

Abstract
Apicomplexan parasites—including the causative agents of malaria (Plasmodium sp.) and toxoplasmosis (Toxoplasma gondii)—harbor a secondary endosymbiotic plastid, acquired by lateral genetic transfer from a eukaryotic alga. The apicoplast has attracted considerable attention, both as an evolutionary novelty and as a potential target for chemotherapy. We report a recombinant fusion (between a nuclear-encoded apicoplast protein, the green fluorescent protein and a rhoptry protein) that targets to the apicoplast but grossly alters its morphology, preventing organellar segregation during parasite division. Apicoplast-deficient parasites replicate normally in the first infectious cycle and can be isolated by fluorescence-activated cell sorting, but die in the subsequent host cell, confirming the ‘delayed death’ phenotype previously described pharmacologically, and validating the apicoplast as essential for parasite viability.
Keywords: Apicomplexa/apicoplast/delayed death phenotype/organelle segregation/Plasmodium falciparum
Introduction
The protozoan phylum Apicomplexa includes many important human and veterinary pathogens, responsible for a wide variety of diseases (Levine, 1988). Plasmodium sp. are the causative agents of malaria (Persidis, 2000), Toxoplasma gondii is a serious congenital infection in humans and sheep (Dubey and Welcome, 1988; Roizen et al., 1995), and both T.gondii and Cryptosporidium parvum are prominent opportunistic pathogens associated with AIDS (Luft and Remington, 1992; Luft et al., 1993). Other apicomplexan parasites infect animals as diverse as cattle, poultry and shellfish, with severe economic impact. Among the Apicomplexa, molecular genetic systems have been developed only for Toxoplasma and Plasmodium; of these, T.gondii provides the more accessible system for ultrastructural analysis (Roos et al., 1999a).
Considerable effort has been invested in devising effective drug treatments for these pathogens, and such studies have gained renewed impetus due to the emergence and spread of drug resistance (Ricketts and Pfefferkorn, 1993; White, 1998; Warhurst, 1999), and therapeutic complications associated with the chronic treatment of AIDS patients (Haberkorn, 1996). The recent discovery of an unexpected organelle—a non-photosynthetic plastid designated the apicoplast—in Toxoplasma, Plasmodium and other apicomplexan parasites has opened a new realm for identifying potential drug targets (Fichera and Roos, 1997; Waller et al., 1998; Jomaa et al., 1999; McFadden and Roos, 1999). These studies have also raised fascinating biological questions as to the origin and function(s) of this unusual organelle (Wilson et al., 1996; Köhler et al., 1997; McFadden and Waller, 1997; Roos et al., 1999b).
The apicomplexan plastid is thought to have arisen by ‘secondary endosymbiosis’, when the ancestor of these parasites engulfed a eukaryotic alga, and retained the (endosymbiotic) algal plastid (Palmer and Delwiche, 1996; Köhler et al., 1997; McFadden et al., 1997; Blanchard and Hicks, 1999; Dzierszinski et al., 1999). Both the phylogeny of the apicoplast genome and the structure of this organelle (surrounded by four membranes) support this hypothesis (McFadden and Roos, 1999). Pharmacological studies suggest that the apicoplast is the target for a variety of antibiotics commonly thought of as antibacterial agents (macrolides, lincosamides, rifamipicins, fluoroquinolones, etc.; Fichera and Roos, 1997). All of these drugs exhibit peculiar kinetics of cell killing: parasite replication is only inhibited after invasion of the subsequent host cell after initiation of treatment (Pfefferkorn et al., 1992; Fichera et al., 1995). Such observations have led to speculation that the apicoplast may be required for the establishment of a functional parasitophorous vacuole, the specialized structure inside infected cells within which parasites replicate until host cell lysis (Suss-Toby et al., 1996; Lingelbach and Joiner, 1998). The mechanistic basis of this ‘delayed death phenotype’ remains unexplained.
Although the apicoplast genome encodes no metabolic enzymes, mining the Plasmodium falciparum genome and T.gondii expressed sequence tag (EST) databases (Ajioka, 1998; Bowman et al., 1999; Gardner, 1999) has identified numerous nuclear-encoded apicoplast genes, implicating this organelle in the biosynthesis of fatty acids and terpenoids (Waller et al., 1998; Jomaa et al., 1999; Roos et al., 1999b). Nuclear-encoded apicoplast proteins are characterized by a bipartite N-terminal sequence, which is both necessary and sufficient for targeting to the organelle (Waller et al., 1998). The extreme N-terminus consists of a secretory signal sequence, mediating co-translational translocation into the endoplasmic reticulum; the subterminal domain functions as a plastid transit peptide (Roos et al., 1999b; Waller et al., 2000). In combination, these domains provide a remarkable scheme for targeting proteins across the multiple membranes surrounding the apicoplast. In the course of studies on apicoplast targeting, we inadvertently generated a fusion protein, which produces a surprising phenotype: mis-segregation of the apicoplast during parasite replication. These mutant parasites provide a cell biological confirmation of the ‘delayed death’ phenotype, validate the apicoplast as a target for drug development, and provide useful tools for dissecting the function of this intriguing organelle.
Results
A recombinant plastid–rhoptry–GFP fusion protein perturbs apicoplast morphology
We have previously demonstrated that a fusion between the nuclear-encoded apicoplast acyl-carrier protein (ACP) and the green fluorescent protein (GFP) results in targeting of the GFP reporter into the apicoplast in living parasites (Waller et al., 1998), as shown in Figure 1A. The parasitophorous vacuole shown in this image contains four T.gondii tachyzoites, the clonal progeny of a single invasion. Each parasite harbors a single apicoplast (green), invariably located just apical to the nucleus in these highly polarized parasites. This pattern is distinct from that observed when GFP is targeted to other apical organelles such as the rhoptries (Striepen et al., 1998), in which one or more green streaks are observed, further towards the apical end of each parasite (Figure 1B; construct ROP1–GFP). Incorporation of a chloramphenicol acetyl transferase (CAT) gene on the transfection plasmid (Kim et al., 1993) permits isolation of stable ACP–GFP or ROP1–GFP transgenic parasites by chloramphenicol selection (Striepen et al., 1998).
