the presence (+) or absence (–) of 2 mM ATP (as described in Materials and methods). Lysates were digitonin solubilized, immunoprecipitated with TAP1 (148.3) mAb and after separation on a 10% SDS–polyacrylamide gel, immunoblotted with TAP1 (R.RING4C) antisera. (B) Peptide-stimulated chemical cross-linking of the TAP heterodimer is inhibited by US6. Membranes from Pala- and US6-transfected Pala cells were incubated in the presence (+) or absence (–) of 10 µM HLA-B27 (SRYWAIRTR) or ICP47 peptides for 1 h at 4°C before cross-linking with EGS. The cross-linked membranes were resolved on a 6% SDS–polyacrylamide gel and immunoblotted with TAP1 (148.3) mAb. Asterisk, an unidentified band.
US6 inhibits conformational changes associated with peptide binding
Peptide binding by TAP has been shown to induce conformational changes in the TAP heterodimer (Lacaille and Androlewicz, 1998; Neumann and Tampe, 1999). Since US6 inhibits peptide translocation but not peptide binding by TAP (Ahn et al., 1997; Hengel et al., 1997), we wanted to determine whether US6 inhibited the conformational rearrangement in TAP promoted by peptide binding. We therefore used the assay developed by Lacaille and Androlewicz (1998) to examine the cross-linking of TAP in membranes prepared from Pala and Pala-US6 cells (Figure 6B). As previously reported (Lacaille and Androlewicz, 1998), in membranes from wild-type Pala cells, a 220 kDa product recognized by the anti-TAP1 (Figure 6B) and anti-TAP2 antibodies (data not shown), was observed in the presence of the cross-linker ethylene glycol bis(succinimidyl succinate) (EGS). This 220 kDa band was enhanced by the TAP-binding peptide (SRYWAIRTR) and abolished by the minimal ICP47 peptide (Figure 6B). However, in marked contrast to Pala cells, in Pala-US6 membranes there was little detectable 220 kDa cross-linking product recognized by the anti-TAP1 (Figure 6B) or anti-TAP2 antibody (data not shown) even in the presence of the TAP-binding peptide (Figure 6B). Together these data are consistent with US6 preventing TAP cross-linking from forming the 220 kDa product, presumably by inhibiting the conformational rearrangement of TAP that normally follows peptide binding.
US6 sequence requirements for TAP inhibition
Previous studies indicate that the lumenal domain of US6 is sufficient to bind to TAP and inhibit MHC class I maturation (Ahn et al., 1997). To define further the minimal US6 sequence required for TAP inhibition, a series of N- and C-terminal deletions of the US6 lumenal domain sequence was constructed.
The predicted site of cleavage of the US6 signal sequence is between residues Gly19 and Glu20. N-terminal deletions were constructed in which 20 and 40 residues C-terminal to this cleavage site were deleted (US6ΔN20FLAG and US6ΔN40FLAG, respectively). Since the US6 signal sequence was removed in these deletions, the two N-terminal deletion mutants were cloned downstream of the human growth hormone signal sequence (Figure 7A). Further N-terminal deletions of 60 and 80 residues were also constructed, but these were extremely unstable and we were unable to generate stable cell lines that expressed detectable levels of these proteins (data not shown). However, stable HeLa-M cell lines expressing US6ΔN20FLAG and US6ΔN40FLAG were generated. Immunoblot analysis of the stable cell lines generated showed expression of both US6ΔN20FLAG and US6ΔN40FLAG (Figure 7B). US6ΔN20FLAG had a similar predicted Mr to full-length US6FLAG due to the additional sequence corresponding to seven amino acids derived from the human growth hormone signal sequence after signal peptidase cleavage, and eight residues from the linker region (Connolly et al., 1994). The US6ΔN40FLAG protein migrated faster, at ∼22 kDa, due to the greater truncation and because the N-linked glycosylation (Asn52) site had been deleted. The levels of expression of both deletions, in particular US6ΔN40FLAG, were low, indicating that the N-terminus of the US6 lumenal domain is involved in the stability of the US6 molecule. These deletions were assessed for the cell surface expression of MHC class I (Figure 7C), and ATP-agarose binding by TAP (Figure 7D). In both assays the US6ΔN20FLAG mutant appeared almost identical to the full-length US6 molecule (US6FLAG). Transfectants expressing US6ΔN40FLAG showed an intermediate cell surface MHC class I level, between those of untransfected HeLa-M and transfectants expressing the US6FLAG molecule. Furthermore, US6ΔN40FLAG caused a partial reduction in ATP-agarose binding by TAP1 in cell lysates prepared from these transfectants (Figure 7D) and TAP-mediated peptide translocation assays (data not shown). The intermediate phenotype, in terms of class I cell surface expression and ATP-agarose binding in US6ΔN40FLAG transfectants, probably results from the low expression level of US6ΔN40FLAG. Therefore, the N-terminal 40 residues of the US6 lumenal domain appear not to be required for inhibition of TAP, but stabilize expression of the molecule.
