

Abstract
Hsp72, a major inducible member of the heat shock protein family, can protect cells against many cellular stresses including heat shock. In our present study, we observed that pretreatment of NIH 3T3 cells with mild heat shock (43°C for 20 min) suppressed UV-stimulated c-Jun N-terminal kinase 1 (JNK1) activity. Constitutively overexpressed Hsp72 also inhibited JNK1 activation in NIH 3T3 cells, whereas it did not affect either SEK1 or MEKK1 activity. Both in vitro binding and kinase studies indicated that Hsp72 bound to JNK1 and that the peptide binding domain of Hsp72 was important to the binding and inhibition of JNK1. In vivo binding between endogenous Hsp72 and JNK1 in NIH 3T3 cells was confirmed by co-immunoprecipitation. Hsp72 also inhibited JNK-dependent apoptosis. Hsp72 antisense oligonucleotides blocked Hsp72 production in NIH 3T3 cells in response to mild heat shock and concomitantly abolished the suppressive effect of mild heat shock on UV-induced JNK activation and apoptosis. Collectively, our data suggest strongly that Hsp72 can modulate stress-activated signaling by directly inhibiting JNK.
Keywords: c-Jun N-terminal kinase/heat shock protein/Hsp72/stress-activated protein kinase
Introduction
The mitogen-activated protein kinases (MAPKs) are typical signaling mediators that transmit intracellular signals initiated by extracellular stimuli to the nucleus. The MAPK signaling cascades regulate a variety of cellular activities, including cell growth, differentiation, survival and death (Minden and Karin, 1997; Ip and Davis, 1998; Schaeffer and Weber, 1999). The family of mammalian MAPKs includes three distinct subgroups: extracellular signal-regulated kinases (ERK), c-Jun N-terminal kinases/stress-activated protein kinases (JNK/SAPK) and p38 (Cano and Mahadevan, 1995). The ERK pathway, which is often activated by mitogenic stimuli such as peptide growth factors, consists of ERK and upstream kinases that include mitogen-activated protein kinase/extracellular signal-regulated kinase kinase1 (MEK1) and Raf-1 (Schaeffer and Weber, 1999). The p38 pathway, which can be stimulated by various stresses, including osmotic stress, consists of p38 and upstream kinases such as MKK3 or MKK6 (Schaeffer and Weber, 1999). Like the p38 pathway, the JNK/SAPK pathway can be activated by a variety of cellular stresses that include genotoxic stress, free radicals, heat shock, osmotic shock, ischemia and proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1β (Minden and Karin, 1997; Ip and Davis, 1998). The activation signal of the JNK pathway passes through MEKK1 or its equivalent MAPK kinase kinases, which activate SEK1/JNKK1/MKK4 or MKK7 (Minden and Karin, 1997; Ip and Davis, 1998). SEK1 and MKK7 in turn activate JNK. When activated, JNK can phosphorylate c-Jun or other transcription factors including ATF-2 and Elk-1 (Gupta et al., 1995; Minden et al., 1995; Yang et al., 1998). c-Jun phosphorylation enhances the transcription-stimulating activity of c-Jun (Ip and Davis, 1998). The physiological function of JNK is not fully understood, but JNK is thought be associated with the cellular machinery of stress-activated responses that under certain conditions can induce cell death (Minden and Karin, 1997; Ip and Davis, 1998). In fact, the JNK pathway has been shown to mediate intracellular signals for apoptotic cell death initiated by a variety of stress factors including heat shock, UV, oxidative stress and many others (Xia et al., 1995; Chen et al., 1996; Verheij et al., 1996; Zanke et al., 1996; Seimiya et al., 1997).
Many cellular stresses such as heat shock, ischemia and UV radiation not only stimulate stress-activated MAPKs, but also induce expression of heat shock proteins. Heat shock proteins function as chaperones or regulate protein translocation into organelles (Chirico et al., 1988; Murakami et al., 1988; Beckmann et al., 1990; Shi and Thomas, 1992). Heat shock protein 72 (Hsp72) is the major inducible heat shock protein (Wu et al., 1985). It plays a central role in protein synthesis, folding and translocation into organelles (Langer and Neupert, 1991; Amaral et al., 1993). In response to cellular stresses that generate denaturation of proteins, Hsp72 prevents protein aggregation and induces refolding of damaged proteins into the native state (Frydman and Hartl, 1994; Freeman et al., 1995; Minami et al., 1996). Hsp72 has ATPase activity combined with ATP and polypeptide binding activities (Milarski and Morimoto, 1989; Freeman et al., 1995). Either ATP or ADP can bind to the N-terminal domain of Hsp72, whereas polypeptide substrates bind to the C-terminal domain. The two binding domains are functionally coupled in such a way that ATP-bound Hsp72 has a lower affinity for polypeptide substrates than the ADP-bound protein (Palleros et al., 1993; Schmid et al., 1994). On the other hand, binding of polypeptide substrates to Hsp72 stimulates its ATPase activity (Flynn et al., 1989). The chaperone function of Hsp72 can also be modulated by other proteins. Hsp40/Hdj1 and Hip enhance the protein folding activity of Hsp72 (Freeman et al., 1995; Hohfeld et al., 1995; Minami et al., 1996), while Bag-1 functions as a negative regulator of Hsp72 (Takayama et al., 1997; Bimston et al., 1998).
Many lines of evidence point to constitutively overexpressed Hsp72 as being able to block apoptotic cell death initiated by cellular stress including heat shock, ceramide, ethanol, ionizing irradiation, TNF-α and ischemia (Jaattela, 1993; Simon et al., 1995; Bellmann et al., 1996; Samali and Cotter, 1996; Gabai et al., 1997; Liossis et al., 1997; Mosser et al., 1997; Meriin et al., 1998; Sharp et al., 1999). Moreover, Hsp72, when induced in a cellular response to apoptotic stress, exerts an anti-apoptotic effect in various types of cells (Samali and Cotter, 1996; Gordon et al., 1997), and it is frequently expressed at higher levels in human cancer cells (Li et al., 1995). Importantly, it has been discovered that Hsp70 can modulate the functions of several major components, including the JNK pathway and caspase-3, of the apoptotic program (Gabai et al., 1997, 2000; Mosser et al., 1997; Jaattela et al., 1998; Meriin et al., 1998). The JNK signaling cascade, in particular, appears to be the major target of the anti-apoptotic activity of Hsp72 (Gabai et al., 1997, 2000; Meriin et al., 1998, 1999).
In order to understand better the mechanism by which Hsp72 protects cells from apoptosis induced by cellular stress, we investigated the effects of Hsp72 actions on the components of the JNK signaling pathway. We report here that Hsp72 interacts physically with JNK, thereby suppressing both JNK activation and JNK-mediated cell death. Our study, using deletion mutants of Hsp72, also revealed that Hsp72 inhibits JNK activation independently of its chaperone activity. Taken together, our data suggest that Hsp72 is a natural inhibitor of JNK and that the direct interaction with JNK may be important to the mechanism by which Hsp72 suppresses the JNK signaling cascade.
Results
Hsp72 suppresses the JNK signaling pathway
To identify a possible role for Hsp72 in the regulation of intracellular signaling cascades, we examined whether Hsp72 could modulate the JNK signaling pathway. Expression of Hsp72 protein was induced in NIH 3T3 cells by exposing the cells to mild heat (43°C) for 20 min followed by further incubation for 16 h at 37°C (Figure 1A). While UV radiation stimulated JNK1 activity in untreated NIH 3T3 cells, UV-stimulated JNK1 activity was repressed in the heat-treated cells. In order to test whether Hsp72 was involved in the mechanism underlying JNK1 inhibition following mild heat treatment, we stably transfected NIH 3T3 cells with either the empty vector (named NIH 3T3-neo) or a vector constitutively expressing Hsp72 (named NIH 3T3-Hsp72). The UV-induced stimulation of JNK1 activity was markedly reduced in NIH 3T3-Hsp72 cells in comparison with NIH 3T3-neo control cells (Figure 1B). The forced expression of Hsp72 did not affect the intracellular level of endogenous JNK1 protein in the transfected NIH 3T3 cells (data not shown). Hsp72 also suppressed JNK1 activity stimulated by ΔMEKK1, an active form of MEKK1 (Figure 1C). Taken together, these data suggest that Hsp72 induces suppression of the JNK signaling pathway. These results were in good agreement with the previous reports (Gabai et al., 1997, 2000; Mosser et al., 1997; Meriin et al., 1998, 1999). We therefore looked at the molecular mechanism of the suppression of the JNK pathway by Hsp72 in more detail.
