

Abstract
Twinfilin is a ubiquitous and abundant actin monomer–binding protein that is composed of two ADF-H domains. To elucidate the role of twinfilin in actin dynamics, we examined the interactions of mouse twinfilin and its isolated ADF-H domains with G-actin. Wild-type twinfilin binds ADP-G-actin with higher affinity (KD = 0.05 μM) than ATP-G-actin (KD = 0.47 μM) under physiological ionic conditions and forms a relatively stable (koff = 1.8 s−1) complex with ADP-G-actin. Data from native PAGE and size exclusion chromatography coupled with light scattering suggest that twinfilin competes with ADF/cofilin for the high-affinity binding site on actin monomers, although at higher concentrations, twinfilin, cofilin, and actin may also form a ternary complex. By systematic deletion analysis, we show that the actin-binding activity is located entirely in the two ADF-H domains of twinfilin. Individually, these domains compete for the same binding site on actin, but the C-terminal ADF-H domain, which has >10-fold higher affinity for ADP-G-actin, is almost entirely responsible for the ability of twinfilin to increase the amount of monomeric actin in cosedimentation assays. Isolated ADF-H domains associate with ADP-G-actin with rapid second-order kinetics, whereas the association of wild-type twinfilin with G-actin exhibits kinetics consistent with a two-step binding process. These data suggest that the association with an actin monomer induces a first-order conformational change within the twinfilin molecule. On the basis of these results, we propose a kinetic model for the role of twinfilin in actin dynamics and its possible function in cells.
INTRODUCTION
The actin cytoskeleton plays a fundamental role in diverse cell biological processes, such as endocytosis, exocytosis, cell motility, and cytokinesis. Each of these processes requires accurate regulation of the structure and dynamics of actin filaments by a large number of actin-binding proteins. These proteins interact with G-actin or filaments and regulate different aspects of actin filament turnover (for review, see Pollard et al., 2000).
Twinfilin is a small, 40-kDa, actin monomer–binding protein originally isolated from yeast, Saccharomyces cerevisiae (Goode et al., 1998; Lappalainen et al., 1998). More recently, homologues of yeast twinfilin were identified in mammals, Drosophila melanogaster, Caenorhabditis elegans, and Schizosaccharomyces pombe, suggesting that twinfilins are ubiquitous in eukaryotes from yeast to mammals (Vartiainen et al., 2000; Wahlström et al., 2001). Twinfilins are composed of two ADF/cofilin-like (ADF-H) domains connected by a short linker region and followed by a C-terminal tail (Palmgren et al., 2002). ADF/cofilins are ubiquitous proteins that interact with both G-actin and filaments and promote rapid actin dynamics by increasing the depolymerization rate at the minus end of actin filaments (for review, see Bamburg et al., 1999).
Unlike ADF/cofilins, twinfilins do not interact with F-actin or promote filament depolymerization. All twinfilins characterized to date are actin monomer–binding proteins that prevent actin filament assembly (Goode et al., 1998; Vartiainen et al., 2000; Wahlström et al., 2001). Twinfilin forms a 1:1 complex with G-actin, based on F-actin sedimentation assays, in which twinfilin shifts G-actin to the supernatant in a 1:1 molar ratio (Goode et al., 1998). Furthermore, the migration of twinfilin-actin complex in a sucrose gradient is consistent with a 1:1 molar ratio complex (Vartiainen et al., 2000). Yeast twinfilin inhibits the spontaneous nucleotide exchange of G-actin in a manner similar to that of ADF/cofilins (Hawkins et al., 1993; Hayden et al., 1993; Goode et al., 1998). Native gel electrophoresis demonstrated that yeast twinfilin forms a stronger complex with ADP-G-actin than ATP-G-actin in low-salt conditions (Palmgren et al., 2001). However, there is currently no information about the affinity of twinfilin for G-actin under physiological conditions or about the role of its two ADF-H domains in actin binding.
Twinfilin appears to be involved in actin-based cytoskeletal activities in cells. It is ubiquitously expressed in mouse and Drosophila tissues and is involved in cytoskeletal remodeling during development (Vartiainen et al., 2000; Wahlström et al., 2001). In cells, twinfilin shows diffuse cytoplasmic localization but is also concentrated to actin filament structures (Vartiainen et al., 2000; Palmgren 2001; Wahlström et al., 2001). Deletion of the twinfilin gene in yeast results in abnormal cortical actin patches, defects in bipolar bud-site selection pattern, and a synthetic lethality with certain cofilin and profilin mutations (Goode et al., 1998; Wolven et al., 2000). Mutation in the Drosophila twinfilin gene results in small adult size, rough eye phenotype, and aberrant bristle morphology. These phenotypes arise from uncontrolled polymerization of actin filaments in the absence of twinfilin, demonstrating that twinfilin is intimately involved in the regulation of actin filament assembly in cells (Wahlström et al., 2001). Although the mechanism by which twinfilin regulates actin dynamics is not known, studies on budding yeast suggest that twinfilin may contribute to actin filament turnover by localizing G-actin to the sites of rapid actin filament assembly. The localization of twinfilin to the sites of rapid actin filament assembly is mediated through direct interactions between twinfilin and capping protein (Palmgren et al., 2001).