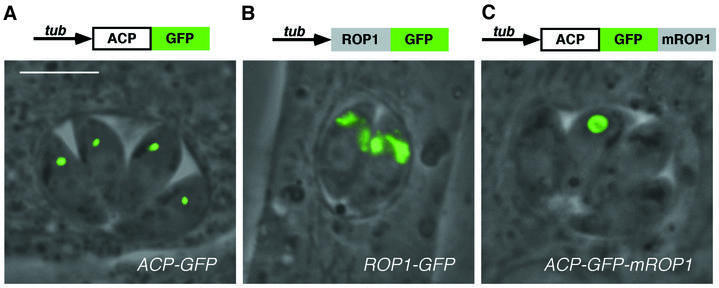
Fig. 1. Distinct targeting patterns displayed by ACP–GFP, ROP1–GFP and ACP–GFP–mROP1. Fusion of the nuclear-encoded apicoplast protein ACP to GFP targets this recombinant reporter molecule into the apicoplast in stable transgenic parasites (A). Similarly, stable expression of the rhoptry protein ROP1 fused to GFP results in targeting to the rhoptries in every parasite (B). Transient expression of the ACP–GFP–mROP1 construct reveals a ring-like staining pattern in only one parasite per vacuole (C). All panels show superimposed fluorescence and phase-contrast images of living parasites. Bar = 5 µm.
In order to study targeting sequences involved in the import of proteins into the apicoplast, we have generated a variety of constructs based on the ACP–GFP reporter. In one such construct, a truncated fragment of ROP1 containing rhoptry targeting sequences (Soldati et al., 1998) was inadvertently retained at the C-terminus of ACP–GFP (see Materials and methods). Transient transfection with this construct (ACP–GFP–mROP1) resulted in an unusual pattern of fluorescence, often appearing as a large, ring-like structure (Figure 1C). This staining pattern was usually observed in the apical juxtanuclear region expected for the apicoplast, but was also occasionally found at the posterior end of the parasite (not shown). Stranger still, this ring-like structure was usually found in only one parasite per vacuole; other parasites within the same vacuole were not labeled, in contrast to previous observations that every parasite contains a single apicoplast (Köhler et al., 1997).
To determine whether this ring-like structure is indeed the apicoplast, we turned to electron microscopic analysis. The morphology of the parasite nucleus, mitochondrion, Golgi, rhoptries, micronemes, dense granules, etc. appears normal in ACP–GFP–mROP1-transfected parasites, but these studies revealed very unusual apicoplast architecture, as shown in Figure 2. The apicoplast is readily identifiable by the presence of four delimiting membranes (Köhler et al., 1997; McFadden and Roos, 1999), but rather than the typical pattern of a relatively clear internal lumen within the organelle (Figure 2A), numerous inclusions were observed, surrounded by multiple membranes and enclosing material morphologically similar to the parasite cytoplasm (including cytoplasmic ribosomes). This peculiar ultrastructure was clearly attributable to transfection with the ACP–GFP–mROP1 construct, as apicoplast morphology was normal in parasites transfected with ACP–GFP or ROP1–GFP (not shown). Immunolabeling using polyclonal anti-GFP and gold-conjugated secondary antibodies confirmed that the ring-like structure was indeed the apicoplast (Figure 2F). No GFP staining was observed in the rhoptries or elsewhere in these parasites.
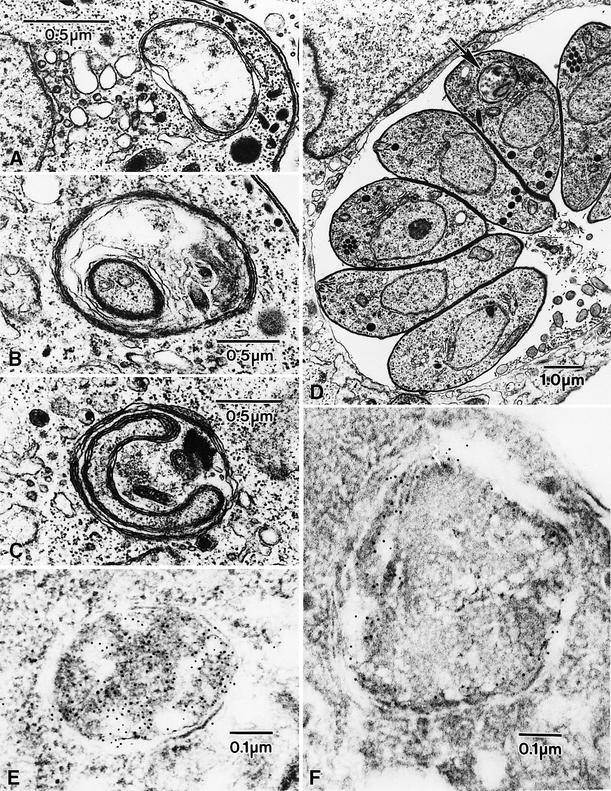
Fig. 2. Morphological changes in the apicoplast of parasites transfected with ACP–GFP–mROP1. (A) Normal apicoplast morphology, revealing four membranes surrounding a relatively electron-lucent interior. (B–D) Membranous inclusions are clearly visible within the apicoplast of parasites transfected with ACP–GFP–mROP1; these inclusions are typically surrounded by four membranes, and appear to enclose cytoplasmic material. The apicoplast in ACP–GFP–mROP1-transfected parasites is also larger than controls. Note that only a single abnormal apicoplast (arrow) is visible in the parasites seen within the single vacuole in (D). (E and F) Immunogold staining localizes GFP throughout the apicoplast in ACP–GFP transgenics (E), but only near the periphery of the apicoplast in parasites expressing ACP–GFP–mROP1 (F).
Transient expression of ACP–GFP–mROP1 inhibits apicoplast segregation
Each T.gondii tachyzoite establishes an independent para sitophorous vacuole upon host cell invasion (Suss-Toby et al., 1996; Lingelbach and Joiner, 1998), and mitotic replication proceeds synchronously, producing 2, 4, 8, … parasites within a single vacuole (Ogino and Yoneda, 1966; Fichera et al., 1995), ultimately lysing the host cell. The observation of only a single apicoplast per vacuole in parasites expressing ACP–GFP–mROP1 (cf. Figures 1C and 2D) therefore suggests that expression of this construct (and consequent disruption of apicoplast morphology) inhibits plastid segregation during parasite cell division. To examine this possibility, we used time-lapse video microscopy to follow the process of parasite division in living parasites expressing ACP–GFP–mROP1. Figure 3 (top) shows a vacuole containing two parasites (at time t = 0), only one of which contains an apicoplast. These parasites grow and ultimately divide, but the apicoplast fails to segregate, yielding a vacuole with four parasites but only a single apicoplast. Incubation of ACP–GFP– mROP1-transfected parasites for even longer periods produces vacuoles with up to 128 parasites, containing only a single giant plastid (Figure 3, bottom).