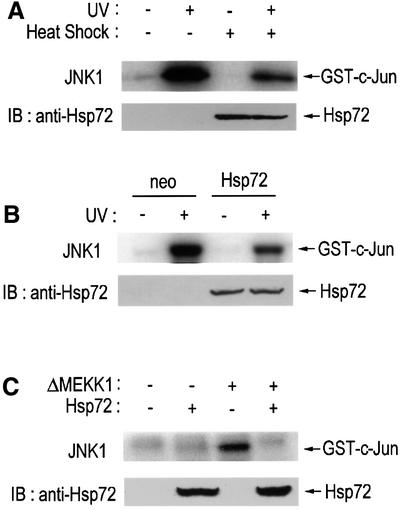
Fig. 1. Hsp72 suppresses the JNK signaling pathway. (A) NIH 3T3 cells were exposed to mild heat (43°C) for 20 min, and then incubated at 37°C for 16 h. The cells were irradiated with UV light (60 J/m2), further incubated for 1 h at 37°C and assayed for JNK1 activity by immunocomplex kinase assay. (B) NIH 3T3-neo or NIH 3T3-Hsp72 cells were exposed to UV light (60 J/m2), incubated at 37°C for 1 h and assayed for JNK1 activity by immunocomplex kinase assay as in (A). (C) NIH 3T3 cells were transiently transfected with plasmids expressing ΔMEKK1, HA-JNK1 or Hsp72 as indicated. After 50 h of transfection, cells were assayed for JNK1 activity by immunocomplex kinase assay. (A–C) In the immunocomplex kinase assays, cell lysates were subjected to immunoprecipitation with anti-JNK1 (A and B) or anti-HA monoclonal antibody (C). The resultant immunopellets were assayed for JNK1 activity using GST–c-Jun(1–79) as substrate. Expression of Hsp72 protein was analyzed by immunoblot using anti-Hsp72 monoclonal antibody. IB, immunoblot.
Hsp72 does not affect MEKK1 or SEK1 activity
The JNK signaling cascade is composed of JNK and its upstream kinases, which include MAPKKs such as SEK1, JNKK1, MKK4 and a MAPKKK such as MEKK1 (Verheij et al., 1996; Ip and Davis, 1998). In order to manifest a mechanism for JNK inhibition by Hsp72, we examined the effect of Hsp72 on either SEK1 or MEKK1. UV radiation induced the stimulation of SEK1 or MEKK1 activity in both NIH 3T3-neo and NIH 3T3-Hsp72 cells (Figure 2). Interestingly, ectopic Hsp72 expression did not affect the UV-induced stimulation of SEK1 or MEKK1. These data suggested that Hsp72 might suppress the JNK signaling cascade by acting on a site(s) downstream of MEKK1 or SEK1. In the following experiments, therefore, we investigated the possibility that JNK itself may be the target of Hsp72.
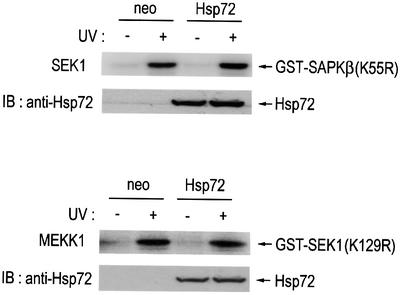
Fig. 2. Hsp72 does not affect SEK1 or MEKK1 activity in NIH 3T3 cells. NIH 3T3-Hsp72 and NIH 3T3-neo cells were exposed to UV light (60 J/m2) and then incubated further at 37°C for 1 h. Cell lysates were subjected to immunoprecipitation with either anti-SEK1 or anti-MEKK1 antibody. Immunopellets were assayed for SEK1 or MEKK1 activity using GST–SAPKβ(K55R) or GST–SEK1(K129R) as substrate, respectively. IB, immunoblot.
Hsp72 inhibits JNK1 in vitro
In order to test whether JNK1 was a direct target of Hsp72, we first examined the action of Hsp72 on JNK1 in vitro. An active JNK1 was obtained by immunoprecipitation of UV-exposed NIH 3T3 cells using anti-JNK1 antibody. In the in vitro kinase assay, pretreatment of active JNK1 with Hsp72 protein resulted in inhibition of JNK1 activity (Figure 3A). In comparison, Hsp72 pretreatment had little effect on the enzymic activity of either SEK1 or MEKK1 in vitro (Figure 3B). Thus, our data suggest that JNK1 was the major target protein of Hsp72 in the MEKK1-SEK1-JNK signaling cascade. Furthermore, Hsp72 pretreatment did not affect either ERK or p38 activity in vitro (Figure 3A).
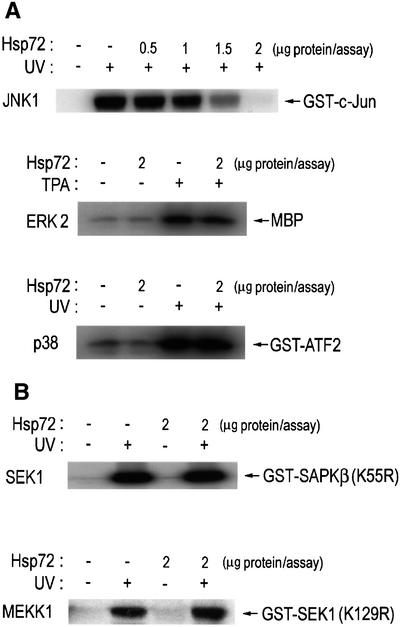
Fig. 3. Hsp72 suppresses JNK1 activity in vitro. NIH 3T3 cells were irradiated with UV light (60 J/m2), incubated for 1 h at 37°C and then subjected to immunoprecipitation using anti-JNK1, anti-p38, anti-SEK1 or anti-MEKK1 antibody, where indicated. In the ERK2 experiment, NIH 3T3 cells were exposed to 100 nM 12-O-tetradecanoylphorbol 13-acetate (TPA) for 15 min at 37°C and then subjected to immunoprecipitation using anti-ERK2 antibody. The immunopellets were incubated with purified human Hsp72 protein in 50 µl of HEPES buffer pH 7.4, for 1 h at room temperature, washed twice with HEPES buffer and then assayed for JNK1, ERK2, p38, SEK1 or MEKK1 activity using GST–c-Jun(1–79), myelin basic protein (MBP), GST–ATF2(1–109), GST–SAPKβ(K55R) or GST–SEK1(K129R) as substrate, respectively.
Hsp72 associates physically with JNK1
In order to confirm that Hsp72 acted directly on JNK1, we assessed the physical interaction between Hsp72 and JNK1. In the in vitro binding studies, we mixed His-Hsp72 with glutathione S-transferase (GST) fusion proteins that were immobilized on glutathione–agarose beads (Figure 4A). The immunoblot analysis with anti-Hsp72 antibody showed that His-Hsp72 bound to GST–JNK1, but not to the GST control or GST–SEK1 (Figure 4A). Next, we did another in vitro binding study in which GST, GST–JNK1 or GST–SEK1 was applied to His-Hsp72 immobilized on Ni2+–agarose beads. The immunoblot analysis using anti-GST antibody showed that GST–JNK1, but not the GST control or GST–SEK1, interacted with His-Hsp72 on the beads (Figure 4B). Furthermore, in a pull-down binding experiment using NIH 3T3 cell lysates, His-Hsp72 interacted with JNK1 but not with ERK2 or p38 (Figure 4C).