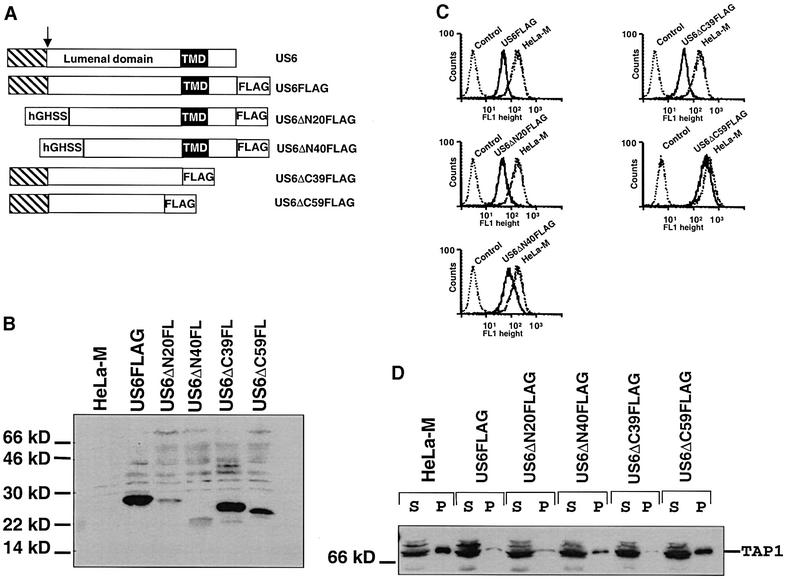
Fig. 7. The C-terminus of the US6 lumenal domain is essential for function. (A) Schematic representation of the construction of the US6 N- and C-terminal deletions. The US6 N-terminal signal sequence is indicated by a hatched box, the arrow indicates the site of signal peptide cleavage and the transmembrane domain is indicated by a black box. The human growth hormone signal sequence is indicated by hGHSS. (B) Expression of the deletion constructs. Cell lysates were prepared and analysed by immunoblotting with the FLAG mAb M2. (C) Cytofluorometric analysis of cell surface MHC class I expression in HeLa-M cells expressing the US6 deletions. Cells were stained with the monoclonal mAb W6/32 and a FITC-conjugated secondary antibody. In the control, HeLa-M cells were only stained with the secondary antibody. (D) ATP-agarose binding of TAP1 in the HeLa-M cell transfectants. Digitonin extracts of HeLa-M cells were incubated with ATP-agarose for 1 h, and ATP-agarose bound pellet (P) and unbound supernatant (S) fractions separated on a 10% SDS–polyacrylamide gel, transferred onto PVDF membranes and probed with TAP1 (148.3) antibodies.
C-terminal deletions of US6 were also constructed. The C-terminal 39 residues of US6, which comprise the cytoplasmic tail and membrane-spanning domain, were removed, such that only the lumenal domain of US6 was expressed (US6ΔC39FLAG) (Figure 7A). This truncated US6 remains ER localized as determined by immunofluorescence and endo H sensitivity (data not shown). A further 20 residues were deleted to generate US6ΔC59FLAG (Figure 7A). Immunoblotting analysis of HeLa-M cell lines stably expressing each truncation indicates that unlike the N-terminal deletions both US6ΔC39FLAG and US6ΔC59FLAG were expressed at levels comparable to full-length US6 (Figure 7B). Consistent with previous reports, expression of the US6ΔC39FLAG (US6 lumenal domain) construct in HeLa-M cells reduced cell surface class I expression to levels comparable to US6FLAG transfectants (Ahn et al., 1997) (Figure 7C). Furthermore, US6ΔC39FLAG inhibited ATP binding by TAP (Figure 7D). This is an important observation because it demonstrates that the US6 ER lumenal domain is sufficient to inhibit ATP binding by the cytoplasmic NBDs of TAP. In marked contrast, US6ΔC59FLAG expression had no significant effect on either cell surface class I expression (Figure 7C) or ATP binding by TAP (Figure 7D). Therefore, whereas the N-terminal 40 residues of the US6 lumenal domain appear to be required for stable expression of US6, the C-terminal 20 residues of the lumenal domain are essential for the inhibition of TAP.
Discussion
HMCV expresses a cohort of proteins that are involved in the evasion of the MHC class I antigen presentation pathway (Tortorella et al., 2000). One of these proteins, US6, associates with TAP to inhibit peptide transport into the lumen of the ER (Ahn et al., 1997; Hengel et al., 1997; Lehner et al., 1997). Unlike the other characterized TAP inhibitor, the HSV protein ICP47, US6 does not affect peptide binding by TAP (Ahn et al., 1997; Hengel et al., 1997). In this study we find that US6 has a novel mechanism of action. US6 causes TAP1 to assume a conformation that is unable to bind ATP, resulting in inhibition of peptide transport.
Three different but complementary approaches were used to determine the effect of US6 upon ATP binding by TAP. ATP binding was assayed with both ATP-agarose and photoaffinity analogues of ATP. US6 inhibited the binding to ATP-agarose by TAP and its associated proteins, tapasin and MHC class I. This was a direct effect on TAP because the photolabelling of TAP itself, with two different 8-azidoATP analogues, [α-32P]8N3ATP and 8N3ATPγbiotin, was also inhibited by US6. These results are in marked contrast to the mode of action of the HSV-derived viral TAP inhibitor ICP47, which has no significant effect upon [α-32P]8N3ATP (Ahn et al., 1996; Tomazin et al., 1996) or ATP-agarose (data not shown) binding by TAP. Therefore, US6 and ICP47 inhibit TAP by very different mechanisms.
