

Abstract
Transforming growth factor-β (TGF-β) signals through membrane-bound serine/threonine kinase receptors, which upon stimulation phosphorylate Smad proteins and thereby trigger their nuclear translocation and transcriptional activity. Although the three mammalian isoforms of TGF-β are highly homologous at the level of sequence, analysis of their in vivo function by gene knockouts revealed striking differences, suggesting no significant functional redundancy between TGF-β1, -2 and -3. While signal transduction by TGF-β1 has been well characterized, receptor binding and activation by the TGF-β2 isoform is less well understood. Here, we show that TβRII-B, an alternatively spliced variant of the TGF-β type II receptor, is a TGF-β2 binding receptor, which mediates signalling via the Smad pathway in the absence of any TGF-β type III receptor (TβRIII). L6 cells lacking endogenous TβRIII as well as TβRII-B do not respond to TGF-β2. Transfection of these cells with TβRII-B restores TGF-β2 sensitivity. The expression of TβRII-B is restricted to cells originating from tissues such as bone where the isoform TGF-β2 has a predominant role. This reflects the importance of this receptor in TGF-β isoform-specific signalling.
Keywords: alternative splicing/osteoblast TGF-β/TGF-β receptor
Introduction
Transforming growth factor-β (TGF-β) is a member of a large family of structurally related cytokines. The family consists of >30 ligand proteins regulating a wide variety of biological processes, such as proliferation, differentiation and cell death (Roberts and Sporn, 1993). The phenotypes resulting from the knockout of the three mammalian TGF-β isoforms TGF-β1, TGF-β2 and TGF-β3 are very distinct and not overlapping. TGF-β1 null mice have an autoimmune-like inflammatory disease (Schull et al., 1992; Kulkarni and Karlsson, 1993; Diebold et al., 1995), TGF-β2 knockout mice exhibit perinatal mortality and severe developmental defects (Sanford et al., 1997) and TGF-β3-deficient mice have cleft palate and are defective in lung development (Kaartinen et al., 1995; Proetzel et al., 1995). This indicates that these ligands have isoform-specific activities that cannot be compensated by other family members.
Signalling via TGF-β1 is initiated by binding of TGF-β1 to the constitutive active serine/threonine kinase receptor TβRII (TGF-β type II receptor). Upon ligand binding, the TGF-β type I receptor (TβRI) is recruited into the hetero-oligomeric signalling complex and subsequently TβRII activates TβRI by transphosphorylation at its cytoplasmic GS box (Wrana et al., 1994a). Activated TβRI transiently associates with cytoplasmic effectors, the Smad proteins, which become phosphorylated at their C-terminus and dissociate from the receptor. Upon complex formation with Smad4, these hetero-oligomeric Smad complexes are translocated into the nucleus to regulate transcription (Piek et al., 1999; Massagué and Chen, 2000).
In contrast to TGF-β1, signalling by TGF-β2 seems to have a different mode of receptor activation, since TβRII has a low intrinsic affinity to this isoform (Cheifetz and Massagué, 1991; Lin et al., 1995). The requirement of the type III receptor (TβRIII) for responsiveness to TGF-β2 has been described in different cell types (Lopez-Casillas et al., 1993; Sankar et al., 1995; Brown et al., 1999). TβRIII binds the ligand TGF-β2 and presents it to TβRII upon oligomerization of both receptor types (Lopez-Casillas et al., 1993). However, it is still unclear why direct binding of TGF-β1 to TβRII does not have the same effect. Therefore, it was proposed that TGF-β2 alters the composition or activity of TβRII–TβRI complexes in order to activate a unique set of downstream signalling molecules that result in specific TGF-β2 effects (Brown et al., 1999).
Here we describe and functionally characterize an isoform of the type II receptor, TβRII-B, which binds and signals directly via the TGF-β2 isoform without the requirement for TβRIII. TβRII-B is an alternatively spliced variant of TβRII resulting in N-terminal alterations of the mature receptor. Unlike TβRII this splicing variant shows a restricted expression pattern. The site of predominant expression includes osteoblasts and mesenchymal precursor cells, which correlates with the unique expression of TGF-β2 in chondrocytes and osteocytes (Pelton et al., 1991).
Results
Isolation of the TβRII-B cDNA clone
RT–PCR was used to screen for variants of the TGF-β type II receptor showing alterations in the extracellular domain. Upon amplification of cDNA from the human hepatoma cell line Hep3B, an additional PCR product with lower mobility was detected (Figure 6A, lane 9). Sequence analysis revealed that this PCR product is identical to TβRII-B, an alternatively spliced variant of TβRII described previously (Nikawa, 1994; Hirai and Fijita, 1996). The alternative splicing causes an insertion of 26 amino acids at the N-terminus of the mature receptor, replacing Val32 (Figure 1A).
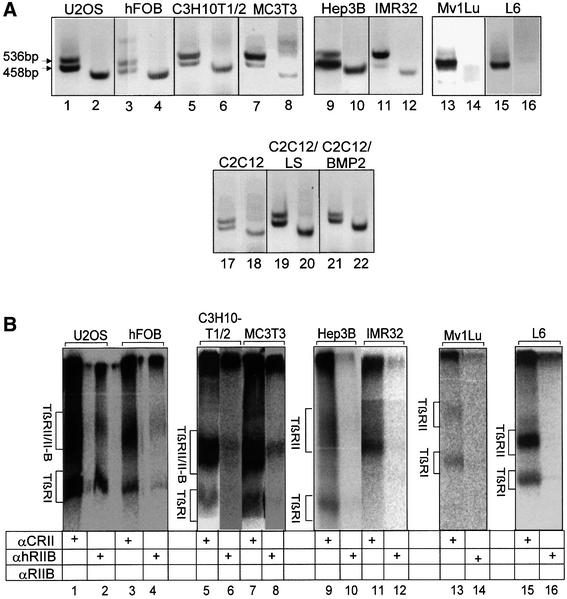
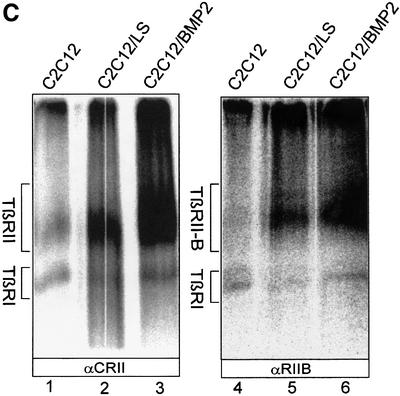
Fig. 6. Restricted expression pattern of TβRII-B. (A) RT–PCR analysis of TβRII-B mRNA in different cell lines. The cDNAs were prepared from human osteosarcoma cells (U2OS), human fetal osteoblasts (hFOB), murine mesenchymal precursor cells MC3T3, C3H10T1/2 cells and C2C12 myoblasts, the human hepatoma cell line Hep3B, human neuroblastoma cells (IMR32), Mv1Lu cells and rat myoblasts (L6). PCR products were obtained using the primers P1 and P5 (odd lane numbers) or the TβRII-B-specific primers Pins combined with P5 (even lane numbers). Two PCR products using P1/P5 (for example, lane 1) as well as a single PCR product using Pins/P5 (for example, lane 2) indicate the presence of TβRII-B mRNA. A single PCR product using P1/P5 (lanes 13 and 15) and no PCR product using Pins/P5 (lanes 14 and 16) indicate the expression of only TβRII mRNA. C2C12 cells were analysed either undifferentiated (lanes 17 and 18) or after differentiation in low serum (LS; lanes 19 and 20) or in LS containing 40 nM BMP-2 (lanes 21 and 22). (B) Endogenous expression of TβRII and TβRII-B at the cell surface of different cell lines was detected by affinity labelling with [125I]TGF-β1. Cell lysates were immunoprecipitated either with α-CRII (odd lane numbers), the antibody specific for the human TβRII-B, α-hRIIB (lanes 2, 4, 10 and 12) or α-RIIB, which recognizes also the murine TβRII-B (lanes 6, 8, 14 and 16). Hep3B, IMR32, Mv1Lu and L6 cells do not show any TβRII-B protein at the cell surface. (C) Upregulation of TGF-β receptors during differentiation of C2C12 cells. Cell surface expression of TGF-β type II receptors (lanes 1–3) and specifically TβRII-B (lanes 4–6) was determined after affinity labelling using iodinated TGF-β1 on C2C12 cells, which are either undifferentiated (lanes 1 and 4), differentiated into multinucleated myotubes (lanes 2 and 5) or differentiated into the osteoblast lineage by BMP-2 (lanes 3 and 6).