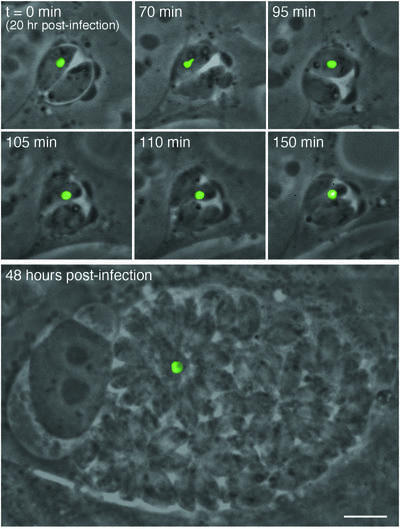
Fig. 3. Unequal segregation of the apicoplast induced by ACP–GFP–mROP1. RH parasites were transiently transfected with ACP–GFP–mROP1 and inoculated into an HFF cell monolayer. Top: beginning 24 h post-transfection/infection (at which point expression of the transgene becomes apparent), parasite division was monitored by time-lapse video microscopy (time points indicated in each frame). Bottom: a different vacuole at 48 h post-transfection, containing ∼64 parasites, only one of which contains an apicoplast. All images are at the same magnification. Bar = 5 µm.
Several experiments were carried out to confirm that transfection with the ACP–GFP–mROP1 construct results in mis-segregation of the entire apicoplast organelle (rather than the ACP–GFP–mROP1 reporter protein alone). Exploiting the ability to visualize multiple fluorescent proteins simultaneously in living cells, stable parasite transgenics expressing ACP–GFP in the apicoplast (Waller et al., 1998) were transfected with an ACP–red fluorescent protein (RFP)–mROP1 fusion construct (Figure 4, top panels). (Note that in contrast to the previous experiment, where the ACP–GFP–mROP1 construct was responsible for disrupting apicoplast morphology, in this experiment GFP serves simply as a marker for the apicoplast lumen; disruption is mediated by the RFP construct.) Untransfected tachyzoites express only the GFP reporter, and the apicoplast can be seen as a single green fluorescent dot in every parasite. Parasites expressing the ACP–RFP–mROP1 fusion construct, however, exhibit only one apicoplast in one parasite per vacuole, containing both GFP and RFP. GFP or RFP labeling in other parasites within the same vacuole is virtually undetectable, and dispersed throughout the apical region of these cells (not shown). Thus, the apicoplast is completely absent in these ‘dark’ parasites. Immuno labeling with antibody directed against a native apicoplast protein also confirms the presence of only a single apicoplast per vacuole (data not shown).
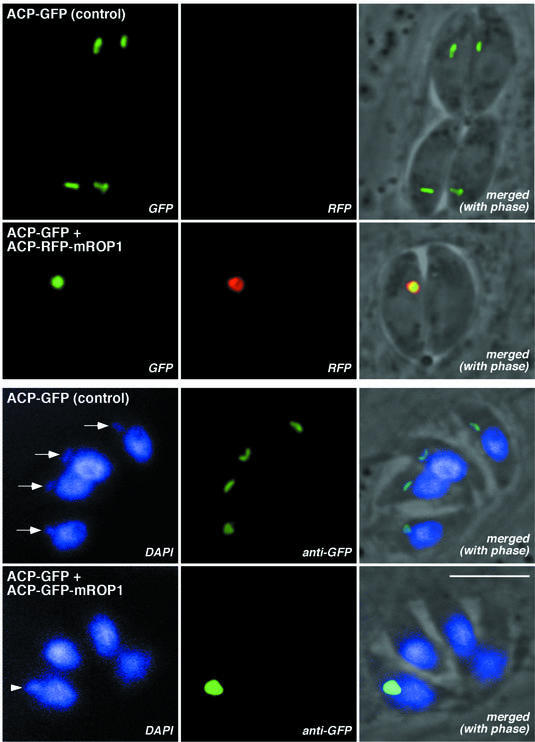
Fig. 4. Loss of the entire apicoplast in parasites expressing ACP–GFP–mROP1 or ACP–RFP–mROP1. Top (native fluorescence of living parasites): the apicoplast lumen fluoresces green in a stable ACP–GFP transgenic line (note that every parasite contains an apicoplast). Transfection with ACP–RFP–mROP1 results in mis-segregation of both the RFP fusion protein and the apicoplast lumenal marker (GFP). Bottom: the apicoplast genome is visible as a distinct extranuclear dot just apical to the nucleus in fixed parasites stained with DAPI (arrows), and this dot co-localizes with GFP in ACP–GFP transgenics. Transfection with ACP–GFP–mROP1 results in mis-segregation of both the apicoplast genome (arrowhead) and the apicoplast marker (GFP). Bar = 5 µm.
In a further experiment, parasites were fixed and stained with 4′,6-diamidino-2-phenylindole (DAPI) (Figure 4, bottom panels), permitting visualization of the 35 kb apicoplast genome (Fichera and Roos, 1997; Köhler et al., 1997). In control parasites expressing ACP–GFP, apicoplast DNA can be seen as a faint dot in every parasite, just apical to the much larger nucleus. Apicoplast DNA co-localizes precisely with the apicoplast itself (stained with polyclonal anti-GFP). In parasites transfected with ACP–GFP–mROP1, however, only one apicoplast can be seen per vacuole, using either DAPI or anti-GFP. Note that this apicoplast is very large and stains very brightly with DAPI (compare with controls), indicating that the apicoplast genome continues to replicate even in the absence of organellar segregation.
Stable transgenic parasites expressing GFP in the apicoplast, rhoptries or other organelles are readily isolated (cf. Figure 1), and transient expression of ACP–GFP–mROP1 was observed in a high proportion of transfected parasites. We were unable to obtain stable transformants expressing ACP–GFP–mROP1, however, despite several attempts using different selection methods. Unfortunately, systems for inducible expression are not currently available for T.gondii (Roos et al., 1994).
Apicoplast segregation mutants confirm that this is an essential organelle and validate the ‘delayed death’ phenotype
The ability of parasites without an apicoplast to grow and divide (cf. Figure 3) indicates that this organelle is not required for short-term survival, calling into question arguments that the apicoplast is an essential component of the parasite (Fichera and Roos, 1997; Rogers et al., 1998; Jomaa et al., 1999; Sullivan et al., 2000). However, the inability to isolate stable transformants suggests that the apicoplast may be essential in the longer term. These observations are reminiscent of the ‘delayed death’ phenotype, in which the killing effect of drugs thought to target the apicoplast is observed only after escape from the first host cell and invasion into a second cell (Pfefferkorn et al., 1992; Fichera et al., 1995). The ability to produce plastid-deficient parasites by transient expression of ACP–GFP–mROP1 provides an opportunity to test the ‘delayed death’ hypothesis.