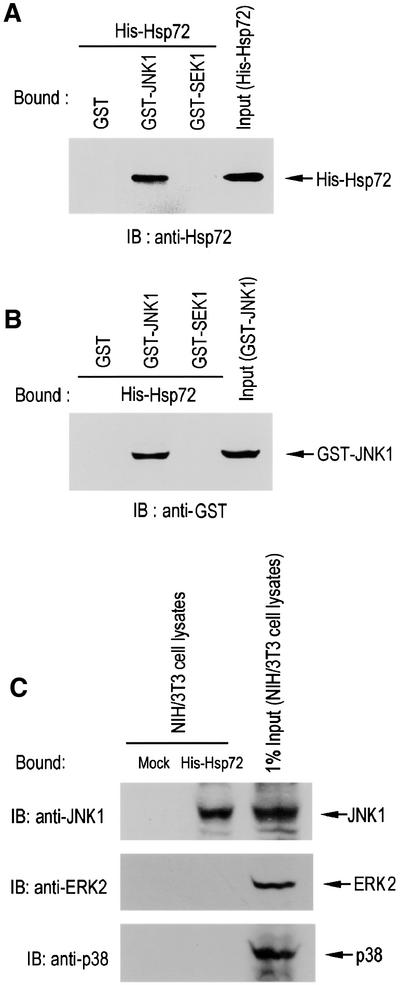
Fig. 4. Hsp72 interacts directly with JNK1 in vitro. (A) Purified His-Hsp72 protein was added to recombinant GST control, GST–JNK1 or GST–SEK1 protein immobilized on glutathione–agarose beads. Bound proteins were extensively washed, eluted, separated by SDS–PAGE on a 10% polyacrylamide gel and then analyzed by immunoblot using mouse anti-Hsp72 monoclonal antibody. (B) GST control, GST–JNK1 or GST–SEK1 protein was added to His-Hsp72 immobilized on Ni2+-NTA–agarose beads. Bead-bound proteins were then washed and eluted from the beads. The eluted proteins were subjected to SDS–PAGE on a 10% polyacrylamide gel, and detected by immunoblotting using mouse anti-GST monoclonal antibody. (C) NIH 3T3 cell lysates were applied to His-Hsp72 that was immobilized on Ni2+–agarose beads. Bound proteins were extensively washed and eluted from the beads. The eluted proteins were separated by SDS–PAGE and then analyzed by immunoblotting probed with mouse monoclonal anti-JNK1, rabbit polyclonal anti-ERK2 or mouse monoclonal anti-p38 antibody.
Next, we examined whether Hsp72 could associate physically with JNK1 in intact cells. NIH 3T3-neo or NIH 3T3-Hsp72 cells were subjected to immunoprecipitation with anti-JNK1 antibody, and the resultant immunopellets were examined by immunoblot analysis using anti-Hsp72 antibody (Figure 5). The immunoblot data indicated that overexpressed Hsp72 interacted directly with endogenous JNK1 in NIH 3T3-Hsp72 cells. We also examined whether exposure of cells to mild heat shock would induce the physical interaction between the two endogenous proteins JNK1 and Hsp72, in intact cells. The immunoblot data show that the exposure of NIH 3T3 cells to a mild heat shock (43°C for 20 min) induced the accumulation of endogenous Hsp72. Immunoblot analysis of protein complexes immunoprecipitated with anti-JNK1 antibody demonstrated that Hsp72 was associated physically with JNK1 in the heat-treated NIH 3T3 cells (Figure 5). These findings strongly suggest that a physical interaction between the two endogenous proteins, Hsp72 and JNK1, does occur in intact cells in response to mild heat shock.
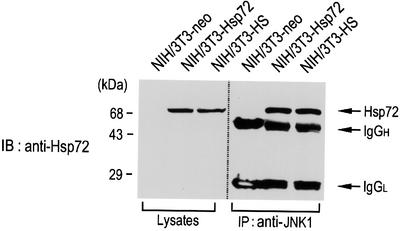
Fig. 5. Hsp72 associates physically with JNK1 in intact cells. Cell lysates from NIH 3T3-neo, NIH 3T3-Hsp72 or heat shock (HS)-treated NIH 3T3 cells were subjected to immunoprecipitation using mouse anti-JNK1 monoclonal antibody. The resultant immunopellets were subjected to SDS–PAGE on a 10% polyacrylamide gel and then analyzed by immunoblotting using mouse anti-Hsp72 monoclonal antibody. Cell lysates were also subjected to immunoblot analysis with mouse anti-Hsp72 monoclonal antibody. NIH 3T3-HS denotes NIH 3T3 cells that were pretreated with a mild heat shock at 43°C for 20 min. IgGH, the heavy chain of immunoglobulin G; IgGL, the light chain of immunoglobulin G.
The peptide binding domain of Hsp72 is critical for the inhibitory effect on JNK
Hsp72 has an ATP binding and a polypeptide binding domain in its structure (Milarski and Morimoto, 1989; Freeman et al., 1995). We investigated which of the domains might be involved in the interaction of Hsp72 with JNK. We used various Hsp72 deletion mutants. Hsp72ΔABD, Hsp72ΔPBD or Hsp72ΔN lack amino acids 120–428 (ATP binding domain), 436–618 (peptide binding domain) or 1–120 (N-terminal region), respectively. We carried out an in vitro binding assay in which in vitro-translated 35S-labeled JNK1 was applied to His-Hsp72 that was immobilized on Ni2+–agarose beads (Figure 6A). Our data show that the 35S-labeled JNK1 bound to full-length Hsp72, to Hsp72ΔABD and to Hsp72ΔN, but not to Hsp72ΔPBD. We then examined the inhibitory activities of the Hsp72 proteins on JNK1 activity in vitro (Figure 6B). The JNK1 activity was almost completely suppressed by full-length Hsp72, Hsp72ΔABD and Hsp72ΔN, but not by Hsp72ΔPBD. These data, therefore, suggest that the peptide binding domain of Hsp72 is critical for the Hsp72 interaction with JNK1 and its inhibitory effect on JNK1. These results were in excellent agreement with a previous report, demonstrating that a Hsp72 mutant lacking the ATP binding domain could inhibit JNK activation in transfected cells (Yaglom et al., 1999).
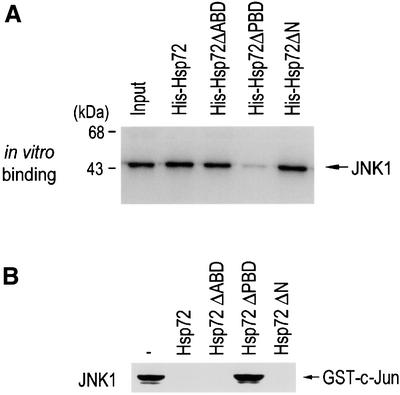
Fig. 6. The peptide binding domain of Hsp72 is critical for the suppression of JNK1 by Hsp72. (A) The peptide binding domain is essential for Hsp72 binding to JNK1 in vitro. In vitro-translated 35S-labeled JNK1 was applied to His-Hsp72 proteins that were bound to Ni2+-NTA–agarose beads. Bead-bound proteins were extensively washed, eluted and analyzed by SDS–PAGE on a 10% polyacrylamide gel and autoradiography. The input 35S-labeled protein (20%) is also shown. (B) The peptide binding domain is essential for Hsp72 to inhibit JNK1. NIH 3T3 cells were exposed to UV (60 J/m2) irradiation, lysed and subjected to immunoprecipitation with anti-JNK1 antibody. The immunopellets were incubated with 2 µg of wild-type Hsp72 or each mutant protein in 50 µl of HEPES buffer pH 7.4 for 1 h at room temperature, washed twice with HEPES buffer and then assayed for JNK1 activity.
Hsp72 suppresses the phosphorylation of JNK by SEK1
As shown in Figures 4 and 5, our data indicate that Hsp72 interacts directly with JNK both in vitro and in vivo. Concomitantly, Hsp72 inhibited the enzymic activity of JNK in vitro. These findings suggest that one possible mechanism by which Hsp72 suppresses JNK by interacting directly with the enzyme is that Hsp72 may inhibit a catalytic activity of JNK. Another possible mechanism is that Hsp72, through binding to JNK, may block the JNK phosphorylation catalyzed by a JNK kinase such as SEK1 or MKK7. We investigated this possibility by examining the effect of Hsp72 protein on the in vitro phosphorylation of JNK by SEK1 (Figure 7A). JNK3(K55R), a kinase-inactive JNK3 mutant lacking autophosphorylation activity, was used as a substrate for SEK1 in the in vitro kinase assay. Our data demonstrated that Hsp72 did not affect the SEK1-catalyzed phosphorylation of myelin basic protein, suggesting that Hsp72 did not inhibit a catalytic activity of SEK1. Interestingly, Hsp72 inhibited the JNK phosphorylation by SEK1. These data are consistent with the proposed model in which Hsp72, through binding to JNK, may interfere with the phosphorylation of JNK by SEK1. In order to test this model further, we examined the action of Hsp72 on the interaction between JNK and SEK1 in intact cells. Immunoblot analysis of the SEK1 immunoprecipitates using anti-JNK1 antibody showed in vivo binding between JNK1 and SEK1 in NIH 3T3-neo cells (Figure 7B). Ectopic expression of Hsp72 resulted in a dramatic decrease in in vivo binding between JNK1 and SEK1 in NIH 3T3-Hsp72 cells. Based on these results, it may be proposed that Hsp72, through binding to JNK, may prevent the interaction between JNK and SEK1, thereby inhibiting SEK1-catalyzed JNK phosphorylation. Similarly, ectopic expression of Hsp72 inhibited the interaction between JNK1 and MKK7 in cotransfected cells (Figure 7C). We also investigated whether Hsp72 could block the interaction between JNK1 and c-Jun in intact cells (Figure 7D). The cell lysates from NIH 3T3-neo or NIH 3T3-Hsp72 cells were immunoprecipitated with anti-c-Jun antibody, and the resultant immunopellets were analyzed by immunoblotting probed with anti-JNK1 antibody. The immunoblot data show that the physical interaction between JNK1 and its substrate, c-Jun, was reduced in NIH 3T3-Hsp72 cells, compared with NIH 3T3-neo cells.