Inhibition of ATP binding to TAP by US6 was not absolute, as at high concentrations of 8N3ATPγbiotin binding to TAP was observed. US6 therefore appears to reduce the overall affinity of TAP for ATP. Examination of the labelling of the two subunits of the TAP heterodimer, TAP1 and TAP2, provided further insight into this effect. In wild-type Pala cell lysates the TAP1 NBD was preferentially labelled with 8N3ATPγbiotin, and little labelling of TAP2 was observed. Expression of TAP NBDs in Escherichia coli and insect cells has also shown that the NBD of TAP1 is labelled more readily than that of TAP2 (Muller et al., 1994; van Endert, 1999; Lapinski et al., 2000). We do not know why TAP2 labels poorly with ATP, although nucleotide occlusion or poor solvent accessibility are possible explanations. In marked contrast to the wild-type situation, in the presence of US6 even high concentrations of 8N3ATPγbiotin did not label TAP1, but TAP2 labelling was seen. Therefore, US6 caused a marked change in the behaviour of the NBDs of TAP. The overall affinity of TAP for ATP was reduced, presumably by inhibiting ATP binding to TAP1. US6 does, however, allow binding of ATP by TAP2, albeit at high 8N3ATPγbiotin concentrations.
In parallel to and probably directly related to the inhibition of ATP binding by TAP, US6 also affects the conformational stability of TAP. Incubation of TAP at 37°C in the absence of nucleotide causes a conformational change (Russ et al., 1995) with loss of antibody reactivity (van Endert, 1999). This effect is reversed by tri- and dinucleotides, which, by binding to TAP1, appear to stabilize the transporter (van Endert, 1999). In this study we find that US6 stabilizes the conformation of TAP at 37°C, but does not require exogenous nucleotide. Thus, US6-associated TAP resembles TAP stabilized by exogenous ATP. Although it is not possible to determine directly whether there is nucleotide bound by TAP when associated with US6, the behaviour of TAP in Pala-US6 cells at 37°C is consistent with either an ATP or ADP in stable association with the TAP1 NBD. A nucleotide bound to the TAP1 NBD, which had a low off-rate or was solvent inaccessible would also prevent further nucleotide binding. This may then explain the loss of ATP binding to TAP1 caused by US6.
Despite US6 inhibiting ATP binding by the TAP1 subunit we could demonstrate no association of US6 with singly expressed TAP1. US6 probably requires both subunits of the TAP heterodimer, but may bind TAP2 alone. Although the site on TAP with which US6 interacts is unknown, a direct interaction with the NBDs is not predicted, as the lumenal domain of US6 (US6ΔC39FLAG) is sufficient to inhibit ATP binding by TAP. This result implies that US6 binds to the ER-exposed loops of the membrane domains of TAP rather than to the cytoplasmic NBDs. Since the lumenal domain of US6 is sufficient to modulate the behaviour of the NBDs of TAP, this suggests that the membrane domains communicate with the NBDs via conformational coupling or signalling and that US6 interferes with this process.
Signalling between the membrane domains and the NBDs has been observed in several ABC transporters, and there are many data to support a role for both ATP-dependent and substrate-induced conformational changes. For example, in P-glycoprotein (Pgp), substrate binding by the membrane domains promotes ATP hydrolysis by the NBDs (Ambudkar et al., 1992; Sarkadi et al., 1992; Sharom et al., 1993). In TAP, the binding of the substrate to the membrane domains has been shown to promote conformational changes (Lacaille and Androlewicz, 1998; Neumann and Tampe, 1999). Peptide binding by TAP appears to involve a subtle rearrangement of TAP, which can be detected by the increased chemical cross-linking of TAP1 and TAP2 (Lacaille and Androlewicz, 1998). We find that US6, like ICP47, also inhibits the formation of this cross-linking TAP1–TAP2 product. Since US6 does not prevent peptide binding by TAP, our result implies that US6 inhibits the conformational rearrangement that must follow peptide binding. This may represent the point at which US6 blocks peptide translocation. Consistent with these observations, US6 prevents changes in the lateral mobility of TAP associated with peptide binding, which also suggests a conformational effect (Reits et al., 2000). The precise nature of the conformational rearrangement in TAP is unknown. However, in separate studies we have shown that peptide enhances ATP binding by TAP, most likely TAP1 (J.T.Karttunen, P.J.Lehner, S.Sen Gupta, E.W.Hewitt and P.Cresswell, in preparation), and presumably via an effect on the NBDs. Therefore, given the profound effect of US6 on the behaviour of the NBDs, US6 may well inhibit such peptide-induced conformational changes in the NBDs.