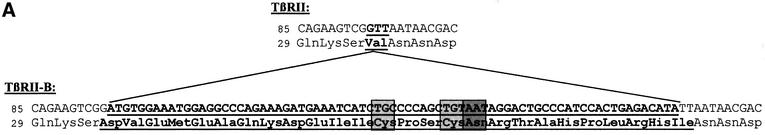
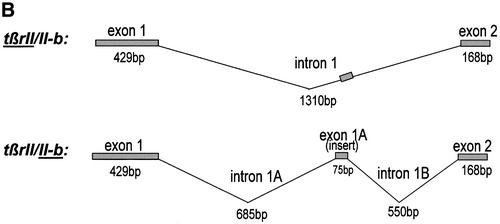
Fig. 1. TGFβ type II-B receptor is an alternatively spliced form of TβRII. (A) The amino acid sequence of TβRII-B compared with TβRII contains an insert of 26 amino acids after Ser31, replacing Val32 of TβRII. The insertion sequence of human TβRII-B is underlined. A potential N-linked glycosylation site (Asn48) and two Cys residues (Cys44, Cys47) are shown in shaded boxes. (B) Schematic outline of alternative splicing of the tβrII-intron 1 resulting in an additional exon, exon 1A.
In order to analyse the exon–intron structure of tβrII-b, PCR analysis of genomic DNA from human placenta was performed using insert-specific primers (data not shown). We were able to localize the insert as an additional exon (exon 1A) within intron 1 (Figure 1B).
Unlike TβRII, TβRII-B binds all three TGF-β isoforms
TβRII is known to bind the isoforms TGF-β1 and TGF-β3. Binding of these ligands causes recruitment of the type I receptor (TβRI) into a signalling receptor complex followed by activation of TβRI through transphosphorylation (Wrana et al., 1992; Moustakas et al., 1993; Wrana et al., 1994a; ten Dijke et al., 1996; Wells et al., 1999). The isoform TGF-β2, however, does not follow this mode of receptor binding and oligomerization, at least not by using these receptors. TβRII does not bind the isoform TGF-β2 when expressed alone (Lin et al., 1995).
To study binding of different TGF-β isoforms to TβRII-B we performed binding and crosslinking analysis of radiolabelled ligands on COS-7 cells transfected with either TβRII or TβRII-B. The receptors were immunoprecipitated from cell lysates using the antiserum α-CRII, which detects both type II receptors (Figure 2, lanes 1–7 and 9). Both receptors bind the isoforms TGF-β1 and TGF-β3 indistinguishably. However, binding of the β2 isoform is strikingly different. TβRII-B binds TGF-β2 even in the absence of TβRI or TβRIII (Figure 2, lane 4), which suggests distinct binding properties of TGF-β2. This is different to the cooperative binding mode postulated for TGF-β2 via preformed complexes of TβRII with TβRI or TβRIII (Rodriguez et al., 1995; Massagué, 1998). Accordingly, other studies have shown that the majority of the type I and type II receptors for TGF-β exist as homodimers and not hetero-oligomers at the cell surface in the absence of ligand (Gilboa et al., 1998; Wells et al., 1999).
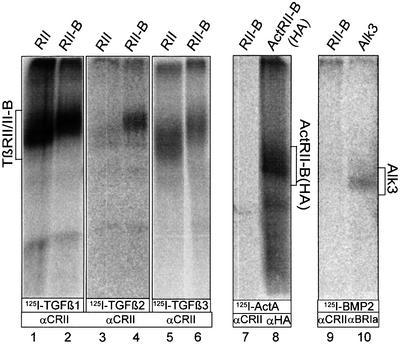
Fig. 2. All three TGFβ isoforms bind TβRII-B. COS-7 cells transfected with TβRII or TβRII-B were affinity labelled with [125I]TGF-β1 (lanes 1 and 2), [125I]TGF-β2 (lanes 3 and 4) or [125I]TGF-β3 (lanes 5 and 6), crosslinked and immunoprecipitated with α-CRII, an antibody raised against the C-terminus of both type II receptors. Unlike TβRII, TβRII-B binds the isoform TGF-β2, when expressed singly in COS-7 cells (lanes 4 and 3). In contrast, iodinated activin A (lane 7) or BMP-2 (lane 9) does not bind to TβRII-B, but do bind to their respective high-affinity receptors ActRII-B (lane 8) and ALK3 (lane 10).
TβRII-B forms complexes with TβRI, TβRII and TβRIII
It has been shown before that addition of ligand induces hetero-oligomeric complexes of the known TGF-β receptors (TβRI–TβRII, TβRIII–TβRII) (Wrana et al., 1992, 1994a; Henis et al., 1994; Gilboa et al., 1998; Wells et al., 1999). In order to analyse complex formation of TβRII-B with the known TGF-β receptors at the cell surface, we performed ligand binding and crosslinking experiments in transiently transfected COS-7 cells expressing various combinations of TGF-β receptors. TβRII-B interacts with TβRI in the presence of each of the three TGF-β isoforms (Figure 3A, lanes 2, 4 and 6). The interaction of TβRII-B with TβRIII through TGF-β1 and TGF-β2 is shown in Figure 3B (lanes 2 and 6). Even though TβRII-B is not dependent on complexes with TβRIII for its binding of TGF-β2, hetero-oligomers of both receptor types are detected. In contrast, TβRII binds TGF-β2 only when co-expressed with TβRIII (compare lanes 3 and 5). This is observed as well in cells expressing endogenous TGF-β receptors. The cell line R1b/L17 lacks endogenous TβRI (Laiho et al., 1990) and, as we show later, also TβRII-B. Binding of TGF-β2 to TβRII (Figure 3C, lane 2) results from complex formation with TβRIII. These complexes are essential for TGF-β2 binding to TβRII. Since Mv1Lu cells do not express any TβRII-B (Figure 6A and B, lanes 13 and 14) the existence of TβRIII in these cells seems to be absolutely necessary for binding and signalling via TGF-β2.
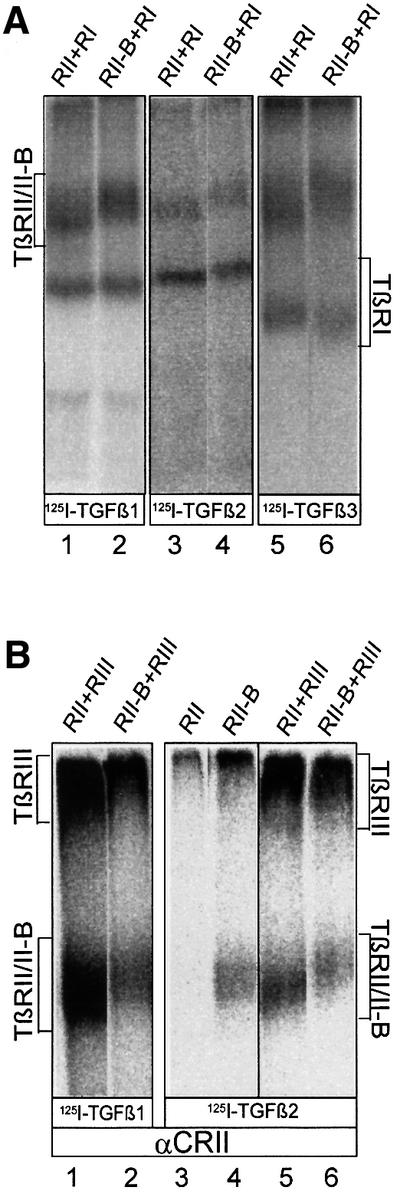
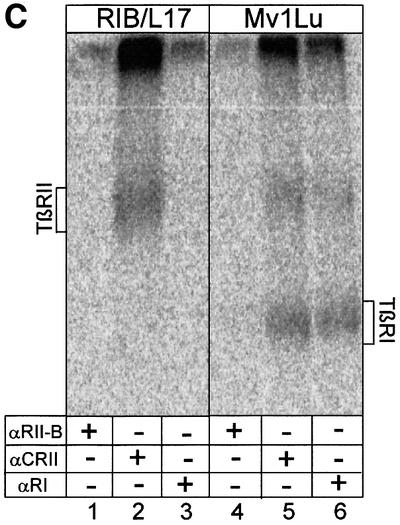
Fig. 3. Binding and complex formation of TβRII-B upon co-expression with TβRI or TβRIII. (A) Receptor complexes containing TβRII and TβRI or TβRII-B and TβRI were detected after binding and crosslinking of [125I]TGF-β1 (lanes 1 and 2), [125I]TGF-β2 (lanes 3 and 4) and [125I]TGF-β3 (lanes 5 and 6) by immunoprecipitation with α-CRII. Receptor combinations are indicated above each lane. (B) COS-7 cells were transiently transfected with plasmids encoding TβRII or TβRII-B alone, or cotransfected with TβRIII (indicated above each lane). After affinity labelling with [125I]TGF-β1 (lanes 1 and 2) or [125I]TGF-β2 (lanes 3–6) receptors were detected by immunoprecipitation with α-CRII. The positions of ligand-bound TβRII, TβRII-B and TβRIII are indicated. Both type II receptors interact with TβRIII in the presence of TGF-β1 (lanes 1 and 2) or TGF-β2 (lanes 5 and 6). TβRII can bind to TGF-β2 only if co-expressed with TβRIII (lane 5), but not without any associated receptor (lane 3). TGF-β2 binding to TβRII-B is not dependent on the formation of receptor complexes (lane 4). (C) Binding of [125I]TGF-β2 to TGF-β receptors at the cell surface of Mv1Lu and R1b/L17 cells. Immunoprecipitations were performed using the TβRII-B-specific antibody, α-RII-B (lanes 1 and 4), the RII/RII-B antibody, α-CRII (lanes 2 and 5) and the type I receptor antibody, α-RI (lanes 3 and 6). R1b/L17 cells lack TβRI, whereas Mv1Lu-cells do not (lanes 3 and 6).