Parasites stably expressing ACP–GFP were transiently transfected with the ACP–GFP–mROP1 construct and permitted to infect host cells. Despite mis-segregation of the apicoplast, these parasites replicated normally, ultimately lysing the host cell monolayer as rapidly as controls (data not shown). Extracellular parasites were then harvested and fractionated using a fluorescence-activated cell sorter (FACS). The GFP fluorescence profile reveals three populations (Figure 5). (i) A peak of dark parasites (left-most shaded area in Figure 5) contains tachyzoites that lost the apicoplast due to mis-segregation. These parasites exhibit fluorescence comparable to untransfected wild-type parasites (black line) rather than the parental ACP–GFP transgenics from which they were derived (green line). Western blotting (not shown) indicates that protein is still expressed from the stable ACP–GFP transgene, but in the absence of an apicoplast (Figure 4), this protein is not processed, fluoresces poorly, and may be secreted from the parasite (Roos et al., 1999b; Waller et al., 2000). (ii) A large central peak (middle shaded area in Figure 5) coincides with the staining of untransfected parasites, which are green due to the expression of a stable ACP–GFP transgene (green line). This population consists of parasites that failed to take up or express the ACP–GFP–mROP1 marker. (iii) A small shoulder of ‘super-bright’ tachyzoites (right-most shaded area in Figure 5) exhibiting stronger fluorescence than the brightest untransfected parasites. This peak contains those few parasites that retained the apicoplast during replication, accumulating ACP–GFP and ACP–GFP– mROP1 in a large, non-segregating organelle (Figures 3 and 4). Microscopic observation of sorted parasites confirms these interpretations (not shown, but see below).
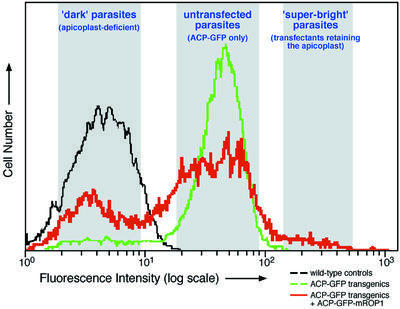
Fig. 5. Apicoplast-free parasites and parasites containing non-segregating apicoplasts can be isolated by FACS. The red line indicates the fluorescence intensity distribution of ACP–GFP transgenic parasites transiently transfected with ACP–GFP–mROP1. Approximately 60% of parasites did not take up and/or express the ACP–GFP–mROP1 plasmid (central shaded area), and exhibit fluorescence comparable to the parental ACP–GFP transgenics (green line). More than 35% of parasites were comparable in intensity to wild-type controls lacking any GFP (black line). This ‘dark’ population (left-most shaded area) consists of parasites that lost their apicoplast (see Figures 1–4 and text). Less than 5% of the population (right-most shaded area) were brighter than any parasites seen in untransfected parasites expressing ACP–GFP (green line). This ‘super-bright’ population consists of parasites that retained the single giant apicoplast during mis-segregation (see Figures 1 and 3). Parasites were isolated from each of the shaded regions for further experiments (see text and Figure 6).
FACS-sorted parasites from each of these populations were inoculated into fresh host cell cultures, and assayed for invasion, ACP expression/localization, intracellular growth rates and plaque-forming ability (Figure 6). Parasites that failed to express the ACP–GFP–mROP1 fusion all contained a normal apicoplast, and replicated intracellularly with a doubling time of ∼9.7 h (filled squares in Figure 6), exactly as seen for wild-type controls (open squares and left-hand micrograph). These parasites also formed plaques as efficiently as wild type (∼7 days).
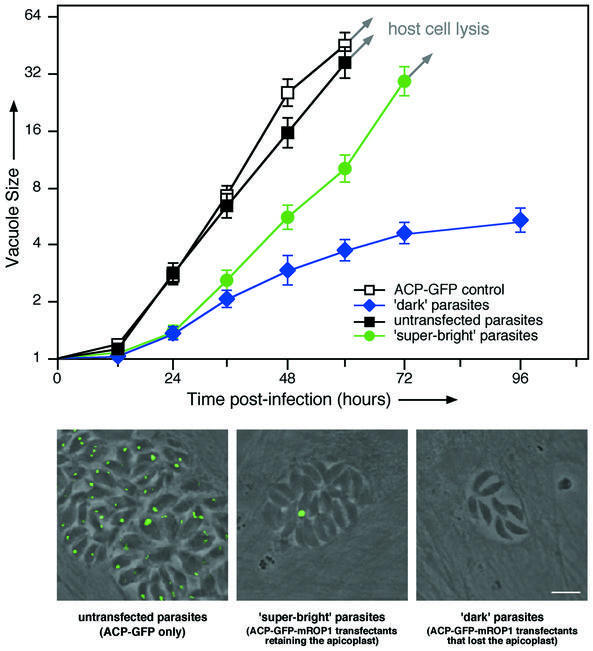
Fig. 6. Replication of FACS-sorted plastid segregation mutants in the second infectious cycle validates the ‘delayed death’ phenotype. Parasites collected from each of the fluorescence intensity gatings shown in Figure 5 were inoculated into fresh human foreskin fibroblast (HFF) cell cultures, and replication rates were followed over the subsequent 96 h. Each symbol indicates the average number of parasites per parasitophorous vacuole—the progeny of a single clonal infection (error bars indicate SE; N = 50). Untransfected wild-type parasites (open squares) and ACP–GFP transgenics that failed to take up or express ACP–GFP–mROP1 (filled squares) replicated with a doubling time of 9.7 h. Parasites expressing ACP–GFP–mROP1 that retained the apicoplast (‘super-bright’ population from Figure 5) exhibited comparable replication rates (green circles; doubling time ∼10.6 h). Parasites that lost the apicoplast (‘dark’ population from Figure 5) grew very slowly, and ultimately died (blue diamonds). Immunofluorescent staining of ACP at 48 h post-infection confirms these interpretations (images at bottom; see text for further discussion). Bar = 5 µm.
‘Super-bright’ parasites (green circles) were able to grow and lyse out from their host cells, but contained only a single large apicoplast (central micrograph), just as seen for transfected parasites in the first infectious cycle (Figure 3). Doubling times for these parasites (∼10.5 h) were slightly slower than controls, possibly due to the metabolic cost of maintaining a gigantic apicoplast. A few plaques were obtained following infection with ‘super-bright’ parasites after long-term incubation (≥10 days), but these parasites no longer contained any ACP–GFP– mROP1 plasmid and exhibited wild-type apicoplast morphology, possibly due to fragmentation of the giant apicoplast during parasite division at some point after loss of the episomal ACP–GFP–mROP1 construct (see Discussion).