We do not know whether US6 traps TAP in a conformational intermediate of its catalytic cycle or causes TAP to assume an aberrant conformation. Although our data do not discriminate between these two possibilities, there is a precedent for the trapping of a conformational intermediate of an ABC transporter. The monoclonal antibody UIC2 binds to the extracellular portion of Pgp and inhibits substrate translocation (Mechetner et al., 1997). It has been proposed that UIC2 traps Pgp in a transient conformation in which no ATP is bound, and blocks further ATP binding (Mechetner et al., 1997). The conformation of US6-associated TAP does not appear to be grossly aberrant, as shown by binding of the TAP-associated tapasin and MHC class I proteins, as well as peptides (Ahn et al., 1997; Lehner et al., 1997). In addition, stabilization of TAP at 37°C by US6, together with the reversal of inhibition of ATP binding with Triton X-100 detergent, all suggest that US6 does not induce a grossly aberrant conformation in TAP. Therefore, we favour the model in which US6 traps TAP as a conformational intermediate. The ability to bind peptide but not to reach the conformation that normally follows peptide binding would suggest that TAP is held at the stage of peptide binding. If this model is correct, our data imply that, at the point of peptide binding, the NBD of TAP2 is solvent accessible and can bind to ATP.
In conclusion, this study describes the mechanism of immune evasion by the HMCV gene product US6. US6 has a profound effect upon the behaviour of the NBDs of TAP. By trapping TAP in a conformation that inhibits binding of ATP, US6 effectively prevents utilization of TAP’s energy source and inhibits peptide translocation. This not only provides evidence for the key role played by the NBDs in the regulation of peptide transport by TAP, but also suggests an intimate association between the NBDs and the conformation of TAP.
Materials and methods
Constructs
pCDNA3.1(–)Pac. The puromycin resistance gene cassette (Pac) was cut out of pPUR (Clonetech) with AvrII and a partial digest with BsmI. A 1017 bp fragment comprising the Pac cassette was cloned into the AvrII and BsmI sites of pCDNA3.1(–) (Invitrogen) such that the neomycin resistance gene was replaced.
pCDNA3.1(–)Pac-US6FLAG. A FLAG (DYKDDDDK) epitope tag was fused to the C-terminus of US6 by amplifying the US6 open reading frame (ORF) by PCR with the primers US6-5′- and US6-3′-FLAG (see Oligonucleotides section). The resultant US6FLAG ORF was cloned into pCDNA3.1(–)Pac as a KpnI–EcoRI fragment.
pCDNA3.1(–)Pac-US6ΔN20FLAG and -US6ΔN40FLAG. The N-terminal signal sequence of US6 was removed in these deletions. Therefore, to facilitate their targeting to the ER they were cloned downstream of the human growth hormone signal sequence in the plasmid pCDNA3.1(–)Pac-hGHSS. This plasmid was generated by cloning the human growth hormone signal sequence ORF from pRK5-ssHRP-cmyc (Connolly et al., 1994) as a SmaI–EcoRI fragment into the XhoI (blunt-ended with Klenow fragment and dNTPs) and EcoRI sites of pCDNA3.1(–)Pac. US6ΔN20FLAG was amplified by PCR with the primers ΔN20-US6 and US6-3′-FLAG, whereas US6ΔN40FLAG was amplified with the primers ΔN40-US6 and US6-3′-FLAG. The PCR products were cloned as EcoRI–KpnI fragments into the same sites in pCDNA3.1(–)Pac-hGHSS.
pCDNA3.1(–)Pac-US6ΔC39FLAG and -US6ΔC59FLAG. C-terminal truncations were generated in which a FLAG epitope tag was fused to their C-terminus. US6ΔC39FLAG was amplified by PCR with the primers US6-5′ and ΔC39-US6, and US6ΔC59FLAG was amplified with the primers US6-5′ and ΔC59-US6. The PCR products were cloned as KpnI–EcoRI fragments into the same sites in pCDNA3.1(–)Pac.
pMSCV-NEO-US6FLAG. The US6FLAG ORF was cloned from pCDNA3.1(–)US6FLAG as an AflII (blunt-ended with Klenow fragment and dNTPs)–EcoRI fragment into pMSCV-NEO (Stevenson et al., 2000) that had been cut with XhoI (blunt-ended with Klenow fragment and dNTPs)–EcoRI.
Oligonucleotides. US6-5′, TATATCTAGAATGGATCTCTTGATTCGTCTC; US6-3′-FLAG, ATATGGTACCTATTTATCATCATCATCTTTGTAATCGGAGCCACAACGTCGAATCCC; ΔN20-US6, ACACGAATTCGTTCCGAGAACGAAGTCGCAC; ΔN40-US6, ACACGAATTCGCAGATGATAGCTGGAAACAG; ΔC39-US6, TATAGAATTCTTATTTATCATCATCATCTTTGTAATCACCTCCGTGTCGTTCTATCAAGCG; ΔC59-US6, ATATGGTACCTTATTTATCATCATCATCTTTGTAATCCATAAGCGTTAACAACCGGTG.