To study the oligomerization of the two TGF-β type II receptors TβRII and TβRII-B, we used HA-epitope-tagged TβRII cotransfected with untagged TβRII-B. Each of these receptors carry in addition to the common epitope (detected by α-CRII) at least one specific epitope (recognized by α-hRIIB or by α-RIIB for TβRII-B, and by α-HA for HA-TβRII). Sequential immunoprecipitations from cell lysates were performed after binding and crosslinking with the indicated iodinated ligands to show that TβRII and TβRII-B form complexes in the presence of either isoform (Figure 4, lanes 4, 7 and 8). While heteromeric complexes were detected using first α-hRIIB and then α-HA, we were unable to detect these with the reverse experimental set-up (first α-HA, second α-hRIIB; see Figure 4, lane 3). One possible explanation is that the antibody α-hRIIB does not recognize its epitope under the conditions used in the second immunoprecipitation (Figure 4, lanes 5 and 6). There is no cross-reactivity of the antisera α-hRIIB or α-RIIB with TβRII, as tested by immunoprecipitations of affinity-labelled TβRII (data not shown).
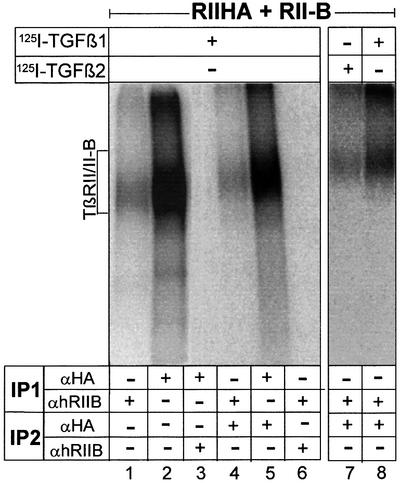
Fig. 4. Type II/II-B receptor hetero-oligomers are detected at the cell surface after ligand binding. COS-7 cells were cotransfected with HA-epitope-tagged TβRII and non-tagged TβRII-B. Binding and crosslinking were performed with [125I]TGF-β1 (lanes 1–6 and 8) and [125I]TGF-β2 (lane 7). The heteromeric complex of TβRII and TβRII-B was detected by sequential immunoprecipitations (IPs) using the human TβRII-B-specific antibody α-hRIIB in the first IP and the α-HA antibody in second IP (lanes 4, 7 and 8).
We also showed that TβRII/TβRII-B heteromers bind TGF-β2 (Figure 4, lane 7). In this case one TβRII-B receptor chain is enough to facilitate binding of TGF-β2 to both TβRII and TβRII-B, whereas the homomeric form of TβRII is not.
In conclusion, we have demonstrated that TβRII-B interacts with TβRI, TβRII and TβRIII at the cell surface via TGF-β1 and TGF-β2.
Neither alternative disulfide bond formation nor N-glycosylation influences binding properties of TβRII-B to TGF-β2
As illustrated in the sequence of the TβRII-B insert (Figure 1A), two additional cysteines (Cys44 and Cys47) are present in the extracellular domain of TβRII-B. This might enable additional or alternative disulfide bond formation. We have mutated the cysteines to alanines by PCR mutagenesis either individually or both (TβRII-BC44A, TβRII-BC47A, TβRII-BC44AC47A). All constructs were expressed in COS-7 cells and tested for their binding properties. No difference between the mutants and the wild-type TβRII-B was seen with respect to binding of TGF-β2 (Figure 5A, lanes 1–4) or TGF-β1 and to interaction with TβRI (data not shown).
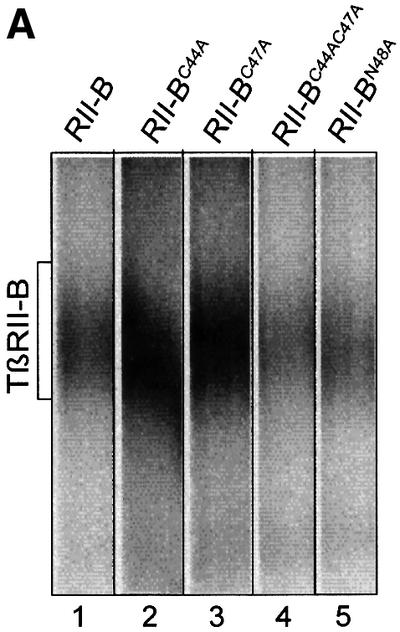
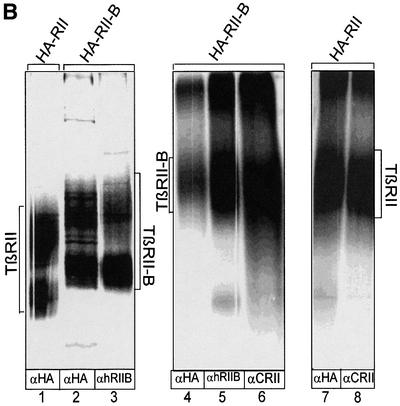
Fig. 5. Neither alternative disulfide bond formation nor N-glycosylation, but addition of N-terminal epitope tags, influences ligand binding to TβRII-B. (A) COS-7 cells were transiently transfected with the wild-type TβRII-B (lane 1) and mutant forms of TβRII-B, where Cys44 (lane 2), Cys47 (lane 3) or Cys44 and Cys47 (lane 4) or Asn48 (lane 5) were mutated to alanine. After binding and crosslinking with [125I]TGF-β2, receptors were immunoprecipitated with α-CRII. The position of ligand-bound TβRII-B is indicated. All four mutants of TβRII-B are able to bind TGF-β2. (B) COS-7 cells were transfected with HA-tagged TβRII or TβRII-B. After metabolic labelling with [35S]cysteine/methionine (lanes 1–3) or binding and crosslinking using [125I]TGF-β1 (lanes 4–8), the receptors were immunoprecipitated using antibodies as indicated. Control immunoprecipitations were carried out with α-hRIIB (lane 5) and α-CRII (lanes 6 and 8).
In addition, the sequence of the insert in TβRII-B shows a potential N-glycosylation site at Asn48 (Figure 1A). Deglycosylation of TβRII by tunicamycin treatment of transfected COS-7 cells has been shown not to affect binding of this receptor to TGF-β1 (Wells et al., 1997). To exclude potential glycosylation at Asn48 of TβRII-B, which might cause binding of TGF-β2 to this receptor, we mutated this residue to alanine, resulting in the mutant TβRII-BN48A. No difference was seen in binding TGF-β2 compared with the wild-type receptor (Figure 5A, lanes 1 and 5). Similar results were obtained using tunicamycin-treated COS-7 cells transfected with the TβRII-B construct (data not shown).
Next, we tagged TβRII-B N-terminal of the insertion with an HA-epitope and examined whether this modification alters ligand binding or whether ligand binding interferes with recognition by the α-HA antibody. Figure 5B (lanes 5 and 6) shows that addition of the epitope does not inhibit ligand binding, but bound and crosslinked TGF-β1 interferes with the accessibility of the epitope for the α-HA antibody (Figure 5B, lane 4). This is not the case for TβRII, if an epitope tag is added also to the very N-terminus (Figure 5B, lane 7). Controls without the ligand (Figure 5B, lanes 1–3) show that the HA-epitope (lane 2) as well as the insert epitope (lane 3) at the N-terminus of TβRII-B are equally accessible to their antibodies. This suggests that the N-terminus of TβRII-B makes major contributions to the binding pocket of TGF-β isoforms.