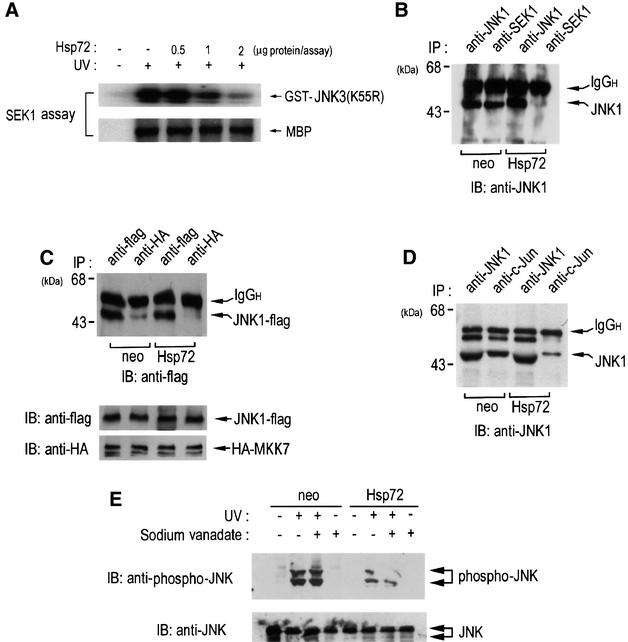
Fig. 7. Hsp72 inhibits JNK phosphorylation by SEK1. (A) NIH 3T3 cells were exposed to 60 J/m2 UV radiation, incubated further for 1 h at 37°C and then subjected to immunoprecipitation using mouse anti-SEK1 monoclonal antibody. In vitro phosphorylation of GST–JNK3(K55R) or myosin basic protein (MBP) by the SEK1 immunopellets was performed in the absence or presence of recombinant human Hsp72 protein. (B) NIH 3T3-neo or NIH 3T3-Hsp72 cells were subjected to immunoprecipitation using mouse anti-SEK1 or mouse anti-JNK1 antibody. The immunoprecipitates were subjected to SDS–PAGE and analyzed by immunoblotting using mouse anti-JNK1 antibody. IgGH, the heavy chain of immunoglobulin G. (C) NIH 3T3-neo and NIH 3T3-Hsp72 cells were transiently cotransfected with pcDNA3-JNK1-Flag and pcDNA3-HA-MKK7. After 48 h of transfection, the cell lysates were subjected to immunoprecipitation using mouse monoclonal anti-HA or anti-Flag antibody. The immunoprecipitates were analyzed by immunoblotting probed with anti-Flag antibody. The cell lysates were also immunoblotted with anti-HA or anti-Flag antibody. (D) NIH 3T3-neo or NIH 3T3-Hsp72 cells were immunoprecipitated with mouse monoclonal anti-c-Jun or mouse monoclonal anti-JNK1 antibody. The resultant immunopellets were further analyzed by immunoblotting probed with anti-JNK1 antibody. (E) NIH 3T3-neo or NIH 3T3-Hsp72 cells were pretreated with 2 mM sodium vanadate for 15 min, then irradiated with UV light (60 J/m2) and further incubated for 1 h at 37°C. Cell lysates were analyzed by immunoblotting probed with either anti-phospho-JNK antibody or anti-JNK antibody.
It is noteworthy that a recent study proposed that Hsp72 might suppress JNK activation by enhancing JNK dephosphorylation by a JNK phosphatase(s), whose activity was sensitive to protein phosphatase inhibitors, including sodium vanadate (Meriin et al., 1999). We therefore examined whether sodium vanadate could mitigate Hsp72-induced suppression of JNK activation in NIH 3T3-Hsp72 cells. As shown in Figure 7E, UV radiation induced the stimulation of JNK phosphorylation in NIH 3T3-neo control cells, and UV-stimulated JNK phosphorylation was decreased in NIH 3T3-Hsp72 cells. Pretreatment of cells with sodium vanadate failed to reverse the Hsp72-induced inhibition of the JNK phosphorylation in NIH 3T3-Hsp72 cells.
Hsp72 suppresses the JNK-dependent transactivating activity of c-Jun
One of the immediate downstream events of JNK signaling is the phosphorylation of c-Jun. When activated by its upstream kinases, JNK phosphorylates c-Jun and the c-Jun phosphorylation increases the transcription-stimulating activity of c-Jun (Derijard et al., 1994). We therefore tried to determine whether Hsp72 could modulate the JNK-dependent transcription-stimulating activity of c-Jun. We used a luciferase reporter system and the results are shown in Figure 8. Overexpression of ΔMEKK1 enhanced c-Jun-mediated luciferase reporter activity. The ΔMEKK1-induced transcription-stimulating activity of c-Jun was blocked by SEK1(K129R), a dominant-negative mutant of SEK1. The data thus indicate that ΔMEKK1 induced stimulation of the transcriptional activity of c-Jun through the JNK signaling pathway. Hsp72 suppressed ΔMEKK1-induced stimulation of the c-Jun-mediated luciferase reporter activity (Figure 8). These results suggest that Hsp72 inhibits the JNK-dependent transactivating activity of c-Jun.
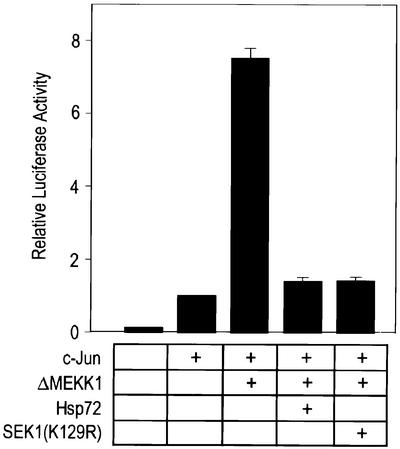
Fig. 8. Hsp72 suppresses the transactivating activity of c-Jun stimulated by the JNK pathway. NIH 3T3 cells were transiently transfected with a luciferase reporter plasmid pFR-Luc, pFA2-c-jun, pFC-MEKK1, pCMV-Hsp72 or pcDNA3-SEK1(K129R) as indicated. pcDNA3-β-gal was included in all transfections. After 50 h, the transfectants were lysed and assayed for luciferase activity. Luciferase reporter activity in each sample was normalized according to the β-galactosidase activity measured in the same sample.
Hsp72 suppresses JNK-dependent cell death
JNK has been shown to play a key role in apoptotic cell death under certain conditions (Xia et al., 1995; Chen et al., 1996; Johnson et al., 1996; Verheij et al., 1996; Park et al., 1997). We therefore examined the effect of Hsp72 on JNK-dependent cell death. Overexpression of ΔMEKK1, a constitutively active form of MEKK1, resulted in an increase in apoptotic cell death in NIH 3T3 cells, and this cell death could be repressed by SEK1(K129R) (Figure 9A). The data thus suggest that ΔMEKK1-induced apoptotic cell death may occur through the MEKK1-SEK1-JNK pathway. The DAPI-staining and TUNEL-staining data indicated that ectopically expressed Hsp72 suppressed ΔMEKK1-induced cell death in NIH 3T3-Hsp72 cells (Figure 9A). We then tested the effects of various Hsp72 mutants on ΔMEKK1-induced cell death in 293 HEK cells transiently transfected with plasmids expressing ΔMEKK1 and the Hsp72 proteins (Figure 9B). Our data show that ΔMEKK1-dependent cell death was repressed by full-length Hsp72, Hsp72ΔABD or Hsp72ΔN, but not by Hsp72ΔPBD. These findings suggest that the peptide binding domain may be critical for the Hsp72 suppression of apoptosis induced upon activation of the JNK pathway.