‘Dark’ parasites invaded the host cell monolayer as efficiently as wild type, but showed no intracellular fluorescence (right-hand micrograph), grew very slowly and eventually died (blue diamonds). Initial doubling times were ∼26.1 h, but growth slowed thereafter. These parasites never produced vacuoles containing >16 tachyzoites, never managed to lyse the host cells and never produced plaques. The death of plastid-deficient parasites in this second infectious cycle confirms the ‘delayed death’ phenotype observed when the apicoplast is eliminated by pharmacological means (Fichera and Roos, 1997; Sullivan et al., 2000).
Discussion
In the course of studies on apicoplast targeting and purification in the protozoan parasite T.gondii, we generated a fusion construct containing the apicoplast protein ACP, the fluorescent protein reporter GFP and a fragment of the rhoptry protein ROP1 (ACP–GFP–mROP1). Expression of this recombinant ‘poison’ protein in transiently transfected T.gondii tachyzoites results in specific targeting to the apicoplast, but produces an unusual phenotype: grossly aberrant apicoplast morphology, containing numerous complicated inclusions (Figures 1 and 2). Remarkably, apicoplast segregation is abolished during mitosis in these parasites (Figure 3). This serendipitous discovery greatly facilitates efforts to elucidate the function of the apicomplexan plastid.
Apicoplast segregation mutants
Wild-type parasites contain only a single apicoplast per cell (Köhler et al., 1997). Failure of the apicoplast to divide during parasite replication therefore means that the organelle is partitioned into only one of the two daughter cells formed during mitosis, one of the four granddaughters, etc. (Striepen et al., 2000). Both apicoplast-containing and -deficient parasites continue to replicate within the parasitophorous vacuole (Figure 3), at a rate only slightly reduced in comparison to wild-type controls. These vacuoles may grow to contain 64 or more parasites (the synchronously replicating, clonal progeny of a single infection), only one of which contains an apicoplast. The apicoplast can grow to an immense size in those parasites that retain the organelle, indicating a lack of cell cycle checkpoints specifically regulating apicoplast segregation. The apicoplast also continues to import nuclear-encoded apicoplast proteins, and continues to replicate the 35 kb organellar genome (Wilson et al., 1996; J.C.Kissinger, web site: http://www.sas.upenn.edu/∼jkissing/toxomap.html; Figure 4).
The ability to express GFP in T.gondii transgenics, to target this reporter to the apicoplast (Waller et al., 1998) and to isolate GFP-expressing parasites by FACS (Striepen et al., 1998) facilitates examination of apicoplast segregation mutants in living parasites (Figure 5). After lysis of the host cell, both plastid-containing and -deficient parasites are able to infect new host cells and establish a parasitophorous vacuole. Those parasites that retain the apicoplast go on to produce plaques, ultimately lysing the entire host cell monolayer. While parasites that lack an apicoplast replicate normally within the initial parasitophorous vacuole (Figure 3), these parasites replicate very slowly within the second host cell, ultimately dying without escaping from that cell (Figure 6). Apicoplast-deficient parasites fail to form plaques even after prolonged cultivation. The death of these parasites validates the apicoplast as an essential organelle in T.gondii and, by extension, most other apicomplexan parasites as well (Clough et al., 1997; Fichera and Roos, 1997; Rogers et al., 1998; Sullivan et al., 2000; Zhu et al., 2000).
The mechanism of apicoplast mis-segregation
The ROP1 protein (which constitutes the C-terminal portion of the ACP–GFP–mROP1 construct responsible for apicoplast mis-segregation) exhibits several interesting characteristics, behaving as a soluble protein within the rhoptries, but rapidly associating with membranes after secretion (Saffer et al., 1992). The predicted amino acid sequence of ROP1 contains no obvious transmembrane domains, but exhibits weak similarity to rat salivary gland proteins involved in protein complex assembly (Ossorio et al., 1992). It is possible that fusion of this protein to ACP–GFP (which on its own is efficiently targeted to the apicoplast; Waller et al., 1998) prevents complete translocation across the multiple membranes surrounding the apicoplast (McFadden and Roos, 1999; Roos et al., 1999b). Consistent with this interpretation, immunolocalization typically reveals a peripheral staining pattern (Figure 2F). Preliminary studies also suggest that ACP–GFP–mROP1 associates with the membrane pellet after cell disruption and sodium carbonate extraction, while ACP–GFP is soluble (data not shown). In other systems, fusion proteins whose tertiary structure prevents unfolding are known to block efficient organellar translocation (Endo and Schatz, 1988).
Precisely how expression of ACP–GFP–mROP1 causes the dramatic changes observed in apicoplast morphology remains unknown. It is possible that getting stuck during translocation across the apicoplast membranes interferes with import mechanisms (although the import and processing of native ACP appear normal in parasites expressing ACP–GFP–mROP1 and retaining the plastid; not shown). Import of the ACP domain of the fusion protein could result in internalization of apicoplast membranes into the organelle in association with the mROP1 domain, producing the membranous inclusions observed by electron microscopy (Figure 2).
Disruption of apicoplast segregation may be a consequence of the presence of multiple large inclusions within the organelle (Robertson et al., 1995). The apicoplast is closely associated with the centriole in the apical juxtanuclear region of parasite cells (Striepen et al., 2000). The centriole divides early in the mitotic process, and the apicoplast becomes elongated as the two daughter centrioles migrate to opposite sides of the nucleus, associated with the ends of the intranuclear spindle. Anchored to the centrioles, the apicoplast is ‘cut’ in two (without any obvious fission ring) by the microtubule-dependent growth of daughter parasite pellicles within the mother. Time-lapse imaging of parasites expressing ACP–GFP–mROP1 reveals early stages of apicoplast elongation in dividing parasites (Figure 3; t = 70 min), but the organelle never becomes fully elongated, ultimately associating with only one of the daughter parasites. In extreme cases, the partially elongated (but non-segregating) apicoplast is pushed all the way to the posterior end of a dividing parasite as the daughter cell pellicle assembles, a localization never seen in control parasites.
ACP–GFP–mROP1 expression peaks ∼20 h after transient transfection, declining as the non-replicating plasmid is diluted and/or degraded. Inhibition of apicoplast segregation is maintained for many days, however, presumably because it is difficult to restore normal morphology to a mutant organelle. As noted above, apicoplast-deficient parasites die in the second host cell, while those that retain an apicoplast survive and divide, with the organelle growing larger and larger (Figure 3, bottom). Indefinite growth of the apicoplast is clearly impossible, but microscopic imaging of parasites in the second host cell or later occasionally reveals, in addition to one parasite containing a giant apicoplast, small clusters of parasites that contain an apparently normal apicoplast within some vacuoles. It is likely that the large, amorphous apicoplast occasionally fragments during cell division, producing parasites that are effectively identical to wild type (provided that they acquire at least one apicoplast genome). These parasites grow more rapidly than parasites containing giant apicoplasts (Figure 6), forming plaques and eventually taking over the culture during long-term cultivation.