TβRII-B displays a restricted expression pattern
In order to study the expression of TβRII-B at the RNA and protein level, RT–PCR and binding experiments were performed in cell lines established from different tissues. Surprisingly, depending on the cell type, different scenarios for the expression of TβRII-B were observed: (i) no alternative splicing in Mv1Lu and L6 cells and therefore no TβRII-B expression (Figure 6A, lanes 13–16 and B, lanes 13–16); (ii) alternative splicing but no detectable expression of TβRII-B at the cell surface of Hep3B and IMR32 cells (Figure 6A, lanes 9–12 and B, lanes 9–12); (iii) alternative splicing and expression of TβRII-B at the cell surface of murine mesenchymal precursor cells (MC3T3 and C2C12 cells), human fetal osteoblast (hFOB) and the human osteosarcoma cell line U2OS (Figure 6A, lanes 1–8 and 17–22, B, lanes 1–8 and C).
While TβRII is almost ubiquitously expressed on cells, TβRII-B shows a distinct and specific expression pattern mainly in bone-related cells, such as osteoblasts and mesenchymal precursor cells. The mesenchymal precursor cell line C2C12 can form myotubes when cultivated for 3–5 days in low serum [0.2% fetal calf serum (FCS)]. The addition of 40 nM bone morphogenetic protein (BMP)-2 converts the differentiation of C2C12 cells into the osteoblast lineage (Katagiri et al., 1994). As shown in Figure 6C, TβRII-B is expressed early in the precursor cell line (lanes 1 and 4), but is upregulated during differentiation into myoblasts (lanes 2 and 5) and even more strongly in osteoblasts (lanes 3 and 6).
Taken together, the data here show the restriction of expression of TβRII-B to cells such as osteoblasts, where the TGF-β2 isoform has a specific biological role. In other cell lines such as human hepatoma cells and neuroblastoma cells, the alternative splicing does not result in detectable expression of the receptor at the cell surface. No alternative splicing occurs in a third subset of cells, suggesting a tissue-specific mechanism for splicing.
TβRII-B is a signalling receptor
In order to study signalling of TGF-β2 via the endogenously expressed TβRII-B receptor, we investigated ligand-induced phosphorylation of Smad2, a TGF-β pathway-restricted Smad, which is phosphorylated by activated TβRI (Macias-Silva et al., 1996; Zhang et al., 1996). Two different cell lines have been used, which differ in the composition of their TGF-β receptors. The human osteosarcoma cell line U2OS expresses TβRI, TβRII and TβRII-B (Figure 6A and B), but lacks TβRIII (data not shown). The rat myoblast cell line L6 lacks TβRIII (Wang et al., 1991; Lopez-Casillas et al., 1993) and TβRII-B (Figure 6A and B), while it expresses TβRI and TβRII. It has been shown previously that TβRIII binds all three isoforms with high affinity and is essential for the presentation of TGF-β2 to the signalling complex, i.e. TβRII and TβRI (Wang et al., 1991; Sankar et al., 1995; Brown et al., 1999). Both cell lines were treated with either TGF-β1 or TGF-β2 for 30 min and cell lysates were analysed by western blotting using PS2 antiserum, which recognizes specifically the phosphorylated form of Smad2 (Ishisaki et al., 1999). In L6 cells Smad2 is highly phosphorylated upon stimulation with TGF-β1 (Figure 7A, lane 5) whereas it is phosphorylated to a lesser extent with TGF-β2 (Figure 7A, lane 6). In U2OS cells, however, the additional expression of TβRII-B results in strong phosphorylation of Smad2 after TGFβ2 treatment. This is independent of TβRIII expression (Figure 7A, lane 3).
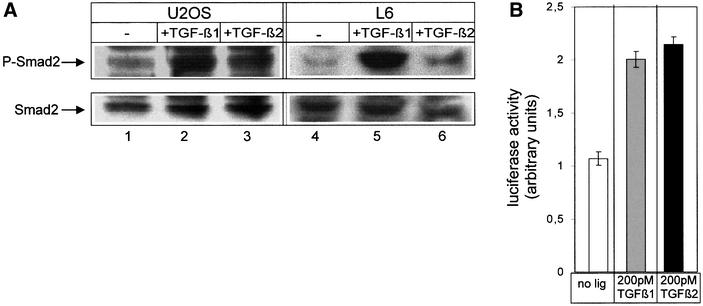
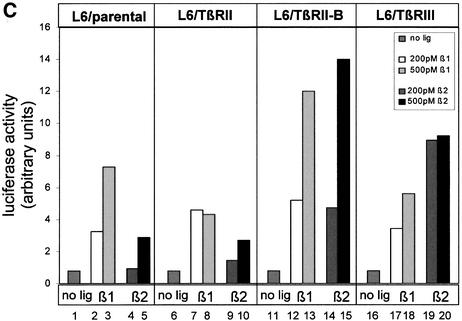
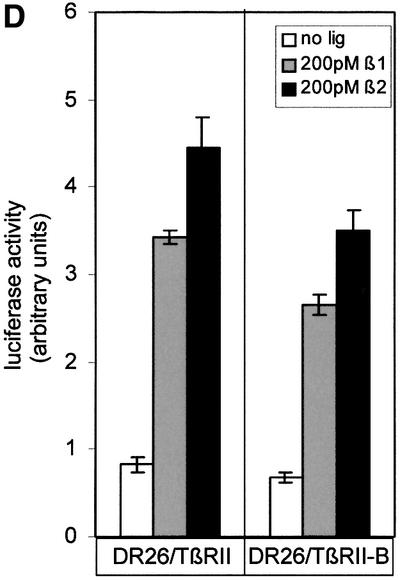
Fig. 7. TβRII-B transduces TGF-β2 signals via Smad2 independently of TβRIII. (A) U2OS cells (lanes 1–3) and L6 cells (lanes 4–6) were treated with 200 pM TGF-β1 (lanes 2 and 5) or 200 pM TGF-β2 (lanes 3 and 6). Total cell lysates were used for western blotting with PS2 antiserum (upper panel). Equal loading was confirmed using α-Smad2 (α-SED) antiserum (lower panel). (B) U2OS cells were transfected with the TGF-β-sensitive reporter plasmid p3TP-luc and pRL-TK for reference. After stimulation with 200 pM TGF-β1 or 200 pM TGF-β2, luciferase activity was measured. Data were normalized to pRL-TK activity to control for transfection efficiency. (C) L6 cells were transfected with the receptors indicated, p3TP-luc and pRL-TK, and then incubated with 200 pM (white), 500 pM TGF-β1 (grey), 200 pM TGF–β2 (dark grey) or 500 pM TGF-β2 (black). Data were normalized to pRL-TK activity and represent the mean of three independent experiments. (D) DR26 cells were transfected with p3TP-luc and pRL-TK together with TβRII-B or TβRII constructs. Luciferase activity was determined as described above.
Next, signalling via both TGF-β isoforms was analysed in reporter gene assays. First, the induction of the TGF-β-responsive reporter gene p3TP-luc was tested in U2OS cells, where TGF-β1 as well as TGF-β2 showed a 2-fold increase in luciferase activity (Figure 7B). Secondly, we analysed L6 cells for their responsiveness to both TGF-β isoforms. The parental cell line does respond to the TGF-β1 isoform, but shows only weak induction by the TGF-β2 isoform (Figure 7C, columns 1–5). This indicates that even though preformed complexes of TβRII and TβRI that could bind the ligand TGF-β2 might exist (Rodriguez et al., 1995; Gilboa et al., 1998; Wells et al., 1999), these complexes induce only minor responsiveness to TGF-β2 in the p3TP-luc reporter gene assay. Interestingly, transfection not only of TβRIII (Figure 7C, columns 19 and 20) but also of TβRII-B (Figure 7C, columns 14 and 15) leads to TGF-β2 response of these cells. Transfection of TβRII (Figure 7C, columns 6–10) shows no increase in responsiveness to TGF-β2. These data, together with the results from U2OS cells (Figure 7A and B), demonstrate for the first time signalling of TGF-β2 independently of the TβRIII.