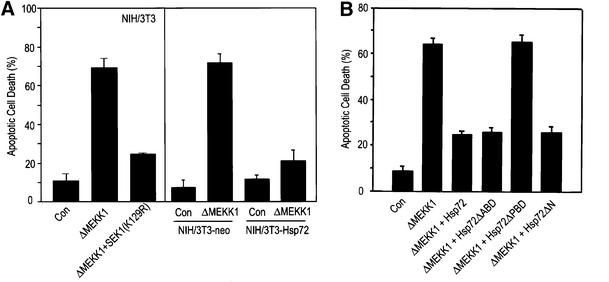
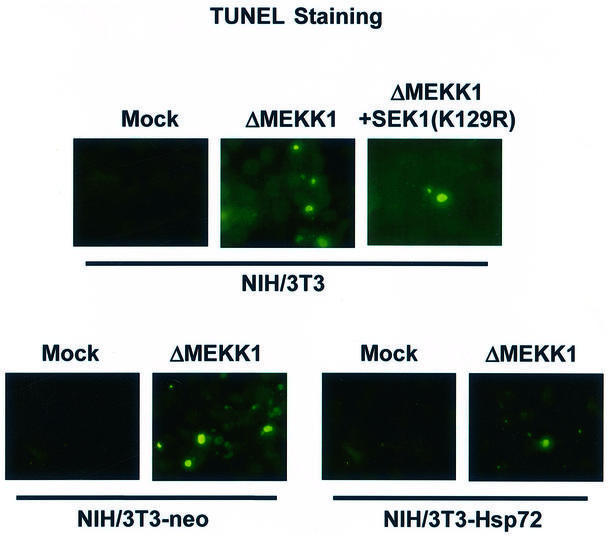
Fig. 9. Hsp72 suppresses JNK-dependent cell death. (A) NIH 3T3 cells were transiently transfected with an empty vector or vector expressing ΔMEKK1 or SEK1(K129R) along with pEGFP. NIH 3T3-neo or NIH 3T3-Hsp72 cells were transiently transfected with pcDNA3 empty vector or pcDNA3-ΔMEKK1 along with pEGFP. After 50 h of transfection, the cells were fixed and stained with DAPI. GFP-positive cells were analyzed for apoptotic nuclei with a fluorescence microscope. For TUNEL staining, the transfected cells were fixed, permeabilized and incubated with terminal deoxynucleotidyl transferase and fluorescein-labeled dUTP. TUNEL-positive cells were examined with a fluorescence microscope. (B) 293 HEK cells were transiently transfected with pcDNA3-ΔMEKK1, pCMV-Hsp72, pCMV-Hsp72ΔABD, pCMV-Hsp72ΔPBD or pCMV-Hsp72ΔN along with pEGFP as indicated. After 50 h, the transfected cells were assayed for apoptotic cell death as in (A). Data shown above are the mean ± average deviation of triplicates from one of two representative experiments.
Hsp72 antisense oligonucleotides abolish the inhibitory effect of mild heat shock on the JNK pathway
As shown in Figure 1, pretreatment of NIH 3T3 cells with a mild heat shock-induced suppression of JNK activation. Because heat shock can induce the expression of a variety of proteins including heat shock proteins, we examined whether Hsp72 induction would be a major requirement of the heat-induced suppression of JNK activation. Exposure of NIH 3T3 cells to a mild heat shock resulted in the accumulation of Hsp72 in the cells (Figure 10A). The Hsp72 induction was blocked when the cells were pretreated with Hsp72 antisense oligonucleotides, while nonsense oligonucleotides did not affect the heat shock-induced expression of Hsp72. A co-immunoprecipitation study also demonstrated that heat shock-induced Hsp72 associated physically with JNK1 in intact cells, and that the interaction between Hsp72 and JNK1 was not observed in cells pretreated with Hsp72 antisense oligonucleotides (Figure 10A). Furthermore, Hsp72 antisense oligonucleotides blocked the suppressive effect of mild heat shock on UV-induced JNK1 activation (Figure 10B) and UV-induced apoptotic cell death (Figure 10C). Taken together, these data strongly suggest that Hsp72, through binding to JNK, mediates the heat shock-induced inhibition of JNK activation and UV-induced apoptosis.
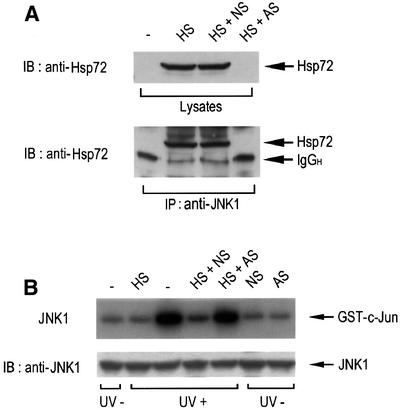
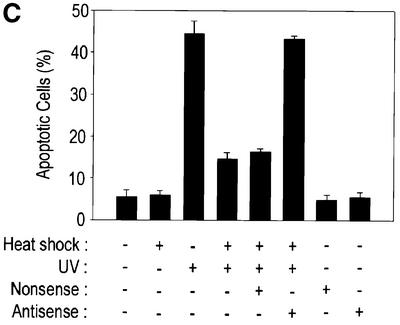
Fig. 10. Hsp72 antisense oligonucleotides block the inhibitory effect of mild heat shock on JNK activation and UV-induced apoptosis. NIH 3T3 cells were transiently transfected with 10 µM Hsp72 antisense (AS) or nonsense (NS) oligonucleotides using GenePORTER 2. After 30 h of transfection, the cells were exposed to mild heat shock (HS) at 43°C for 20 min and then further incubated at 37°C for 16 h. (A) Cell lysates were subjected to immunoblot analysis with mouse anti-Hsp72 monoclonal antibody. Cell lysates were also subjected to immunoprecipitation using mouse anti-JNK1 monoclonal antibody. The immunopellets were then analyzed by immunoblotting probed with mouse anti-Hsp72 antibody. IgGH, the heavy chain of immunoglobulin G. (B) Cells were irradiated with UV light (60 J/m2) and then incubated for 1 h. Cell lysates were then analyzed for JNK1 activity by immunocomplex kinase assay using anti-JNK1 antibody. Intracellular levels of JNK1 were determined by immunoblot analysis with anti-JNK1 antibody. (C) Cells were irradiated with UV (60 J/m2), further incubated for 16 h, and then measured for apoptotic cell death with DAPI staining as in Figure 9. Data shown above are the mean ± average deviation of triplicates from one of two representative experiments.
Discussion
In the present study, we show that heat-induced Hsp72 interacts physically with JNK in intact cells, thereby inhibiting JNK activation and JNK-dependent apoptotic cell death. Thus, our data suggest that Hsp72 suppresses the JNK signaling pathway by acting directly on JNK itself.
JNK has been shown to bind several non-substrate proteins, including p21WAF1, JIP, Rb and GSTp (Shim et al., 1996, 2000; Whitmarsh et al., 1998; Adler et al., 1999). p21, Rb or GSTp inhibits JNK through protein– protein interactions (Shim et al., 1996, 2000; Adler et al., 1999), whereas JIP serves as a scaffolding protein in the JNK pathway (Whitmarsh et al., 1998). We show here that Hsp72 is another intracellular protein that interacts directly with JNK and inhibits JNK activation. Hsp72 contains an ATP binding and a peptide binding domain (Milarski and Morimoto, 1989; Freeman et al., 1995). The ATP binding domain, in particular, plays a pivotal role in the molecular chaperone activity of Hsp72 (Ungewickell, 1985). Interestingly, our results indicate that Hsp72 without the ATP binding domain can inhibit JNK1 activity, whereas Hsp72 lacking the peptide binding domain fails to bind and inhibit JNK1. Thus, the ATP binding domain is not essential for Hsp72 binding and inhibition of JNK1, suggesting that the suppression of JNK1 by Hsp72 occurs independently of its well-characterized chaperone activity. We also observed that Hsp72 is not a substrate for JNK1 in vitro (data not shown).
Hsp72 plays a key role in cell survival under many apoptotic stress conditions including heat shock (Jaattela, 1993; Simon et al., 1995; Bellmann et al., 1996; Samali and Cotter, 1996; Gabai et al., 1997, 2000; Liossis et al., 1997; Mosser et al., 1997; Meriin et al., 1998; Sharp et al., 1999). Hsp72 has also been shown to interact with several apoptosis-regulating proteins such as Apaf-1 and Bcl-2 (Wang et al., 1999; Beere et al., 2000; Saleh et al., 2000). Several recent studies have reported that Hsp72 suppresses the JNK/SAPK signaling pathway (Gabai et al., 1997; Mosser et al., 1997; Buzzard et al., 1998; Gabai et al., 1998, 2000; Meriin et al., 1998, 1999; Volloch et al., 1998; Yaglom et al., 1999). Since this signaling pathway is associated with cellular mechanisms operating in apoptotic cell death under certain conditions (Jaattela, 1993; Simon et al., 1995; Bellmann et al., 1996; Chen et al., 1996; Samali and Cotter, 1996; Verheij et al., 1996; Liossis et al., 1997; Mosser et al., 1997; Nishina et al., 1997; Seimiya et al., 1997; Meriin et al., 1998; Gabai et al., 2000), blockage of the JNK signaling pathway appears to be a major mechanism by which Hsp72 protects cells from stress-induced apoptosis. In the present study, we observed both in vivo and in vitro binding between the two proteins Hsp72 and JNK. Furthermore, Hsp72 inhibited the interaction between JNK and SEK1 in intact cells and SEK1-catalyzed JNK phosphorylation. Based on these results, we propose that Hsp72, through binding to JNK, may prevent JNK activation by a JNK kinase such as SEK1. Our results also suggest that Hsp72, through binding to JNK, may inhibit an enzymic activity of JNK as well as the interaction between JNK and its substrate, c-Jun.