The primary effects of ACP–GFP–mROP1 expression appear to be restricted to the apicoplast. The morphology and segregation of other organelles are normal (Figure 2). There is no evidence that ACP–GFP–mROP1 traffics to the rhoptries (despite the presence of ROP1 coding sequence), and rhoptry protein secretion appears normal in apicoplast-deficient parasites (as assessed by immunostaining; not shown). The ability of the ‘super-bright’ parasites expressing ACP–GFP–mROP1 to infect host cells and grow normally also argues that the rhoptries function normally in these parasites.
The ‘delayed death’ phenotype and function of the apicoplast
Growth assays performed on parasites in which the apicoplast has been inactivated by pharmacological treatment reveal a peculiar kinetics of cell death, in which parasites do not die until after entering the second host cell following drug treatment (Fichera et al., 1995; Sullivan et al., 2000). The apicoplast segregation mutants provide a completely independent cell biological validation of this ‘delayed death phenotype’. Apicoplast-deficient cells expressing ACP–GFP–mROP1 replicate normally in the first host cell, but the growth of FACS-sorted dark parasites slows immediately upon entry into a second host cell. Meanwhile, their green, apicoplast-containing sisters replicate nearly as rapidly as wild-type parasites.
The observation that parasite replication is slowed only upon entry into the next host cell after loss of the apicoplast (induced by either pharmacological or cell biological means) suggests that the apicoplast may be required for establishment of the parasitophorous vacuole. Apicoplast-deficient parasites are clearly capable of parasitophorous vacuole formation (Figure 6), but this vacuole may be dysfunctional in mediating communication between the host and parasite. Previous studies have found no difference in the permeability characteristics of drug-treated parasites relative to wild type, however (Fichera, 1998). Nor have any differences been observed in the production, processing and secretion of rhoptry, microneme or dense granule proteins in plastid-deficient parasites, or in the association of these proteins with the parasitophorous vacuole (data not shown). Interaction of the parasitophorous vacuole with host cell mitochondria and endoplasmic reticulum also appears identical to wild type. Subtle changes may occur in parasitophorous vacuole lipid composition, however, and it is interesting to note that lipid synthesis has been proposed as one metabolic function of the apicoplast (Waller et al., 1998; Jomaa et al., 1999).
The ability to isolate plastid-deficient and ‘super-apicoplast’-containing parasites provides useful reagents for the further characterization of this organelle, including organelle purification, analysis by two-dimensional gel electrophoresis, mass spectroscopy, etc. These reagents are also very helpful for functional studies. For example, the inability of plastid-deficient parasites to proliferate in host cells co-infected with wild-type parasites demonstrates that apicoplast function cannot operate in trans (not shown). Preliminary experiments also indicate that treatment of apicoplast-deficient parasites with ciprofloxacin or clindamycin (antibiotics that specifically target apicoplast DNA and protein synthesis; Fichera and Roos, 1997) fails to exacerbate replication defects observed in the second host cell, providing an assay for testing the function of drugs thought to target the apicoplast specifically. Much current research is devoted to mining of the T.gondii EST and P.falciparum genome databases, with an eye towards identifying nuclear-encoded apicoplast proteins required for the metabolic functions of this organelle (Waller et al., 1998; Jomaa et al., 1999). Comparing apicoplast-containing parasites with apicoplast-deficient mutants will greatly facilitate the analysis of potential metabolic pathways associated with the apicoplast.
Materials and methods
Parasites and host cells
The RH strain of T.gondii tachyzoites was maintained by serial passage in human foreskin fibroblast (HFF) cell monolayers, and plaque assays were carried out as previously described (Roos et al., 1994). To measure the growth rate of intracellular parasites (Fichera et al., 1995), confluent monolayers grown on 1 cm #1 glass coverslips in 24-well plates were inoculated with 2 × 104 parasites/well. After incubation at 37°C for 2 h to allow parasite invasion, cultures were rinsed with fresh medium to remove extracellular parasites. One coverslip was then fixed and stained with anti-ACP (Waller et al., 1998) every 12 h. Parasite growth was assayed by counting the number of parasites per parasitophorous vacuole for at least 50 vacuoles from multiple, randomly selected fields on each coverslip, and the presence/absence of ACP was monitored in parallel. Averages and standard errors were calculated for each sample at each time point (Zar, 1996).
Molecular methods
Plasmids ACP–GFP and ROP1–GFP have been described previously (Striepen et al., 1998; Waller et al., 1998). The ACP–GFP–mROP1 fusion was made by inserting a myc epitope tag followed by sequences encoding ROP1 amino acids 139–338 (the rhoptry targeting domain of this 389 amino acid protein) downstream of GFP coding sequences in the ACP–GFP plasmid. Fifty micrograms of plasmid DNA (Qiagen Maxi-preps) and 107 freshly lysed-out tachyzoites were used for each transfection, using previously described protocols (Roos et al., 1994). In transient assays, fusion protein expression was typically examined 24 h post-transfection. Stable transformants were selected in the presence of 20 µM chloramphenicol (for plasmids expressing CAT) or by co-transfection with plasmid pDHFR-TSc3 (Donald and Roos, 1993) and selection in 1 µM pyrimethamine.
Light microscopy
GFP, RFP, fluorescein isothiocyanate (FITC) and DAPI fluorescence were detected using a Zeiss Axiovert 35 inverted microscope equipped with a 100 W Hg-vapor lamp. For immunofluorescence assays, parasites were fixed with 3.7% paraformaldehyde, permeabilized with 0.25% Triton X-100 and blocked with 1% bovine serum albumin in phosphate-buffered saline (PBS) pH 7.4 at room temperature. GFP was detected using a polyclonal antiserum (Clontech; 1:500 dilution) followed by FITC-conjugated goat anti-rabbit IgG (Sigma; 1:160). Apicoplast DNA was visualized by staining with DAPI (Molecular Probes).
The growth and division of parasites were monitored by video-enhanced microscopy using a digital CCD camera (Hamamatsu). For long-term observation of living samples, parasites and monolayers were maintained at 37°C using a ΔTC3 culture dish system (Bioptechs). Neutral pH was maintained by addition of 10 mM HEPES pH 7.4 (GIBCO-BRL) to the culture medium. In time-lapse experiments, both fluorescence and phase images were taken every 5 min, using an automated program (Openlab; Improvision).
Electron microscopy
Infected cells were fixed in situ with a freshly prepared mixture containing 1% glutaraldehyde (8% stock; Electron Microscopy Sciences) and 1% OsO4 in 50 mM phosphate buffer pH 6.2. Fixative was added at room temperature and samples placed on ice for 45 min. Samples were then rinsed with distilled water to remove excess phosphate, released from the plastic substratum by gentle scraping, pelleted, and stained with 0.5% aqueous uranyl acetate for 6–16 h at 4°C. Following dehydration in acetone and embedding in Epon–Araldite, ultra-thin sections (50–70 nm) were cut and stained with uranyl acetate and lead citrate, and examined using a Philips 200 electron microscope.