Antibodies
The following antibodies were used: 148.3, an anti-TAP1 mAb kindly provided by Dr R.Tampe (Meyer et al., 1994); R.RING4C and R.RING11C, rabbit anti-peptide antibodies raised to the C-terminal region of TAP1 (Ortmann et al., 1994) and TAP2 (TAP2A), respectively; 435.3, an anti-TAP.2 mAb kindly provided by Dr P.van Endert (van Endert et al., 1994); W6/32, an anti-human class I mAb (Parham et al., 1979); HC10, a mAb recognizing free class I heavy chains kindly provided by Dr H.Ploegh (Stam et al., 1986); R.gp48N, a rabbit anti-peptide antibody raised to the N-terminal region of tapasin (Sadasivan et al., 1996); R.US6N, a rabbit anti-peptide antibody raised to residues 1–10 of the N-terminal region of US6. The M2 (anti-FLAG) mAb was purchased from Sigma.
Cell lines and culture
The HeLa-M cervical carcinoma cell line, the EBV-transformed lymphoblastoid cell line Pala and the hybrid lymphoblastoid cell line T2 were grown in RPMI 1640 (Gibco-BRL) supplemented with 10% fetal calf serum.
Transfections and generation of stable cell lines
HeLa-M cells. HeLa-M cells were transfected with 1 µg of DNA using Lipofectamine plus reagent (Gibco-BRL) as per the manufacturer’s instructions. Stable transfectants were selected with 1.2 µg/ml puromycin and subcloned into 96 well plates. Clones expressing the FLAG-tagged constructs were screened by immunoblotting with the anti-FLAG epitope tag mAb M2.
T2-TAP1 cells. T2-TAP1 cells (J.T.Karttunen, P.J.Lehner, S.Sen Gupta, E.W.Hewitt and P.Cresswell, in preparation) were transduced with retrovirally packaged pMSCV-Neo-US6FLAG as described in Stevenson et al. (2000). Transduced cells were selected with 600 µg/ml G418 and subcloned. Clones expressing the FLAG-tagged constructs were screened by immunoblotting with the anti-FLAG epitope tag mAb M2.
Site-specific labelling with [α-32P]8N3ATP and 8N3ATPγbiotin
Cells were harvested by centrifugation at 500 g in a bench top centrifuge, washed once with phosphate-buffered saline (PBS), then lysed at 4°C for 60 min at a density of 108 cells/ml in TBS pH 7.0, 1% digitonin, 5 mM MgCl2, 5 mM phenylmethylsulfonyl fluoride (PMSF) and 5 mM iodoacetamide (IAA). Either [α-32P]8N3ATP or 8N3ATPγbiotin (both from Affinity Labelling Technologies Inc., Lexington, KY) was resuspended in an appropriate volume of TBS pH 7.0. For labelling experiments, 30 µl of lysate (corresponding to 1–2 × 106 cells per reaction) were aliquoted into a 96 well plate at 4°C. Ten microlitres of appropriately diluted 8N3ATP were added to the wells. The plate was irradiated with a hand-held UV lamp at 254 nm for 2.5 min, after which 2 µl of 200 mM dithiothreitol were added and the reactions were left on ice for a further 5 min. The reactions were then diluted into 500 µl of TBS pH 7.0, 0.1% digitonin and 5 mM IAA. In native immunoprecipitations TAP was precipitated with the TAP1 mAb 148.3. In denaturing precipitations the TAP heterodimer was dissociated by incubating in 0.5% SDS at 37°C for 15 min before dilution into 500 µl of TBS pH 7.0, 0.1% digitonin and 5 mM IAA. TAP1 and TAP2 were then precipitated with the 148.3 and 435.3 mAbs, respectively. The immunoprecipitates were resuspended into SDS–PAGE sample buffer, heated at 65°C for 10 min and run on 10% SDS–polyacrylamide gels. Gels with samples labelled with [α-32P]8N3ATP were fixed, dried, and placed on a supersensitive phosphoimager screen (Cyclone, Packard) overnight. Gels with 8N3ATPγbiotin-labelled samples were transferred by western blotting onto PVDF membranes, probed with Extravidin-HRP (Sigma) and visualized by chemiluminescence (NEN Life Sciences).
ATP-agarose binding assay
Cells were harvested by centrifugation at 500 g in a bench top centrifuge and washed once with PBS. 5 × 106 cells were lysed at a density of 2 × 107/ml into TBS pH 7.0, 1% digitonin, 5 mM MgCl2, 5 mM IAA and 1 mM PMSF. After 30 min on ice the lysate was spun in a microfuge at 14 000 g at 4°C for 10 min and the supernatant retained. Twenty microlitres of hydrated N-6 ATP-agarose (Sigma) were added to the supernatant to give a final ATP concentration of 13 µM and the sample mixed by rotation at 4°C for 30–60 min. After rotation the samples were spun in a microfuge at 14 000 g at 4°C for 1 min. The supernatant was retained and the ATP-agarose pellet washed three times with TBS, 0.1% digitonin, 5 mM MgCl2. Four microlitres of 500 mM EDTA were added to the ATP-agarose pellets, and SDS–PAGE sample buffer was added to both the supernatant and ATP-agarose fractions and the samples heated at 65°C for 10 min. Samples were run onto 10 or 12% SDS–polyacrylamide gels and analysed by immunoblotting with specific antibodies.