Exposure of cells to a variety of cellular stresses, including heat shock, can lead to the stimulation of JNK, which may then mediate intracellular signals inducing apoptotic cell death under certain conditions (Xia et al., 1995; Chen et al., 1996; Verheij et al., 1996; Zanke et al., 1996; Seimiya et al., 1997; Shim et al., 2000). Many stress signals activating the JNK pathway can also induce Hsp72 production. A certain stress such as oxidative stress activates JNK, which in turn induces hsp72 gene expression (Lee and Corry, 1998). The induced Hsp72 could then protect the cells from the cytotoxic stress. Our observation that Hsp72 suppresses JNK activation through binding directly to JNK may, therefore, suggest a feedback inhibitory mechanism for the regulation of the JNK signaling pathway in response to cellular stress. In summary, our study highlights that Hsp72 is a natural inhibitory protein of JNK in intact cells.
Materials and methods
Cell culture and transfections
NIH 3T3 cells were routinely maintained at 37°C in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. For a stable transfection, NIH 3T3 cells were transfected with pCMV empty vector or pCMV-Hsp72 by the calcium phosphate method (Chen and Okayama, 1987). After 48 h of transfection, the cells were replated at a 1/20 dilution and maintained in complete medium containing 500 µg/ml G418 (Gibco BRL) to select neomycin-resistant cells.
Cloning and preparation of recombinant Hsp72 proteins
A full-length human hsp72 gene from pH2.3 (ATCC #57495) was subcloned into the pCMV mammalian expression vector or the bacterial expression vector pET30a (Novagen). The Hsp72 deletion mutants constructed for the present study were as follows: Hsp72ΔABD lacking a BglII–BglII fragment (ATP binding domain: amino acid residues 120–428), Hsp72ΔPBD lacking a SmaI–SmaI fragment (peptide binding domain: amino acid residues 436–618) and Hsp72ΔN lacking a NcoI–NcoI fragment (amino acid residues 1–120). Escherichia coli BL21 was transformed with pET30a expressing His6-tagged Hsp72 proteins. Expression of the recombinant His-Hsp72 proteins in the transformed bacteria was induced by 1 mM isopropyl-β-d-thiogalactopyranoside (Sigma). Full-length His-Hsp72 and its mutant proteins were purified using Ni2+-NTA–agarose (Qiagen) according to the manufacturer’s protocol.
Immunocomplex kinase assays
Cultured cells were harvested and lysed in buffer A, containing 50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride (PMSF), 2 µg/ml leupeptin, 2 µg/ml aprotinin, 25 mM glycerophosphate, 0.1 mM sodium orthovanadate, 1 mM sodium fluoride, 1% NP-40, 0.5% deoxycholate and 0.1% SDS. Cell lysates were subjected to centrifugation at 12 000 g for 10 min at 4°C, and the resultant soluble fraction was subjected to immunoprecipitation using an appropriate antibody. The immunopellets were assayed for the protein kinases indicated as described previously (Shim et al., 1996). Phosphorylated substrates were separated by SDS–PAGE, and quantified using a Fuji BAS 2500 phosphorimager. The GST fusion proteins to be used as substrates were expressed in E.coli using pGEX-2T (Pharmacia) and purified using glutathione–Sepharose as described previously (Shim et al., 1996). Antibodies used in the JNK1, ERK2, p38, SEK1 and MEKK1 assays were as follows: mouse monoclonal anti-JNK1 (Pharmingen), rabbit polyclonal anti-ERK2 (Upstate Biotechnology, Inc.), mouse monoclonal anti-p38 (Pharmingen), mouse monoclonal anti-SEK1 (Pharmingen) and rabbit polyclonal anti-MEKK1 (Pharmingen) antibody. Protein concentrations were determined by the Bradford method (Bio-Rad).
Immunoblot analysis
Cultured cells were harvested and lysed in phosphate-buffered saline (PBS) containing 1 mM PMSF, 2 µg/ml leupeptin and 2 µg/ml aprotinin. Cell lysates were clarified by microcentrifugation at 12 000 g for 10 min at 4°C, and the resultant soluble fraction was subjected to SDS–PAGE. After gel electrophoresis, the separated proteins were transferred by electroblotting onto Hybond ECL nitrocellulose (Amersham). The nitrocellulose was then blocked with Tris-buffered saline solution pH 7.4, containing 0.1% Tween 20 and 5% non-fat milk. The blotted proteins were probed with a mouse monoclonal anti-Hsp72 (StressGene) or a mouse monoclonal anti-JNK1 (Pharmingen) antibody. The secondary antibody was a rabbit anti-mouse IgG antibody conjugated to horseradish peroxidase (Amersham). The blots were developed with an enhanced chemiluminescence system (Amersham).
In vitro binding and co-immunoprecipitation
GST–JNK1 and GST–SEK1 were expressed in E.coli using pGEX-4T vector, and purified using glutathione–agarose. His-Hsp72 was bacterially expressed using the pET30a vector and purified using Ni2+-NTA–agarose. Purified His-Hsp72 (1 µg) was incubated at 4°C for 10 h, with each GST fusion protein immobilized on glutathione–agarose beads in 0.5 ml of buffer B, containing 50 mM NaCl, 50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM PMSF, 2 µg/ml leupeptin, 2 µg/ml aprotinin, 25 mM glycerophosphate, 0.1 mM sodium orthovanadate, 1 mM sodium fluoride, 1% NP-40 and 10% glycerol. After the beads had been washed three times with 20 mM HEPES pH 7.4, the bound proteins were eluted from the beads and visualized by SDS–PAGE and immunoblotting using anti-Hsp72 antibody. In a separate binding assay, each GST fusion protein (1 µg) was incubated at 4°C for 10 h with His-Hsp72 immobilized on Ni2+-NTA–agarose beads. The bound proteins were washed, eluted from the beads, separated by SDS–PAGE and visualized by immunoblotting using anti-GST antibody. In a pull-down binding assay, NIH 3T3 cell lysates (2 mg protein) were applied to His-Hsp72 immobilized on Ni2+-NTA–agarose beads. The bound proteins were eluted, separated by SDS–PAGE and analyzed by immunoblotting using anti-JNK1, anti-ERK2 or anti-p38 antibody. In order to look for physical interaction between Hsp72 and JNK1 in intact cells, heat shock-exposed NIH 3T3, NIH 3T3-Hsp72 and NIH 3T3-neo cells were lysed in buffer A, and subjected to immunoprecipitation using mouse monoclonal anti-JNK1 antibody. The immunopellets were then analyzed by SDS–PAGE and immunoblotting using mouse monoclonal anti-Hsp72 antibody.
Luciferase reporter assay of c-Jun-dependent transcription
The transcription-stimulating activity of c-Jun was determined using the PathDetect luciferase reporter kit (Stratagene). NIH 3T3 cells were transiently transfected with pFR-Luc, pFA2-c-jun, pFC-MEKK1, pCMV-Hsp72 and pcDNA3-SEK1(K129R), along with pcDNA3-β-gal by the calcium phosphate method. After 50 h of transfection, the cell lysates were subjected to microcentrifugation at 4°C for 10 min and the soluble fraction was measured for luciferase activity using a luciferase assay kit (Promega). The luciferase reporter activities in the transfected cells were normalized with reference to β-galactosidase activities in the same cells.