For immunoelectron microscopy, infected cells were fixed in situ with a mixture of 4% formaldehyde and 0.5% glutaraldehyde in 100 mM phosphate buffer pH 7.0 for 1–2 h at 4°C, harvested by scraping, pelleted, dehydrated through increasing concentrations of ethanol to 70% and embedded in LR White (Electron Microscopy Sciences). Polymerization was carried out at 37°C for 5 days. Sections (50–70 nm) were cut and mounted on uncoated nickel grids. Sections were incubated with anti-GFP (Seedorf et al., 1999; diluted 1:100 in PBS–glycine), followed by a goat anti-rabbit IgG conjugated to 5 nm gold particles (British BioCell International; 1:100 dilution). Sections were stained with 1% uranyl acetate in 30% methanol and examined as above.
Flow cytometry
Freshly lysed-out tachyzoites were filtered through 3 µm pore size polycarbonate filters (Nuclepore) and centrifuged at 1500 g for 15 min. The parasite pellet was then resuspended in culture medium, counted using a hemocytometer and adjusted to 5 × 106 parasites/ml. FACS assays were performed on a FACStar Plus cell sorter (Becton Dickinson) equipped with CELLQUEST software.
Acknowledgments
Acknowledgements
We wish to thank Andrew Morschauser and Richard Schretzenmair for help with FACS analysis, Ross F.Waller for providing anti-ACP antibodies, John M.Murray and Helen L.Compton for helpful advice, and Martin Fraunholz and Stuart Ralph for critical reading of the manuscript. This work was supported by grants from the NIH; D.S.R. is a Burroughs Wellcome Scholar in Molecular Parasitology.
References
- Ajioka J.W. (1998) Toxoplasma gondii: ESTs and gene discovery. Int. J. Parasitol., 28, 1025–1031. [DOI] [PubMed] [Google Scholar]
- Blanchard J.L. and Hicks,J.S. (1999) The non-photosynthetic plastid in malarial parasites and other apicomplexans is derived from outside the green plastid lineage. J. Eukaryot. Microbiol., 46, 367–375. [DOI] [PubMed] [Google Scholar]
- Bowman S. et al. (1999) The complete nucleotide sequence of chromosome 3 of Plasmodium falciparum. Nature, 400, 532–538. [DOI] [PubMed] [Google Scholar]
- Clough B., Strath,M., Preiser,P., Denny,P. and Wilson,I.R. (1997) Thiostrepton binds to malarial plastid rRNA. FEBS Lett., 406, 123–125. [DOI] [PubMed] [Google Scholar]
- Donald R.G.K. and Roos,D.S. (1993) Stable molecular transformation of Toxoplasma gondii: a selectable dihydrofolate reductase-thymidylate synthase marker based on drug-resistance mutations in malaria. Proc. Natl Acad. Sci. USA, 90, 11703–11707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey J.P. and Welcome,F.L. (1988) Toxoplasma gondii-induced abortion in sheep. J. Am. Vet. Med. Assoc., 193, 697–700. [PubMed] [Google Scholar]
- Dzierszinski F., Popescu,O., Toursel,C., Slomianny,C., Yahiaoui,B. and Tomavo,S. (1999) The protozoan parasite Toxoplasma gondii expresses two functional plant- like glycolytic enzymes. Implications for evolutionary origin of apicomplexans. J. Biol. Chem., 274, 24888–24895. [DOI] [PubMed] [Google Scholar]
- Endo T. and Schatz,G. (1988) Latent membrane perturbation activity of a mitochondrial precursor protein is exposed by unfolding [published erratum appears in EMBO J. (1988), 7, 1915]. EMBO J., 7, 1153–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fichera M.E. (1998) The apicoplast—a novel organelle in apicomplexan parasites is a target for clindamycin and other antibiotics. PhD dissertation, University of Pennsylvania, Philadelphia, PA, pp. 142 + xiv.
- Fichera M.E. and Roos,D.S. (1997) A plastid organelle as a drug target in apicomplexan parasites. Nature, 390, 407–409. [DOI] [PubMed] [Google Scholar]
- Fichera M.E., Bhopale,M.K. and Roos,D.S. (1995) In vitro assays elucidate peculiar kinetics of clindamycin action against Toxoplasma gondii. Antimicrob. Agents Chemother., 39, 1530–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner M.J. (1999) The genome of the malaria parasite. Curr. Opin. Genet. Dev., 9, 704–708. [DOI] [PubMed] [Google Scholar]
- Haberkorn A. (1996) Chemotherapy of human and animal coccidioses: state and perspectives. Parasitol. Res., 82, 193–199. [DOI] [PubMed] [Google Scholar]
- Jomaa H. et al. (1999) Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science, 285, 1573–1576. [DOI] [PubMed] [Google Scholar]
- Kim K., Soldati,D. and Boothroyd,J.C. (1993) Gene replacement in Toxoplasma gondii with chloramphenicol acetyltransferase as selectable marker. Science, 262, 911–914. [DOI] [PubMed] [Google Scholar]
- Köhler S., Delwiche,C.F., Denny,P.W., Tilney,L.G., Webster,P., Wilson,R.J., Palmer,J.D. and Roos,D.S. (1997) A plastid of probable green algal origin in apicomplexan parasites. Science, 275, 1485–1489. [DOI] [PubMed] [Google Scholar]
- Levine N.D. (1988) Progress in taxonomy of the apicomplexan protozoa. J. Protozool., 35, 518–520. [DOI] [PubMed] [Google Scholar]
- Lingelbach K. and Joiner,K.A. (1998) The parasitophorous vacuole membrane surrounding Plasmodium and Toxoplasma: an unusual compartment in infected cells. J. Cell Sci., 111, 1467–1475. [DOI] [PubMed] [Google Scholar]
- Luft B.J. and Remington,J.S. (1992) Toxoplasmic encephalitis in AIDS. Clin. Infect. Dis., 15, 211–222. [DOI] [PubMed] [Google Scholar]
- Luft B.J. et al. (1993) Toxoplasmic encephalitis in patients with the acquired immunodeficiency syndrome. Members of the ACTG 077p/ANRS 009 Study Team. N. Engl. J. Med., 329, 995–1000. [DOI] [PubMed] [Google Scholar]
- McFadden G.I. and Roos,D.S. (1999) Apicomplexan plastids as drug targets. Trends Microbiol., 7, 328–333. [DOI] [PubMed] [Google Scholar]
- McFadden G.I. and Waller,R.F. (1997) Plastids in parasites of humans. BioEssays, 19, 1033–1040. [DOI] [PubMed] [Google Scholar]
- McFadden G.I., Waller,R.F., Reith,M.E. and Lang-Unnasch,N. (1997) Plastids in apicomplexan parasites. In Bhattacharya,D. (ed.), Origins of Algae and their Plastids. Springer-Verlag, New York, NY, pp. 261–287.