TAP stability at 37°C
This procedure was based on a method described by van Endert (1999). Briefly, cell membranes were prepared by freeze-thaw lysis. 1.5 × 106 cells per reaction were frozen as a cell pellet at 80°C. After freezing, the cell pellet was thawed and resuspended at 2 × 107 cells/ml into TBS pH 7.0, 5 mM IAA, 0.5 mM PMSF, and spun at 320 g at 4°C in a microfuge for 10 min. The supernatant was retained and the pellet resupended into 20 mM Tris–HCl pH 7.0, and spun again. The two supernatants were pooled and spun at 100 000 g at 4°C for 45 min in a TLA45 rotor (Beckman). The membrane pellet was washed once with 1 ml of TBS pH 7.0 and resuspended at 1.5 × 107 cells/ml into TBS, and split into 100 µl aliquots. These were incubated for 30 min at either 4 or 37°C with or without 2 mM ATP. The membranes were then solubilized by dilution into TBS pH 7.0, 1% digitonin, 5 mM IAA, 0.5 mM PMSF. TAP was immunoprecipitated with the TAP1 mAb 148.3. The immunoprecipitates were eluted in SDS–PAGE sample buffer, run on 10% SDS–polyacrylamide gels and immunoblotted with the TAP1 antibody R.RING4C.
Peptide-induced cross-linking of TAP
Cross-linking with the homobifunctional cross-linker EGS (Pierce) was performed as described by Lacaille and Androlewicz (1998), except that the membranes were incubated with 10 µM TAP-binding peptide (HLA-B27 peptide, SRYWAIRTR) or HSV-derived ICP47 minimal peptide (residues 1–35, which comprise the active domain of the HSV ICP47 protein; Galocha et al., 1997) on ice for 1 h prior to cross-linking. Samples were resuspended in SDS–PAGE sample buffer, heated at 65°C for 10 min, run on 6% SDS–polyacrylamide gels and analysed by immunoblotting with the anti-TAP1 mAb 148.3.
Flow cytometric analysis
Stable HeLa-M transfectants were stained with the MHC class I mAb W6/32 in PBS, 0.1% bovine serum albumin and visualized with a goat anti-mouse FITC-conjugated secondary antibody (Jackson).
Immunoblotting
Transfer of SDS–polyacrylamide gels onto PVDF membranes and incubation with specific antibodies were performed as described (Lehner et al., 1997).
Acknowledgments
Acknowledgements
We thank Karin Romisch, Jaana Karttunen, Paul MacAry and Philip Stevenson for advice and helpful discussion and Boyd Haley for advice on photolabelling. This work was supported by The Wellcome Trust.
References
- Abele R. and Tampe,R. (1999) Function of the transport complex TAP in cellular immune recognition. Biochim. Biophys. Acta, 1461, 405–419. [DOI] [PubMed] [Google Scholar]
- Ahn K., Meyer,T.H., Uebel,S., Sempe,P., Djaballah,H., Yang,Y., Peterson,P.A., Fruh,K. and Tampe,R. (1996) Molecular mechanism and species specificity of TAP inhibition by herpes simplex virus ICP47. EMBO J., 15, 3247–3255. [PMC free article] [PubMed] [Google Scholar]
- Ahn K., Gruhler,A., Galocha,B., Jones,T.R., Wiertz,E.J., Ploegh,H.L., Peterson,P.A., Yang,Y. and Fruh,K. (1997) The ER-luminal domain of the HCMV glycoprotein US6 inhibits peptide translocation by TAP. Immunity, 6, 613–621. [DOI] [PubMed] [Google Scholar]
- Ambudkar S.V., Lelong,I.H., Zhang,J., Cardarelli,C.O., Gottesman,M.M. and Pastan,I. (1992) Partial purification and reconstitution of the human multidrug-resistance pump: characterization of the drug-stimulatable ATP hydrolysis. Proc. Natl Acad. Sci. USA, 89, 8472–8476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Androlewicz M.J. and Cresswell,P. (1994) Human transporters associated with antigen processing possess a promiscuous peptide-binding site. Immunity, 1, 7–14. [DOI] [PubMed] [Google Scholar]
- Androlewicz M.J., Ortmann,B., van Endert,P.M., Spies,T. and Cresswell,P. (1994) Characteristics of peptide and major histocompatibility complex class I/β2-microglobulin binding to the transporters associated with antigen processing (TAP1 and TAP2). Proc. Natl Acad. Sci. USA, 91, 12716–12720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly C.N., Futter,C.E., Gibson,A., Hopkins,C.R. and Cutler,D.F. (1994) Transport into and out of the Golgi complex studied by transfecting cells with cDNAs encoding horseradish peroxidase. J. Cell Biol., 127, 641–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruh K., Ahn,K., Djaballah,H., Sempe,P., van Endert,P.M., Tampe,R., Peterson,P.A. and Yang,Y. (1995) A viral inhibitor of peptide transporters for antigen presentation. Nature, 375, 415–418. [DOI] [PubMed] [Google Scholar]