Apoptotic cell death
Cultured cells were cotransfected with appropriate plasmid vectors plus pEGFP using Lipofectamine (Gibco BRL). After 50 h of transfection, the cells were fixed with 0.25% glutaraldehyde and stained with 4′,6-diamidino-2-phenyl-indole dihydrochloride (DAPI). The DAPI-stained nuclei in GFP-positive cells were analyzed for apoptotic morphology by fluorescence microscopy. The percentage of apoptotic cells was calculated as the number of GFP-positive cells with apoptotic nuclei divided by the total number of GFP-positive cells. For UV-induced apoptosis, NIH 3T3 cells were exposed to UV light and incubated for an additional 1 h. The cells were then stained with DAPI and examined by fluorescence microscopy. In some experiments, apoptotic cell death was also analyzed by the terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay. The TUNEL staining was carried out using the in situ cell death detection kit (Roche Molecular Biochemicals) according to the manufacturer’s protocol. Briefly, cells in culture were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100/0.1% sodium citrate solution and then incubated for 60 min at 37°C with TUNEL reaction mixture containing terminal deoxynucleotidyl transferase and fluorescein-labeled dUTP. TUNEL-positive cells were detected by fluorescence microscopy.
Antisense oligonucleotides of Hsp72
NIH 3T3 cells were transiently transfected with 10 µM of antisense (5′-CACCTTGCCGTGCTGGAA-3′; nucleotides 61–78) or nonsense (5′-TGGATCCGACATGTCAGA-3′) oligonucleotides of the inducible hsp72 gene as reported previously (Robertson et al., 1999). Transfection was performed using GenePORTER 2 (Gene Therapy Systems). After 8 h of transfection, the cells were washed twice with PBS and again transfected with 10 µM antisense or nonsense oligonucleotides. After 48 h, the cells were used in the experiments indicated.
Acknowledgments
Acknowledgements
We thank R.J.Davis, M.Karin and L.I.Zon for providing JNK1, MEKK1 and SEK1 cDNA clones, respectively, and Dr G.Hoschek for critical reading of the manuscript. This work was supported by the Creative Research Initiatives Program of the Korean Ministry of Science and Technology (E.-J.C.), and in part by a grant (#98-0403-1901-5) from the Korea Science and Engineering Foundation (J.-S.S.).
REFERENCES
- Adler V. et al. (1999) Regulation of JNK signaling by GSTp. EMBO J., 18, 1321–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral M.D., Galego,L. and Rodrigues-Pousada,C. (1993) Heat-shock-induced protein synthesis is responsible for the switch-off of hsp70 transcription in Tetrahymena. Biochim. Biophys. Acta, 1174, 133–142. [DOI] [PubMed] [Google Scholar]
- Beckmann R.P., Mizzen,L.E. and Welch,W.J. (1990) Interaction of Hsp 70 with newly synthesized proteins: implications for protein folding and assembly. Science, 248, 850–854. [DOI] [PubMed] [Google Scholar]
- Beere H.M. et al. (2000) Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nature Cell Biol., 2, 469–475. [DOI] [PubMed] [Google Scholar]
- Bellmann K., Jaattela,M., Wissing,D., Burkart,V. and Kolb,H. (1996) Heat shock protein hsp70 overexpression confers resistance against nitric oxide. FEBS Lett., 391, 185–188. [DOI] [PubMed] [Google Scholar]
- Bimston D., Song,J., Winchester,D., Takayama,S., Reed,J.C. and Morimoto,R.I. (1998) BAG-1, a negative regulator of Hsp70 chaperone activity, uncouples nucleotide hydrolysis from substrate release. EMBO J., 17, 6871–6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzzard K.A., Giaccia,A.J., Killender,M. and Anderson,R.L. (1998) Heat shock protein 72 modulates pathways of stress-induced apoptosis. J. Biol. Chem., 273, 17147–17153. [DOI] [PubMed] [Google Scholar]
- Cano E. and Mahadevan,L.C. (1995) Parallel signal processing among mammalian MAPKs. Trends Biochem. Sci., 20, 117–122. [DOI] [PubMed] [Google Scholar]
- Chen C. and Okayama,H. (1987) High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol., 7, 2745–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.R., Wang,X., Templeton,D., Davis,R.J. and Tan,T.H. (1996) The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and γ radiation. Duration of JNK activation may determine cell death and proliferation. J. Biol. Chem., 271, 31929–31936. [DOI] [PubMed] [Google Scholar]
- Chirico W.J., Waters,M.G. and Blobel,G. (1988) 70K heat shock related proteins stimulate protein translocation into microsomes. Nature, 332, 805–810. [DOI] [PubMed] [Google Scholar]
- Derijard B., Hibi,M., Wu,I.H., Barrett,T., Su,B., Deng,T., Karin,M. and Davis,R.J. (1994) JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell, 76, 1025–1037. [DOI] [PubMed] [Google Scholar]
- Flynn G.C., Chappell,T.G. and Rothman,J.E. (1989) Peptide binding and release by proteins implicated as catalysts of protein assembly. Science, 245, 385–390. [DOI] [PubMed] [Google Scholar]
- Freeman B.C., Myers,M.P., Schumacher,R. and Morimoto,R.I. (1995) Identification of a regulatory motif in Hsp70 that affects ATPase activity, substrate binding and interaction with HDJ-1. EMBO J., 14, 2281–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frydman J. and Hartl,F.-U. (1994) Molecular chaperone functions of hsp70 and hsp60 in protein folding. In Morimoto,R.I., Tissieres,A. and Georgopoulos,C. (eds), The Biology of Heat Shock Proteins and Molecular Chaperones. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 251–283.
- Gabai V.L., Meriin,A.B., Mosser,D.D., Caron,A.W., Rits,S., Shifrin,V.I. and Sherman,M.Y. (1997) Hsp70 prevents activation of stress kinases. A novel pathway of cellular thermotolerance. J. Biol. Chem., 272, 18033–18037. [DOI] [PubMed] [Google Scholar]
- Gabai V.L., Meriin,A.B., Yaglom,J.A., Volloch,V.Z. and Sherman,M.Y. (1998) Role of Hsp70 in regulation of stress-kinase JNK: implications in apoptosis and aging. FEBS Lett., 438, 1–4. [DOI] [PubMed] [Google Scholar]
- Gabai V.L., Yaglom,J.A., Volloch,V., Meriin,A.B., Force,T., Koutroumanis,M., Massie,B., Mosser,D.D. and Sherman,M.Y. (2000) Hsp72-mediated suppression of c-Jun N-terminal kinase is implicated in development of tolerance to caspase-independent cell death. Mol. Cell. Biol., 20, 6826–6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S.A., Hoffman,R.A., Simmons,R.L. and Ford,H.R. (1997) Induction of heat shock protein 70 protects thymocytes against radiation-induced apoptosis. Arch. Surg., 132, 1277–1282. [DOI] [PubMed] [Google Scholar]
- Gupta S., Campbell,D., Derijard,B. and Davis,R.J. (1995) Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science, 267, 389–393. [DOI] [PubMed] [Google Scholar]
- Hohfeld J., Minami,Y. and Hartl,F.U. (1995) Hip, a novel cochaperone involved in the eukaryotic Hsc70/Hsp40 reaction cycle. Cell, 83, 589–598. [DOI] [PubMed] [Google Scholar]
- Ip Y.T. and Davis,R.J. (1998) Signal transduction by the c-Jun N-terminal kinase (JNK)–from inflammation to development. Curr. Opin. Cell Biol., 10, 205–219. [DOI] [PubMed] [Google Scholar]
- Jaattela M. (1993) Overexpression of major heat shock protein hsp70 inhibits tumor necrosis factor-induced activation of phospholipase A2. J. Immunol., 151, 4286–4294. [PubMed] [Google Scholar]
- Jaattela M., Wissing,D., Kokholm,K., Kallunki,T. and Egeblad,M. (1998) Hsp70 exerts its anti-apoptotic function downstream of caspase-3-like proteases. EMBO J., 17, 6124–6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson N.L. et al. (1996) Signal transduction pathways regulated by mitogen-activated/extracellular response kinase kinase kinase induce cell death. J. Biol. Chem., 271, 3229–3237. [DOI] [PubMed] [Google Scholar]
- Langer T. and Neupert,W. (1991) Heat shock proteins hsp60 and hsp70: their roles in folding, assembly and membrane translocation of proteins. Curr. Top. Microbiol. Immunol., 167, 3–30. [DOI] [PubMed] [Google Scholar]