Cells such as the Mv1Lu cells, which express a high amount of TβRIII, facilitate TGF-β2 signalling through this receptor. DR26 cells, which lack functional TβRII, were transiently transfected with either TβRII or TβRII-B. The TGF-β-responsive reporter p3TP-luc (Wrana et al., 1992) was used to measure luciferase activity after TGF-β1 or -β2 addition. Figure 7D shows that there is no significant difference between signalling via the two TGF-β isoforms in these cells. This can be explained by the presence of TβRIII in Mv1Lu cells and derivative cell lines, which compensates for the lack of TGF-β2 binding to the TβRII by presenting the ligand.
TβRII-B interacts with all known type I receptors (ALK1–7) after binding TGF-β1 (data not shown). To investigate signalling via these receptor complexes we have performed reporter gene assays in R1b/L17 cells. Different type I receptor constructs were expressed in R1b/L17 cells either in the presence or absence of TβRII-B. Transcriptional activation of the reporter plasmids p3TP-luc (Wrana et al., 1992) and pSBE-luc (Jonk et al., 1998; data not shown) was determined for both TGF-β1 and TGF-β2. In the case of p3TP-luc, only expression of ALK5 showed induction of the reporter gene; the co-expression of TβRII-B even results in ligand-independent activation (Figure 8). ALK4, the activin type Ib receptor, showed activation of the reporter by the ligand TGF-β1 only when TβRII-B (or TβRII, data not shown) was expressed. Therefore, signalling of TβRII-B via the Smad2/3 pathway is induced primarily through activation of ALK5 (TβRI).
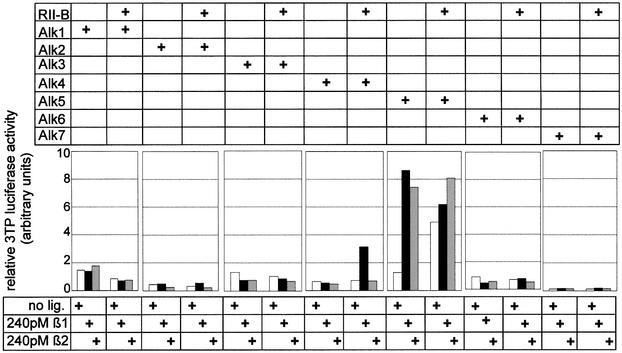
Fig. 8. TβRII-B signals via TβRI (ALK5) to the reporter p3TP-luc. R1b/L17 cells were transiently transfected with ALK1–7 in the absence or presence of TβRII-B (as indicated). Reporter gene activity was measured on p3TP-luc after treatment with either TGF-β1 (black bars) or TGF-β2 (grey bars). Luciferase activity was determined as described.
Taken together, our results demonstrate that TβRII-B is a signalling receptor for the TGF-β2 isoform. Direct binding of this isoform induces TβRIII-independent signalling. This is of particular interest in cells and tissues that lack TβRIII and in which TGF-β2 has a distinct function. In addition to the β2 isoform, TβRII-B also binds and triggers signals from TGF-β1.
Discussion
The three mammalian isoforms TGF-β1, -β2 and -β3 share 71–76% sequence identity and the proteolytically cleaved mature forms are highly conserved between different species. This suggests that during evolution there has been pressure to keep certain differences as well as similarities of these isoforms. This is also reflected in gene targeting experiments that revealed specific phenotypes (Schull et al., 1992; Kulkarni and Karlsson, 1993; Diebold et al., 1995; Kaartinen et al., 1995; Proetzel et al., 1995; Sanford et al., 1997). Even though all isoforms are very similar in sequence and structure, their function in vivo is quite distinct.
While the initial steps of signal transduction by TGF-β1 are well studied, signalling by the TGF-β2 isoform is still less clear. TGF-β1 follows the ‘sequential binding mode’ in order to form the signalling receptor complex at the cell surface (Wrana et al., 1994a; Massagué, 1998). It first binds to the TβRII, causing the recruitment of TβRI into the signalling complex and its subsequent activation by transphosphorylation. Since TβRII does not bind the TGF-β2 isoform (Lin et al., 1995), it was proposed that signalling via TGF-β2 requires the co-expression of TβRIII. TβRIII binds TGF-β2 with high affinity and presents the ligand to TβRII by receptor oligomerization (Lopez-Casillas et al., 1993; Henis et al., 1994). This suggested that cells lacking TβRIII will not respond to TGF-β2 (Sankar et al., 1995), which has been confirmed recently in endocardial cells requiring functional TβRIII for transformation in the heart (Brown et al., 1999). However, it is still unclear why TGF-β1, which would directly activate the TβRII–TβRI complex, does not result in transformation of these ventricular endothelial cells.
Here we describe TβRII-B, an alternatively spliced TβRII, as a receptor that not only binds TGF-β2 directly (Figure 2), but also activates the Smad pathway (Figure 7). Binding and signalling are independent of the co-expression of TβRIII (Figures 2 and 7).
The alternative splicing results in an insertion of 26 amino acids in exchange for Val32 at the extracellular domain of the receptor. This structural alteration leads to a new binding site for TGF-β2 without abolishing binding to the other isoforms, TGF-β1 and -β3 (Figure 2). We have also shown that binding of antibodies close to this new binding site interferes with TGF-β1 binding, probably due to steric hindrance (Figure 5B). Amino acids potentially important for structural characteristics favouring this new binding site are two cysteines in the human TβRII-B (one in the murine TβRII-B) and Asn48, as a potential N-glycosylation site. Mutations of these residues to alanine, however, led to no significant differences in binding of the TGF-β2 isoform compared with the wild-type TβRII-B (Figure 5A).
Co-immunoprecipitation studies revealed that TβRII-B interacts with all known TGF-β receptors (TβRI, TβRII and TβRIII, see Figures 3 and 4) in the presence of ligand. We even show that hetero-oligomeric complexes of TβRII/TβRII-B enable TβRII to bind the TGF-β2 isoform without any TβRIII, suggesting that one subunit of TβRII-B is enough to get any binding of TGF-β2 to TβRII. In cells that endogenously express both TβRII and TβRII-B, the functional significance of TβRII/TβRII-B hetero-oligomers is unknown. Homodimers of TGF-β isoforms are abundant, but TGF-β1.2 and TGF-β2.3 heterodimers have been identified in vivo. In view of the existence of heterodimeric ligands, it is conceivable that two different type II receptors constitute a signalling receptor complex, thereby creating combinatorial signalling (Cheifetz et al., 1987; Ogawa et al., 1992). It is of interest to investigate further the signals originating from TβRII and TβRII-B homo-oligomers versus TβRII/TβRII-B hetero-oligomers.
Previous publications on TβRII-B did not address in detail binding and signalling via different TGF-β isoforms (Hirai and Fijita, 1996). With our studies it became clear that competition with an excess of cold TGF-β2 in a TGF-β1 binding assay does not prove the inability of this receptor to bind TGF-β2. We have shown direct binding of TGF-β2 and this further implies that TGF-β2, in contrast to TGF-β1, uses a different binding site at the type II receptor. We, as well as Hirai and Fijita (1996), have shown that in DR26 cells overexpression of either TβRII-B or TβRII does not show any difference (Figure 7D). This is due to the presence of TβRIII, which binds TGF-β2 and presents it to TβRII (Lopez-Casillas et al., 1993). From our data it is evident now that it is not possible to distinguish between TGF-β2 signalling via TβRIII/TβRII, TβRIII/TβRII-B or directly via TβRII-B, if the reporter gene p3TP-luc is used (Figure 7D).
Therefore, we decided to analyse TGF-β2 signalling in cells lacking TβRIII. Indeed it had been demonstrated before that cells not expressing TβRIII, such as various vascular endothelial and haematopoietic progenitor cell lines, show a relative resistance to TGF-β2 (Ohta et al., 1987; Ottmann and Pelus, 1988; Sankar et al., 1995). Various muscle myoblast cell lines including L6 express TβRII and TβRI but lack TβRIII. We showed that there is no expression of TβRII-B in these cells (Figure 6A and B). For this reason the L6 cell line is the ideal cell system to compare signalling via TβRIII and TβRII-B. In transfection experiments we have shown that signalling via TGF-β2 is restored in these cells in the presence of either TβRIII or TβRII-B (Figure 7C). However, TβRII does not induce TGF-β2 sensitivity. To analyse TGF-β2 signalling through endogenous TβRII-B, independently of TβRIII, we performed the same assays in the osteosarcoma cell line U2OS, which lacks TβRIII but still expresses TβRII-B. These cells respond to TGF-β2, as we show by Smad2-phosphorylation experiments (Figure 7A) and reporter gene assays (Figure 7B). This signalling is facilitated via TβRI and not via ALK1–4 or ALK6 and ALK7 (Figure 8).