- Furukawa H. et al. (1999) Splice acceptor site mutation of the transporter associated with antigen processing-1 gene in human bare lymphocyte syndrome. J. Clin. Invest., 103, 755–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galocha B., Hill,A., Barnett,B.C., Dolan,A., Raimondi,A., Cook,R.F., Brunner,J., McGeoch,D.J. and Ploegh,H.L. (1997) The active site of ICP47, a herpes simplex virus-encoded inhibitor of the major histocompatibility complex (MHC)-encoded peptide transporter associated with antigen processing (TAP), maps to the NH2-terminal 35 residues. J. Exp. Med., 185, 1565–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengel H., Koopmann,J.O., Flohr,T., Muranyi,W., Goulmy,E., Hammerling,G.J., Koszinowski,U.H. and Momburg,F. (1997) A viral ER-resident glycoprotein inactivates the MHC-encoded peptide transporter. Immunity, 6, 623–632. [DOI] [PubMed] [Google Scholar]
- Hill A., Jugovic,P., York,I., Russ,G., Bennink,J., Yewdell,J., Ploegh,H. and Johnson,D. (1995) Herpes simplex virus turns off the TAP to evade host immunity. Nature, 375, 411–415. [DOI] [PubMed] [Google Scholar]
- Holland I.B. and Blight,M.A. (1999) ABC-ATPases, adaptable energy generators fuelling transmembrane movement of a variety of molecules in organisms from bacteria to humans. J. Mol. Biol., 293, 381–399. [DOI] [PubMed] [Google Scholar]
- Kelly A. et al. (1992) Assembly and function of the two ABC transporter proteins encoded in the human major histocompatibility complex. Nature, 355, 641–644. [DOI] [PubMed] [Google Scholar]
- Knittler M.R., Alberts,P., Deverson,E.V. and Howard,J.C. (1999) Nucleotide binding by TAP mediates association with peptide and release of assembled MHC class I molecules. Curr. Biol., 9, 999–1008. [DOI] [PubMed] [Google Scholar]
- Koopmann J.O., Post,M., Neefjes,J.J., Hammerling,G.J. and Momburg,F. (1996) Translocation of long peptides by transporters associated with antigen processing (TAP). Eur. J. Immunol., 26, 1720–1728. [DOI] [PubMed] [Google Scholar]
- Krmpotic A., Messerle,M., Crnkovic-Mertens,I., Polic,B., Jonjic,S. and Koszinowski,U.H. (1999) The immunoevasive function encoded by the mouse cytomegalovirus gene m152 protects the virus against T cell control in vivo. J. Exp. Med., 190, 1285–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacaille V.G. and Androlewicz,M.J. (1998) Herpes simplex virus inhibitor ICP47 destabilizes the transporter associated with antigen processing (TAP) heterodimer. J. Biol. Chem., 273, 17386–17390. [DOI] [PubMed] [Google Scholar]
- Lapinski P.E., Miller,G.G., Tampe,R. and Raghavan,M. (2000) Pairing of the nucleotide binding domains of the transporter associated with antigen processing. J. Biol. Chem., 275, 6831–6840. [DOI] [PubMed] [Google Scholar]
- Lehner P.J. and Trowsdale,J. (1998) Antigen presentation: coming out gracefully. Curr. Biol., 8, R605–R608. [DOI] [PubMed] [Google Scholar]
- Lehner P.J., Karttunen,J.T., Wilkinson,G.W. and Cresswell,P. (1997) The human cytomegalovirus US6 glycoprotein inhibits transporter associated with antigen processing-dependent peptide translocation. Proc. Natl Acad. Sci. USA, 94, 6904–6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechetner E.B., Schott,B., Morse,B.S., Stein,W.D., Druley,T., Davis,K.A., Tsuruo,T. and Roninson,I.B. (1997) P-glycoprotein function involves conformational transitions detectable by differential immunoreactivity. Proc. Natl Acad. Sci. USA, 94, 12908–12913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer T.H., van Endert,P.M., Uebel,S., Ehring,B. and Tampe,R. (1994) Functional expression and purification of the ABC transporter complex associated with antigen processing (TAP) in insect cells. FEBS Lett., 351, 443–447. [DOI] [PubMed] [Google Scholar]
- Muller K.M., Ebensperger,C. and Tampe,R. (1994) Nucleotide binding to the hydrophilic C-terminal domain of the transporter associated with antigen processing (TAP). J. Biol. Chem., 269, 14032–14037. [PubMed] [Google Scholar]
- Neefjes J.J., Momburg,F. and Hammerling,G.J. (1993) Selective and ATP-dependent translocation of peptides by the MHC-encoded transporter. Science, 261, 769–771. [DOI] [PubMed] [Google Scholar]
- Neumann L. and Tampe,R. (1999) Kinetic analysis of peptide binding to the TAP transport complex: evidence for structural rearrangements induced by substrate binding. J. Mol. Biol., 294, 1203–1213. [DOI] [PubMed] [Google Scholar]
- Nijenhuis M. and Hammerling,G.J. (1996) Multiple regions of the transporter associated with antigen processing (TAP) contribute to its peptide binding site. J. Immunol., 157, 5467–5477. [PubMed] [Google Scholar]