- Lee Y.J. and Corry,P.M. (1998) Metabolic oxidative stress-induced HSP70 gene expression is mediated through SAPK pathway. Role of Bcl-2 and c-Jun NH2-terminal kinase. J. Biol. Chem., 273, 29857–29863. [DOI] [PubMed] [Google Scholar]
- Li G.C., Mivechi,N.F. and Weitzel,G. (1995) Heat shock proteins, thermotolerance, and their relevance to clinical hyperthermia. Int. J. Hypertherm., 11, 459–488. [DOI] [PubMed] [Google Scholar]
- Liossis S.N., Ding,X.Z., Kiang,J.G. and Tsokos,G.C. (1997) Overexpression of the heat shock protein 70 enhances the TCR/CD3- and Fas/Apo-1/CD95-mediated apoptotic cell death in Jurkat T cells. J. Immunol., 158, 5668–5675. [PubMed] [Google Scholar]
- Meriin A.B., Gabai,V.L., Yaglom,J., Shifrin,V.I. and Sherman,M.Y. (1998) Proteasome inhibitors activate stress kinases and induce Hsp72. Diverse effects on apoptosis. J. Biol. Chem., 273, 6373–6379. [DOI] [PubMed] [Google Scholar]
- Meriin A.B., Yaglom,J.A., Gabai,V.L., Mosser,D.D., Zon,L. and Sherman,M.Y. (1999) Protein-damaging stresses activate c-Jun N-terminal kinase via inhibition of its dephosphorylation: a novel pathway controlled by HSP72. Mol. Cell. Biol., 19, 2547–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milarski K.L. and Morimoto,R.I. (1989) Mutational analysis of the human HSP70 protein: distinct domains for nucleolar localization and adenosine triphosphate binding. J. Cell Biol., 109, 1947–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami Y., Hohfeld,J., Ohtsuka,K. and Hartl,F.U. (1996) Regulation of the heat-shock protein 70 reaction cycle by the mammalian DnaJ homolog, Hsp40. J. Biol. Chem., 271, 19617–19624. [DOI] [PubMed] [Google Scholar]
- Minden A. and Karin,M. (1997) Regulation and function of the JNK subgroup of MAP kinases. Biochim. Biophys. Acta, 1333, F85–F104. [DOI] [PubMed] [Google Scholar]
- Minden A., Lin,A., Claret,F.X., Abo,A. and Karin,M. (1995) Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell, 81, 1147–1157. [DOI] [PubMed] [Google Scholar]
- Mosser D.D., Caron,A.W., Bourget,L., Denis-Larose,C. and Massie,B. (1997) Role of the human heat shock protein hsp70 in protection against stress-induced apoptosis. Mol. Cell. Biol., 17, 5317–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami H., Pain,D. and Blobel,G. (1988) 70-kD heat shock-related protein is one of at least two distinct cytosolic factors stimulating protein import into mitochondria. J. Cell Biol., 107, 2051–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishina H. et al. (1997) Stress-signalling kinase Sek1 protects thymocytes from apoptosis mediated by CD95 and CD3. Nature, 385, 350–353. [DOI] [PubMed] [Google Scholar]
- Palleros D.R., Reid,K.L., Shi,L., Welch,W.J. and Fink,A.L. (1993) ATP-induced protein–Hsp70 complex dissociation requires K+ but not ATP hydrolysis. Nature, 365, 664–666. [DOI] [PubMed] [Google Scholar]
- Park J., Kim,I., Oh,Y.J., Lee,K.-w., Han,P.-L. and Choi,E.-J. (1997) Activation of c-Jun N-terminal kinase antagonizes an anti-apoptotic action of Bcl-2. J. Biol. Chem., 272, 16725–16728. [DOI] [PubMed] [Google Scholar]
- Robertson J.D., Datta,K., Biswal,S.S. and Kehrer,J.P. (1999) Heat-shock protein 70 antisense oligomers enhance proteasome inhibitor-induced apoptosis. Biochem. J., 344, 477–485. [PMC free article] [PubMed] [Google Scholar]
- Saleh A., Srinivasula,S.M., Balkir,L., Robbins,P.D. and Alnemri,E.S. (2000) Negative regulation of the Apaf-1 apoptosome by Hsp70. Nature Cell Biol., 2, 476–483. [DOI] [PubMed] [Google Scholar]
- Samali A. and Cotter,T.G. (1996) Heat shock proteins increase resistance to apoptosis. Exp. Cell Res., 223, 163–170. [DOI] [PubMed] [Google Scholar]
- Schaeffer H.J. and Weber,M.J. (1999) Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol. Cell. Biol., 19, 2435–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid D., Baici,A., Gehring,H. and Christen,P. (1994) Kinetics of molecular chaperone action. Science, 263, 971–973. [DOI] [PubMed] [Google Scholar]
- Seimiya H., Mashima,T., Toho,M. and Tsuruo,T. (1997) c-Jun NH2-terminal kinase-mediated activation of interleukin-1β converting enzyme/CED-3-like protease during anticancer drug-induced apoptosis. J. Biol. Chem., 272, 4631–4636. [DOI] [PubMed] [Google Scholar]
- Sharp F.R., Massa,S.M. and Swanson,R.A. (1999) Heat-shock protein protection. Trends Neurosci., 22, 97–99. [DOI] [PubMed] [Google Scholar]
- Shi Y. and Thomas,J.O. (1992) The transport of proteins into the nucleus requires the 70-kilodalton heat shock protein or its cytosolic cognate. Mol. Cell. Biol., 12, 2186–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim J., Lee,H., Park,J., Kim,H. and Choi,E.-J. (1996) A non-enzymatic p21 protein inhibitor of stress-activated protein kinases. Nature, 381, 804–806. [DOI] [PubMed] [Google Scholar]
- Shim J. et al. (2000) Rb protein downregulates the stress-activated signals through inhibiting c-Jun N-terminal kinase/stress-activated protein kinase. J. Biol. Chem., 275, 14107–14111. [DOI] [PubMed] [Google Scholar]
- Simon M.M., Reikerstorfer,A., Schwarz,A., Krone,C., Luger,T.A., Jaattela,M. and Schwarz,T. (1995) Heat shock protein 70 overexpression affects the response to ultraviolet light in murine fibroblasts. Evidence for increased cell viability and suppression of cytokine release. J. Clin. Invest., 95, 926–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama S., Bimston,D.N., Matsuzawa,S., Freeman,B.C., Aime-Sempe,C., Xie,Z., Morimoto,R.I. and Reed,J.C. (1997) BAG-1 modulates the chaperone activity of Hsp70/Hsc70. EMBO J., 16, 4887–4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungewickell E. (1985) The 70-kd mammalian heat shock proteins are structurally and functionally related to the uncoating protein that releases clathrin triskelia from coated vesicles. EMBO J., 4, 3385–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheij M. et al. (1996) Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature, 380, 75–79. [DOI] [PubMed] [Google Scholar]
- Volloch V., Mosser,D.D., Massie,B. and Sherman,M.Y. (1998) Reduced thermotolerance in aged cells results from a loss of an hsp72-mediated control of JNK signaling pathway. Cell Stress Chaperones, 3, 265–271. [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Knowlton,A.A., Christensen,T.G., Shih,T. and Borkan,S.C. (1999) Prior heat stress inhibits apoptosis in adenosine triphosphate-depleted renal tubular cells. Kidney Int., 55, 2224–2235. [DOI] [PubMed] [Google Scholar]
- Whitmarsh A.J., Cavanagh,J., Tournier,C., Yasuda,J. and Davis,R.J. (1998) A mammalian scaffold complex that selectively mediates MAP kinase activation. Science, 281, 1671–1674. [DOI] [PubMed] [Google Scholar]
- Wu B., Hunt,C. and Morimoto,R. (1985) Structure and expression of the human gene encoding major heat shock protein HSP70. Mol. Cell. Biol., 5, 330–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z., Dickens,M., Raingeaud,J., Davis,R. and Greenberg,M. (1995) Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science, 270, 1326–1331. [DOI] [PubMed] [Google Scholar]
- Yaglom J.A., Gabai,V.L., Meriin,A.B., Mosser,D.D. and Sherman,M.Y. (1999) The function of HSP72 in suppression of c-Jun N-terminal kinase activation can be dissociated from its role in prevention of protein damage. J. Biol. Chem., 274, 20223–20228. [DOI] [PubMed] [Google Scholar]
- Yang S.H., Whitmarsh,A.J., Davis,R.J. and Sharrocks,A.D. (1998) Differential targeting of MAP kinases to the ETS-domain transcription factor Elk-1. EMBO J., 17, 1740–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanke B.W., Boudreau,K., Rubie,E., Winnett,E., Tibbles,L.A., Zon,L., Kyriakis,J., Liu,F.F. and Woodgett,J.R. (1996) The stress-activated protein kinase pathway mediates cell death following injury induced by cis-platinum, UV irradiation or heat. Curr. Biol., 6, 606–613. [DOI] [PubMed] [Google Scholar]