- Ogino N. and Yoneda,C. (1966) The fine structure and mode of division of Toxoplasma gondii. Arch. Ophthalmol., 75, 218–227. [DOI] [PubMed] [Google Scholar]
- Ossorio P.N., Schwartzman,J.D. and Boothroyd,J.C. (1992) A Toxoplasma gondii rhoptry protein associated with host cell penetration has unusual charge asymmetry. Mol. Biochem. Parasitol., 50, 1–15. [DOI] [PubMed] [Google Scholar]
- Palmer J.D. and Delwiche,C.F. (1996) Second-hand chloroplasts and the case of the disappearing nucleus. Proc. Natl Acad. Sci. USA, 93, 7432–7435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persidis A. (2000) Malaria. Nature Biotechnol., 18, 111–112. [DOI] [PubMed] [Google Scholar]
- Pfefferkorn E.R., Nothnagel,R.F. and Borotz,S.E. (1992) Parasiticidal effect of clindamycin on Toxoplasma gondii grown in cultured cells and selection of a drug-resistant mutant. Antimicrob. Agents Chemother., 36, 1091–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricketts A.P. and Pfefferkorn,E.R. (1993) Toxoplasma gondii: susceptibility and development of resistance to anticoccidial drugs in vitro. Antimicrob. Agents Chemother., 37, 2358–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson E.J., Pyke,K.A. and Leech,R.M. (1995) arc6, an extreme chloroplast division mutant of Arabidopsis also alters proplastid proliferation and morphology in shoot and root apices. J. Cell Sci., 108, 2937–2944. [DOI] [PubMed] [Google Scholar]
- Rogers M.J., Cundliffe,E. and McCutchan,T.F. (1998) The antibiotic micrococcin is a potent inhibitor of growth and protein synthesis in the malaria parasite. Antimicrob. Agents Chemother., 42, 715–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roizen N. et al. (1995) Neurologic and developmental outcome in treated congenital toxoplasmosis. Pediatrics, 95, 11–20. [PubMed] [Google Scholar]
- Roos D.S., Donald,R.G.K., Morrissette,N.S. and Moulton,A.L.C. (1994) Molecular tools for genetic dissection of the protozoan parasite Toxoplasma gondii. Methods Cell Biol., 45, 27–63. [DOI] [PubMed] [Google Scholar]
- Roos D.S., Crawford,M.J., Donald,R.G.K., Fohl,L.M., Hager,K.M., Kissinger,J.C., Reynolds,M.G., Striepen,B. and Sullivan,W.J.,J. (1999a) Transport and trafficking: Toxoplasma as a model for Plasmodium. Novartis Found. Symp., 226, 176–195. [DOI] [PubMed] [Google Scholar]
- Roos D.S., Crawford,M.J., Donald,R.G.K., Kissinger,J.C., Klimczak,L.J. and Striepen,B. (1999b) Origin, targeting and function of the apicomplexan plastid. Curr. Opin. Microbiol., 2, 426–432. [DOI] [PubMed] [Google Scholar]
- Saffer L.D., Mercereau-Puijalon,O., Dubremetz,J.F. and Schwartzman,J.D. (1992) Localization of a Toxoplasma gondii rhoptry protein by immunoelectron microscopy during and after host cell penetration. J. Protozool., 39, 526–530. [DOI] [PubMed] [Google Scholar]
- Seedorf M., Damelin,M., Kahana,J., Taura,T. and Silver,P.A. (1999) Interactions between a nuclear transporter and a subset of nuclear pore complex proteins depend on Ran GTPase. Mol. Cell. Biol., 19, 1547–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldati D., Lassen,A., Dubremetz,J.F. and Boothroyd,J.C. (1998) Processing of Toxoplasma ROP1 protein in nascent rhoptries. Mol. Biochem. Parasitol., 96, 37–48. [DOI] [PubMed] [Google Scholar]
- Striepen B., He,C.Y., Matrajt,M., Soldati,D. and Roos,D.S. (1998) Expression, selection and organellar targeting of the green fluorescent protein in Toxoplasma gondii.Mol. Biochem. Parasitol., 92, 325–338. [DOI] [PubMed] [Google Scholar]
- Striepen B., Crawford,M.J., Shaw,M.K., Tilney,L.G., Seeber,F. and Roos,D.S. (2000) The plastid of Toxoplasma gondii is divided using the mitotic spindle. J. Cell Biol., 151, 1423–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan M., Li,J., Kumar,S., Rogers,M.J. and McCutchan,T.F. (2000) Effects of interruption of apicoplast function on malaria infection, development and transmission. Mol. Biochem. Parasitol., 109, 17–23. [DOI] [PubMed] [Google Scholar]
- Suss-Toby E., Zimmerberg,J. and Ward,G.E. (1996) Toxoplasma invasion: the parasitophorous vacuole is formed from host cell plasma membrane and pinches off via a fission pore. Proc. Natl Acad. Sci. USA, 93, 8413–8418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller R.F. et al. (1998) Nuclear-encoded proteins target to the plastid in Toxoplasma gondii and Plasmodium falciparum. Proc. Natl Acad. Sci. USA, 95, 12352–12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller R.F., Reed,M.B., Cowman,A.F. and McFadden,G.I. (2000) Protein trafficking to the plastid of Plasmodium falciparum is via the secretory pathway. EMBO J., 19, 1794–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warhurst D.C. (1999) Drug resistance in Plasmodium falciparum malaria. Infection, 27, S55–S58. [DOI] [PubMed] [Google Scholar]
- White N.J. (1998) Drug resistance in malaria. Br. Med. Bull., 54, 703–715. [DOI] [PubMed] [Google Scholar]
- Wilson R.J. et al. (1996) Complete gene map of the plastid-like DNA of the malaria parasite Plasmodium falciparum. J. Mol. Biol., 261, 155–172. [DOI] [PubMed] [Google Scholar]
- Zar J.H. (1996) Biostatistical Analysis, 3rd edn. Prentice Hall, Upper Saddle River, NJ.
- Zhu G., Marchewka,M.J. and Keithly,J.S. (2000) Cryptosporidium parvum appears to lack a plastid genome. Microbiology, 146, 315–321. [DOI] [PubMed] [Google Scholar]