- Nijenhuis M., Schmitt,S., Armandola,E.A., Obst,R., Brunner,J. and Hammerling,G.J. (1996) Identification of a contact region for peptide on the TAP1 chain of the transporter associated with antigen processing. J. Immunol., 156, 2186–2195. [PubMed] [Google Scholar]
- Ortmann B., Androlewicz,M.J. and Cresswell,P. (1994) MHC class I/β2-microglobulin complexes associate with TAP transporters before peptide binding. Nature, 368, 864–867. [DOI] [PubMed] [Google Scholar]
- Ortmann B. et al. (1997) A critical role for tapasin in the assembly and function of multimeric MHC class I–TAP complexes. Science, 277, 1306–1309. [DOI] [PubMed] [Google Scholar]
- Pamer E. and Cresswell,P. (1998) Mechanisms of MHC class I-restricted antigen processing. Annu. Rev. Immunol., 16, 323–358. [DOI] [PubMed] [Google Scholar]
- Parham P., Barnstable,C.J. and Bodmer,W.F. (1979) Use of a monoclonal antibody (W6/32) in structural studies of HLA-A,B,C, antigens. J. Immunol., 123, 342–349. [PubMed] [Google Scholar]
- Reits E.A., Vos,J.C., Gromme,M. and Neefjes,J. (2000) The major substrates for TAP in vivo are derived from newly synthesized proteins. Nature, 404, 774–778. [DOI] [PubMed] [Google Scholar]
- Russ G., Esquivel,F., Yewdell,J.W., Cresswell,P., Spies,T. and Bennink,J.R. (1995) Assembly, intracellular localization and nucleotide binding properties of the human peptide transporters TAP1 and TAP2 expressed by recombinant vaccinia viruses. J. Biol. Chem., 270, 21312–21318. [DOI] [PubMed] [Google Scholar]
- Sadasivan B., Lehner,P.J., Ortmann,B., Spies,T. and Cresswell,P. (1996) Roles for calreticulin and a novel glycoprotein, tapasin, in the interaction of MHC class I molecules with TAP. Immunity, 5, 103–114. [DOI] [PubMed] [Google Scholar]
- Sarkadi B., Price,E.M., Boucher,R.C., Germann,U.A. and Scarborough,G.A. (1992) Expression of the human multidrug resistance cDNA in insect cells generates a high activity drug-stimulated membrane ATPase. J. Biol. Chem., 267, 4854–4858. [PubMed] [Google Scholar]
- Schneider E. and Hunke,S. (1998) ATP-binding-cassette (ABC) transport systems: functional and structural aspects of the ATP-hydrolyzing subunits/domains. FEMS Microbiol. Rev., 22, 1–20. [DOI] [PubMed] [Google Scholar]
- Sharom F.J., Yu,X. and Doige,C.A. (1993) Functional reconstitution of drug transport and ATPase activity in proteoliposomes containing partially purified P-glycoprotein. J. Biol. Chem., 268, 24197–24202. [PubMed] [Google Scholar]
- Stam N.J., Spits,H. and Ploegh,H.L. (1986) Monoclonal antibodies raised against denatured HLA-B locus heavy chains permit biochemical characterization of certain HLA-C locus products. J. Immunol., 137, 2299–2306. [PubMed] [Google Scholar]
- Stevenson P.G., Efstathiou,S., Doherty,P.C. and Lehner,P.J. (2000) Inhibition of MHC class I-restricted antigen presentation by γ2-herpesviruses. Proc. Natl Acad. Sci. USA, 97, 8455–8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomazin R., Hill,A.B., Jugovic,P., York,I., van Endert,P., Ploegh,H.L., Andrews,D.W. and Johnson,D.C. (1996) Stable binding of the herpes simplex virus ICP47 protein to the peptide binding site of TAP. EMBO J., 15, 3256–3266. [PMC free article] [PubMed] [Google Scholar]
- Tortorella D., Gewurz,B.E., Furman,M.H., Schust,D.J. and Ploegh,H.L. (2000) Viral subversion of the immune system. Annu. Rev. Immunol., 18, 861–926. [DOI] [PubMed] [Google Scholar]
- van Endert P.M. (1999) Role of nucleotides and peptide substrate for stability and functional state of the human ABC family transporters associated with antigen processing. J. Biol. Chem., 274, 14632–14638. [DOI] [PubMed] [Google Scholar]
- van Endert P.M., Tampe,R., Meyer,T.H., Tisch,R., Bach,J.F. and McDevitt,H.O. (1994) A sequential model for peptide binding and transport by the transporters associated with antigen processing. Immunity, 1, 491–500. [DOI] [PubMed] [Google Scholar]
- Vos J.C., Spee,P., Momburg,F. and Neefjes,J. (1999) Membrane topology and dimerization of the two subunits of the transporter associated with antigen processing reveal a three-domain structure. J. Immunol., 163, 6679–6685. [PubMed] [Google Scholar]
- Vos J.C., Reits,E.A., Wojcik-Jacobs,E. and Neefjes,J. (2000) Head-head/tail-tail relative orientation of the pore-forming domains of the heterodimeric ABC transporter TAP. Curr. Biol., 10, 1–7. [DOI] [PubMed] [Google Scholar]