Besides the TGF-β type II receptor, other members of the receptor family exist as alternative forms. The type II receptor for MIS (AMHRII) shows alternative splicing, resulting in a 61-amino-acid insertion at the same site as TβRII (di Clemente et al., 1994), the Dpp receptor Tkv has two alternative N-termini (Brummel et al., 1994; Penton et al., 1994) and there are two alternative extracellular juxtamembrane regions in ATR-I (Wrana et al., 1994b). Moreover, four alternative splicing variants of ActRII-B (Attisano et al., 1992) and a splicing variant of BMP-RII exist (Kawabata et al., 1995; Nohno et al., 1995; Rosenzweig et al., 1995). Although the functional significance of these variants is still unknown, it is of interest that insertions at the N-terminus of the receptors are seen only in ligand-binding subunits (TβRII, AMHRII, Tkv), suggesting that the N-terminus of the ligand binding subunit is important for ligand specificity. Scanning deletion analysis of the extracellular domain of TβRII has revealed that amino acids N-terminal to the first cysteine are not important for binding to TGF-β1 (Pepin et al., 1996); the insertion in TβRII-B, however, seems to add binding properties for an additional isoform.
Of special interest is the expression and signalling capability of TβRII-B in osteoblasts and osteosarcoma cells. Osteoblasts are of mesenchymal origin and synthesize bone matrix, and osteoclasts derived from the haematopoietic system resorb bone. It was shown that TGF-β2 increases osteoblastic activity by the induction of extracellular matrix secretion, inhibition of matrix mineralization and modulation of osteoprogenitor cell proliferation (Centrella et al., 1994; Rosen et al., 1994). Increased expression of TGF-β2 in osteoblasts results in an osteoporosis-like phenotype (Erlebacher and Derynck, 1996). Inhibition of TGF-β receptor signalling in osteoblasts by transgenic expression of cytoplasmically truncated TβRII has recently been shown to result in decreased bone remodelling, which is probably due to decreased bone resorption by osteoclasts (Filvaroff et al., 1999). This is of special importance during metastasis of breast cancer cells to the skeleton, where TGF-β released from the bone matrix by the action of osteoclasts promotes metastatic growth (Yin et al., 1999). While TGF-β stimulates bone resorption by differentiated osteoclasts it inhibits osteoclast differentiation from bone marrow monocytes.
The specific expression of the TβRII-B as a TGF-β2 receptor in osteoblasts suggests a distinct function of this receptor in the process of bone remodelling. Signalling of TGF-β2 or TGF-β1 via this receptor in comparison with signalling via TβRII and TβRIII needs to be further investigated in order to understand the isoform-specific effects of TGF-β.
Materials and methods
Cell culture
COS-7, Mv1Lu, L6, C3H10T1/2, C2C12 and Hep3B cells were obtained from ATCC, MC3T3-E1 cells from P.ten Dijke (Uppsala), IMR 32 cells from K.Unsicker (Heidelberg) and U2OS cells from J.Hoppe (Würzburg). R1b/L17 and DR26 cells were obtained from J.Massagué (New York).
Isolation of the human TβRII-B clone, RT–PCR and mutagenesis
RNA was extracted from different cell lines as described (Chomczynski and Sacchi, 1987) and reverse transcribed using Superscript II (Gibco-BRL) according to the manufacturer’s instructions. For subsequent PCR, Pfu polymerase (Invitrogen) and specific oligonucleotides corresponding to the extracellular domain of TβRII (P1, nucleotides –23 to –4; P5, nucleotides 435–417; Lin et al., 1992) and for TβRII-B the specific primer Pins (nucleotides 106–132) were used in combination with P5. For the isolation of the TβRII-B clone, the TβRII-B-specific fragment was cut by HindIII and BglII to replace the corresponding fragment in TβRII (H20) (Knaus et al., 1996).
All mutations were generated by PCR mutagenesis. The cysteine residues Cys44 and Cys47 in TβRII-B were mutated to alanine individually or in combination. The glycosylation mutant was generated by replacing Asn48 by alanine. The HA-epitope was introduced after Pro26.
Expression plasmids for ALK1–6 were kindly provided by C.H.Heldin (Uppsala), the expression construct for ALK7 by C.Ibanez (Stockholm).
Ligands
Recombinant TGF-β1, TGF-β2, TGF-β3 and activin A were purchased from R&D Systems. Recombinant human BMP-2 was prepared as described (Ruppert et al., 1996).
Antibodies
Polyclonal antibodies directed against two different peptides of the TβRII-B insert were raised in rabbits and are either specific for the mouse and human (α-RIIB) or the human (α-hRIIB) TβRII-B.
The polyclonal antiserum against a peptide corresponding to the C-terminal sequence of the human TβRII (α-CRII) and the polyclonal antiserum specific for a cytoplasmic peptide in human ALK5 (α-RI) have been described previously (Moustakas et al., 1995). The anti-phospho-Smad2 antiserum was kindly provided to us by P.ten Dijke and C.H.Heldin (Uppsala). The 12CA5 antibody against the HA-tag was purchased from BabCo. The polyclonal anti-peptide antibody against the BMP-type Ia receptor (BRIa) was described earlier (Gilboa et al., 2000).
Transient transfections
COS-7 cells were transfected with plasmids encoding receptor cDNAs using the DEAE–dextran method (Aruffo and Seed, 1987). Forty-eight hours after transfection, binding and crosslinking were performed as described below. Aliquots of cell lysates were subjected to immunoprecipitation.
DR26 cells were transfected using DEAE–dextran, L6, R1b/L17 and U2OS cells using Lipofectamine (Life Technologies) according to the manufacturer’s protocol. Cells were lysed 36–48 h after transfection to measure luciferase activity.
Ligand binding and crosslinking
TGF-β1, -β2, -β3 and activin A were iodinated and crosslinked as described in Lin et al. (1992), BMP-2 as described in Gilboa et al. (2000).
Receptor immunoprecipitation
After binding and crosslinking, COS-7 cells were solubilized in lysis buffer (phosphate-buffered saline pH 7.4 containing 1.0% Triton X-100, 1 mM EDTA and including protease inhibitors) at 4°C for 40 min. Receptors were immunoprecipitated from cell extracts by 12CA5 monoclonal antibodies (α-HA) or by using specific rabbit anti-peptide antisera (α-hRIIB, α-RIIB and α-CRII) together with protein A–Sepharose for at least 4 h at 4°C. For single immunoprecipitations, the bound protein was eluted by heating the beads in SDS–PAGE sample buffer containing β-mercaptoethanol (5 min, 95°C). For sequential immunoprecipitations, the bound protein was eluted from the Sepharose beads in 1% SDS, 50 mM dithiothreitol, 10% β-mercaptoethanol (5 min, 95°C). The supernatant was diluted with lysis buffer to a final SDS concentration of <0.1% and the appropriate antibodies were added for the second immunoprecipitation. TGF-β receptors were analysed by 7.5–10% SDS–PAGE followed by exposure to a phosphoimager screen.
Metabolic labelling
COS-7 cells were starved in serum-free Dulbecco’s modified Eagle’s medium (DMEM) minus cysteine and methionine for 90 min at 37°C. The medium was then replaced with fresh medium supplemented with 0.2 mM oxidized glutathione (Boehringer) and 0.5 mCi/ml of [35S]methionine and [35S]cysteine (Dupont) and incubated for 2–3 h at 37°C. Cells were solubilized as described above. Lysates were immunoprecipitated with appropriate antibodies and proteins analysed by SDS–PAGE.
Reporter gene assays
Cells were starved for 12–24 h after transfection in 0.2% FCS for 4–6 h followed by the addition of 200 or 500 pM TGF-β1 or TGF-β2 for 18–24 h. Cells were lysed, and luciferase activity determined by the Dual Luciferase Assay system (Promega).
Smad2 phosphorylation assay
U2OS or L6 cells (5 × 105) were plated on 6 cm Petri dishes. Starvation was performed for 4 h in DMEM containing 0.2% FCS. TGF-β1 and -β2 (200 pM) were added for 30 min. Cells were lysed in cold TNE buffer (20 mM Tris pH 7.4, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA) including protease inhibitors and phosphatase inhibitors. Aliquots of the cleared lysate were submitted to SDS–PAGE followed by immunoblotting. C-terminally phosphorylated Smad2 was detected by a α-phospho-Smad2 (α-PS2) antibody (Ishisaki et al. 1999) and visualized using the ECL detection system. To show equal loading, the antibodies were removed by incubating the nitrocellulose membrane in stripping buffer (5 mM phosphate buffer, 2% SDS and 0.014% β-mercaptoethanol) for 30 min at 60°C. Smad2 protein was detected using an α-Smad2 (α-SED) antibody (Nakao et al., 1997).
Acknowledgments
Acknowledgements
We thank C.H.Heldin and P.ten Dijke (Uppsala) for Smad2 antisera and ALK constructs, and C.Ibanez (Stockholm) for the ALK7 construct. We gratefully acknowledge helpful discussions with Ralf Schreck and Florian Neubauer. We also thank T.Geissendörfer and W.Hädelt for excellent technical assistance. This work was supported by the SFB 465 TP A10 to P.K.
REFERENCES
- Aruffo A. and Seed,B. (1987) Molecular cloning of two CD7 (T-cell leukemia antigen) cDNAs by a COS cell expression system. EMBO J., 6, 3313–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attisano L., Wrana,J.L., Cheifetz,S. and Massagué,J. (1992) Novel activin receptors: distinct genes and alternative mRNA splicing generate a repertoire of serine/threonine kinase receptors. Cell, 68, 97–108. [DOI] [PubMed] [Google Scholar]
- Brown C.B., Boyer,A.S., Runyan,R.B. and Barnett,J.V. (1999) Requirement of type III TGF-β receptor for endocardial cell transformation in the heart. Science, 283, 2080–2082. [DOI] [PubMed] [Google Scholar]
- Brummel T.J., Twombly,V., Marques,G., Wrana,J.L., Newfeld,S.J., Attisano,L., Massagué,J., O’Connor,M.B. and Gelbart,W.M. (1994) Characterization and relationship of Dpp receptors encoded by the saxophone and thick veins genes in Drosophila. Cell, 78, 251–261. [DOI] [PubMed] [Google Scholar]
- Centrella M., Horowitz,M.C., Wozney,J.M. and McCarthy,T.L. (1994) Transforming growth factor-β gene family members and bone. Endocr. Rev., 15, 27–39. [DOI] [PubMed] [Google Scholar]
- Cheifetz S. and Massagué,J. (1991) Isoform-specific transforming growth factor-β binding proteins with membrane attachments sensitive to phosphatidylinositol-specific phospholipase C. J. Biol. Chem., 266, 20767–20772. [PubMed] [Google Scholar]
- Cheifetz S., Weatherbee,J.A., Tsang,M.L.-S., Anderson,J.K., Mole,J.E., Lucas,R. and Massagué,J. (1987) The transforming growth factor-β system, a complex pattern of cross-reactive ligands and receptors. Cell, 48, 409–415. [DOI] [PubMed] [Google Scholar]
- Chomczynski P. and Sacchi,N. (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem., 162, 156–159. [DOI] [PubMed] [Google Scholar]
- di Clemente N. et al. (1994) Cloning, expression and alternative splicing of the receptor for anti-Mullerian hormone. Mol. Endocrinol., 8, 1006–1020. [DOI] [PubMed] [Google Scholar]
- Diebold R.J., Eis,M.J., Yin,M., Ormsby,I., Boivin,G.P., Darrow,B.J., Saffitz,J.E. and Doetschman,T. (1995) Early-onset multifocal inflammation in the transforming growth factor β1-null mouse is lymphocyte mediated. Proc. Natl Acad. Sci. USA, 92, 12215–12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlebacher A. and Derynck,R. (1996) Increased expression of TGF-β2 in osteoblasts results in an osteoporosis-like phenotype. J. Cell Biol., 132, 195–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filvaroff E., Erlebacher,A., Ye,J., Gitelman,S.E., Lotz,J., Heillman,M. and Derynck,R. (1999) Inhibition of TGF-β receptor signaling in osteoblasts leads to decreased bone remodeling and increased trabecular bone mass. Development, 126, 4267–4279. [DOI] [PubMed] [Google Scholar]
- Gilboa L., Wells,R.G., Lodish,H.F. and Henis,Y.I. (1998) Oligomeric structure of type I and type II transforming growth factor β receptors: homo-dimers form in the ER and persist at the plasma membrane. J. Cell Biol., 140, 767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilboa L., Nohe,A., Geissendörfer,T., Sebald,W., Henis,Y.I. and Knaus,P. (2000) Bone morphogenetic protein complexes on the surface of live cells: a new oligomerization mode for serine/threonine kinase receptors. Mol. Biol. Cell, 11, 1023–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henis Y.I., Moustakas,A., Lin,H.Y. and Lodish,H.F. (1994) The types II and III transforming growth factor-β receptors form homo-oligomers. J. Cell Biol., 126, 139–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai R. and Fijita,T. (1996) A human transforming growth factor-β type II receptor that contains an insertion in the extracellular domain. Exp. Cell Res., 223, 135–141. [DOI] [PubMed] [Google Scholar]
- Ishisaki A.,Yamato,K., Hashimoto,S., Nakao,A., Tamaki,K., Nonaka,K., ten Dijke,P., Sugino,H. and Nishihara,T. (1999) Differential inhibition of Smad6 and Smad7 on bone morphogenetic protein- and activin-mediated growth arrest and apoptosis in B cells. J. Cell Biol., 274, 13637–13642. [DOI] [PubMed] [Google Scholar]
- Jonk L.J., Itoh,S., Heldin,C.H., ten Dijke,P. and Kruijer,W. (1998) Identification and functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-β, activin and bone morphogenetic protein-inducible enhancer. J. Biol. Chem., 273, 21145–21152. [DOI] [PubMed] [Google Scholar]
- Kaartinen V., Voncken,J.W., Shuler,C., Warburton,D., Bu,D., Heisterkamp,N. and Groffen,J. (1995) Abnormal lung development and cleft palate in mice lacking TGF-β3 indicates defects of epithelial–mesenchymal interaction. Nature Genet., 11, 415–421. [DOI] [PubMed] [Google Scholar]
- Katagiri T. et al. (1994) Bone morphogenetic protein-2 converts the differentiation pathway of C2C12 myoblasts into the osteoblast lineage. J. Cell Biol., 127, 1755–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata M., Chytil,A. and Moses,H.L. (1995) Cloning of a novel type II serine/threonine kinase receptor through interaction with the type I transforming growth factor-β receptor. J. Biol. Chem., 270, 5625–5630. [DOI] [PubMed] [Google Scholar]
- Knaus P.I., Lindemann,D., DeCoteau,J.F., Perlman,R., Yankelev,H., Hille,M., Kadin,M.E. and Lodish,H.F. (1996) A dominant inhibitory mutant of the type II transforming growth factor β receptor in the malignant progression of a cutaneous T-cell lymphoma. Mol. Cell. Biol., 16, 3480–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni A.B. and Karlsson,S. (1993) Transforming growth factor-β1 knockout mice. A mutation in one cytokine gene causes a dramatic inflammatory disease. Am. J. Pathol., 143, 3–9. [PMC free article] [PubMed] [Google Scholar]
- Laiho M., Weis,M.B. and Massagué,J. (1990) Concomitant loss of transforming growth factor (TGF)-β receptor types I and II in TGF-β-resistant cell mutants implicates both receptor types in signal transduction. J. Biol. Chem., 265, 18518–18524. [PubMed] [Google Scholar]
- Lin H.Y., Wang,X.F., Ng-Eaton,E., Weinberg,R.A. and Lodish,H.F. (1992) Expression cloning of the TGF-β type II receptor, a functional transmembrane serine/threonine kinase. Cell, 68, 775–785. [DOI] [PubMed] [Google Scholar]
- Lin H.Y., Moustakas,A., Knaus,P., Wells,R.G., Henis,Y.I. and Lodish,H.F. (1995) The soluble exoplasmic domain of the type II transforming growth factor (TGF)-β receptor. A heterogeneously glycosylated protein with high affinity and selectivity for TGF-β ligands. J. Biol. Chem., 270, 2747–2754. [DOI] [PubMed] [Google Scholar]
- Lopez-Casillas F., Wrana,J.L. and Massagué,J. (1993) β-glycan presents ligand to the TGFβ signaling receptor. Cell, 73, 1435–1444. [DOI] [PubMed] [Google Scholar]
- Macias-Silva M., Abdollah,S., Hoodless,P.A., Pirone,R., Attisano,L. and Wrana,J.L. (1996) MADR2 is a substrate of the TGFβ receptor and its phosphorylation is required for nuclear accumulation and signaling. Cell, 87, 1215–1224. [DOI] [PubMed] [Google Scholar]
- Massagué J. (1998) TGF-β signal transduction. Annu. Rev. Biochem., 67, 753–791. [DOI] [PubMed] [Google Scholar]
- Massagué J. and Chen Y.-G. (2000) Controlling TGF-β signaling. Genes Dev., 14, 627–644. [PubMed] [Google Scholar]
- Moustakas A., Lin,H.Y., Henis,Y.I., Plamondon,J., O’Connor,M.M. and Lodish,H.F. (1993) The transforming growth factor β receptors types I, II and III form hetero-oligomeric complexes in the presence of ligand. J. Biol. Chem., 268, 22215–22218. [PubMed] [Google Scholar]
- Moustakas A., Takumi,T., Lin,H.Y. and Lodish,H.F. (1995) GH3 pituitary tumor cells contain heteromeric type I and type II receptor complexes for transforming growth factor β and activin-A. J. Biol. Chem., 270, 765–769. [DOI] [PubMed] [Google Scholar]
- Nakao A. et al. (1997) TGF-β receptor-mediated signaling through Smad2, Smad3 and Smad4. EMBO J., 16, 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikawa J. (1994) A cDNA encoding the human transforming growth factor β receptor suppresses the growth defect of a yeast mutant. Gene, 149, 367–372. [DOI] [PubMed] [Google Scholar]
- Nohno T., Ishikawa,T., Saito,T., Hosokawa,K., Noji,S., Wolsing,D.H. and Rosenbaum,J.S. (1995) Identification of a human type II receptor for bone morphogenetic protein-4 that forms differential heteromeric complexes with bone morphogenetic protein type I receptors. J. Biol. Chem., 270, 22522–22526. [DOI] [PubMed] [Google Scholar]
- Ogawa Y., Schmidt,D.K., Dasch,J.R., Chang,R.-J. and Glaser,C.B. (1992) Purification and characterization of transforming growth factor-β2.3 and -β1.2 hetero-dimers from bovine bone. J. Biol. Chem., 267, 2325–2328. [PubMed] [Google Scholar]
- Ohta M., Greenberger,J.S., Anklesaria,P., Bassols,A. and Massagué,J. (1987) Two forms of transforming growth factor-β distinguished by multipotential haematopoietic progenitor cells. Nature, 329, 539–541. [DOI] [PubMed] [Google Scholar]
- Ottmann O.G. and Pelus,L.M. (1988) Differential proliferative effects of transforming growth factor-β on human hematopoietic progenitor cells. J. Immunol., 140, 2661–2665. [PubMed] [Google Scholar]
- Pelton R.W., Saxena,B., Jones,M., Moses,H.L. and Gold,L.I. (1991) Immunohistochemical localization of TGF β1, TGF β2 and TGF β3 in the mouse embryo: expression patterns suggest multiple roles during embryonic development. J. Cell Biol., 115, 1091–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penton A., Chen,Y., Staehling,H.K., Wrana,J.L., Attisano,L., Szidonya,J., Cassill,J.A., Massagué,J. and Hoffmann,F.M. (1994) Identification of two bone morphogenetic protein type I receptors in Drosophila and evidence that Brk25D is a decapentaplegic receptor. Cell, 78, 239–250. [DOI] [PubMed] [Google Scholar]
- Pepin M.C., Beauchemin,M., Plamondon,J. and O’Connor-McCourt,M.D. (1996) Scanning-deletion analysis of the extracellular domain of the TGF-β receptor II. Biochem. Biophys. Res. Commun., 220, 289–293. [DOI] [PubMed] [Google Scholar]
- Piek E., Heldin,C.-H. and ten Dijke,P. (1999) Specificity, diversity and regulation in TGF-β superfamily signaling. FASEB J., 13, 2105–2123. [PubMed] [Google Scholar]
- Proetzel G., Pawlowski,S.A., Wiles,M.V., Yin,M., Boivin,G.P., Howles,P.N., Ding,J., Ferguson,M.W. and Doetschman,T. (1995) Transforming growth factor-β3 is required for secondary palate fusion. Nature Genet., 11, 409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A.B. and Sporn,M.B. (1993) Physiological actions and clinical applications of transforming growth factor-β (TGF-β). Growth Factors, 8, 1–9. [DOI] [PubMed] [Google Scholar]
- Rodriguez C., Chen,F., Weinberg,R.A. and Lodish,H.F. (1995) Cooperative binding of transforming growth factor (TGF)-β2 to the types I and II TGF-β receptors. J. Biol. Chem., 270, 15919–15922. [DOI] [PubMed] [Google Scholar]
- Rosen D., Miller,S.C., DeLeon,E., Thompson,A.Y., Bentz,H., Mathews,M. and Adams,S. (1994) Systemic administration of recombinant transforming growth factor β2 (rTGF-β2) stimulates parameters of cancellous bone formation in juvenile and adult rats. Bone, 15, 355–359. [DOI] [PubMed] [Google Scholar]
- Rosenzweig B.L., Imamura,T., Okadome,T., Cox,G.N., Yamashita,H., ten Dijke,P., Heldin,C.H. and Miyazono,K. (1995) Cloning and characterization of a human type II receptor for bone morphogenetic proteins. Proc. Natl Acad. Sci. USA, 92, 7632–7636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruppert R., Hoffmann,E. and Sebald,W. (1996) Human bone morphogenetic protein 2 contains a heparin-binding site which modifies its biological activity. Eur. J. Biochem., 237, 295–302. [DOI] [PubMed] [Google Scholar]
- Sanford L.P., Ormsby,I., de Gittenberger,G.A., Sariola,H., Friedman,R., Boivin,G.P., Cardell,E.L. and Doetschman,T. (1997) TGFβ2 knockout mice have multiple developmental defects that are non-overlapping with other TGFβ knockout phenotypes. Development, 124, 2659–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankar S., Mahooti-Brooks,N., Centrella,M., McCarthy,T.L. and Madri,J.A. (1995) Expression of transforming growth factor type III receptor in vascular endothelial cells increases their responsiveness to transforming growth factor β2. J. Biol. Chem., 270, 13567–13572. [DOI] [PubMed] [Google Scholar]
- Schull M.M. et al. (1992) Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature, 359, 693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Dijke P., Miyazono,K. and Heldin,C.H. (1996) Signaling via hetero-oligomeric complexes of type I and type II serine/threonine kinase receptors. Curr. Opin. Cell Biol., 8, 139–145. [DOI] [PubMed] [Google Scholar]
- Wang X.F., Lin,H.Y., Ng-Eaton,E., Downward,J., Lodish,H.F. and Weinberg,R.A. (1991) Expression cloning and characterization of the TGF-β type III receptor. Cell, 67, 797–805. [DOI] [PubMed] [Google Scholar]
- Wells R.G., Yankelev,H., Lin,H.Y. and Lodish,H.F. (1997) Biosynthesis of the type I and type II TGF-β receptors. Implications for complex formation. J. Biol. Chem., 272, 11444–11451. [DOI] [PubMed] [Google Scholar]
- Wells R.G., Gilboa,L., Sun,Y., Liu,X., Henis,Y.I. and Lodish,H.F. (1999) Transforming growth factor-β induces formation of a dithiothreitol-resistant type I/type II receptor complex in live cells. J. Biol. Chem., 274, 5716–5722. [DOI] [PubMed] [Google Scholar]
- Wrana J.L., Attisano,L., Carcamo,J., Zentella,A., Doody,J., Laiho,M., Wang,X.F. and Massagué,J. (1992) TGF-β signals through a heteromeric protein kinase receptor complex. Cell, 71, 1003–1014. [DOI] [PubMed] [Google Scholar]
- Wrana J.L., Attisano,L., Wieser,R., Ventura,F. and Massagué,J. (1994a) Mechanism of activation of the TGF-β receptor. Nature, 370, 341–347. [DOI] [PubMed] [Google Scholar]
- Wrana J.L., Tran,H., Attisano,L., Arora,K., Childs,S.R., Massagué,J. and O’Connor,M.B. (1994b) Two distinct transmembrane serine/threonine kinases from Drosophila melanogaster form an activin receptor complex. Mol. Cell. Biol., 14, 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J.J., Selander,K., Chirgwin,J.M., Dallas,M., Grubbs,B.G., Wieser,R., Massagué,J., Mundy,G.R. and Guise,T.A. (1999) TGF-β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J. Clin. Invest., 103, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Feng,X., We,R. and Derynck,R. (1996) Receptor-associated Mad homologues synergize as effectors of the TGF-β response. Nature, 383, 168–172. [DOI] [PubMed] [Google Scholar]