To understand the molecular mechanism by which twinfilin contributes to actin dynamics, it is essential to elucidate how twinfilin and its individual ADF-H domains interact with G-actin. It is still unclear why twinfilin, which forms a 1:1 complex with G-actin, is composed of two ADF-H domains. Here, we show that mouse twinfilin competes with ADF/cofilin in binding to G-actin and forms an ∼10-fold stronger complex with ADP-G-actin than with ATP-G-actin. The strong actin monomer–binding and –sequestering activities reside in the C-terminal ADF-H domain of twinfilin. However, kinetic analyses suggest that the actin monomer may first associate with the N-terminal ADF-H domain and is then delivered to the C-terminal ADF-H domain through a conformational change within the twinfilin molecule. On the basis of these results, we propose a kinetic model for the role of twinfilin in actin filament turnover.
MATERIALS AND METHODS
Construction of Twinfilin Deletion Mutants
The desired fragments of the mouse twinfilin cDNA were amplified by PCR with oligonucleotides that created NcoI and HindIII sites at the 5′ and 3′ ends of the final DNA fragments, respectively. These fragments were ligated into NcoI- and HindIII-digested pGAT2T plasmid backbone (Peränen et al., 1996) to create plasmids pPL81 (Twf1–142, corresponding to twinfilin residues 1–142), pPL82 (Twf1–174), pPL83 (Twf141–350), pPL84 (Twf169–350), pPL89 (Twf141–322), and pPL90 (Twf169–322). Constructs were then sequenced by the chain-termination method to verify the correct sequence.
Protein Expression and Purification
Mouse wild-type twinfilin and deletion proteins were expressed as glutathione-S-transferase (GST)-fusion proteins in Escherichia coli BL21 (DE3) cells as described (Vartiainen et al., 2000). GST-fusion proteins were enriched from the lysis supernatant with glutathione agarose beads (Sigma, St. Louis, MO), twinfilin was cleaved off the GST by 0.05 mg/ml thrombin, and wild-type twinfilin and deletion proteins were then further purified with Q-Sepharose high-performance anion-exchange and Superdex-75 HiLoad gel filtration columns (Amersham Biosciences AB, Uppsala, Sweden). Wild-type twinfilin eluted from the Superdex-75 column in 60 ml, whereas the deletion proteins eluted in 65–75 ml. The peak fractions containing desired proteins were pooled, concentrated in a Centricon 10-kDa cutoff device to a final concentration of ∼200 μM, frozen in liquid N2, and stored at −70°C. Rabbit muscle actin was purified from acetone powder as described by Spudich and Watt (1971) and clarified by spinning at 75,000 rpm for 5 min in a TLA 100.1 rotor before usage. Human cofilin was purified as described by Yeoh et al. (2002).
Actin Filament Sedimentation Assays
For actin monomer sequestering assays, 3.75 μM rabbit muscle actin was polymerized for 30 min in F-buffer (0.1 M KCl, 1 mM MgCl2, 1 mM ATP, 20 mM Tris, pH 7.5). Ten-microliter aliquots of 0, 10, 20, 30, or 60 μM twinfilin/deletion proteins in G-buffer (20 mM Tris, pH 7.5, 0.2 mM ATP, 0.2 mM DTT, 0.2 mM CaCl2) were mixed with 40 μl of the prepolymerized actin filaments and incubated for 30 min. Reactions were then centrifuged in a Beckman Optima MAX Ultracentrifuge in a TLA-100 rotor at 75,000 rpm for 30 min. Equal proportions of supernatants and pellets were fractionated on 12% SDS-PAGE gels, and proteins were visualized by Coomassie staining. All the steps were carried out at room temperature.
Actin Monomer–Binding Assays
The change in the fluorescence of 7-chloro-4-nitrobenz-2-oxa-1,3-diazole (NBD)-labeled G-actin was used to monitor the binding of twinfilin constructs and cofilin to G-actin as described (Carlier et al., 1997). Actin was labeled by NBD-Cl as described in Detmers et al. (1981) and modified by Weeds et al. (1986). The extent of NBD labeling of actin used in these experiments was between 65 and 70%, ∼90% of which is supposed to reside in the lysine-373. ADP-actin was prepared by incubating NBD-actin with hexokinase-agarose beads (Sigma) and 1 mM glucose o/n at +4°C, as described (Pollard, 1986). Experiments were carried out at room temperature in G-buffer [5 mM Tris-HCl, pH 8.0, 0.1 mM CaCl2, 0.2 mM DTT, 0.2 mM ADP (or 0.1 mM ATP), and 0.5 mg/ml BSA] or F-buffer [5 mM Tris-HCl, pH 8.0, 0.08 mM CaCl2, 0.2 mM DTT, 0.2 mM ADP (or 0.1 mM ATP), 0.5 mg/ml BSA, 0.1 M KCl, and 1 mM MgCl2]. The normalized enhancement or decrease of fluorescence,
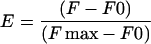 |
was measured with a BioLogic MOS250 fluorometer at each concentration of twinfilin or cofilin with an excitation at 482 nm and emission at 535 nm. The data were analyzed and fitted by use of the equation
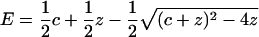 |
where
 |
and
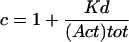 |
In the competition assays, binding was plotted as a function of increasing amounts of competitor in the presence of constant amounts of actin and inhibitor. The data were fitted using the approximated equation
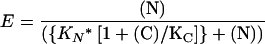 |
where variable [N] is the concentration of the competing N-terminal domain, [C] is the constant concentration of the C-terminal domain, and KN and KC are the respective dissociation constants. The apparent KN values were obtained by fitting the dissociation curve for several concentrations of C-terminal domain while keeping KC = 0.03 μM constant.
Analytical Gel Filtration Assay
Cofilin, twinfilin, and NBD-actin (in F-buffer containing 0.2 mM ADP) were mixed in various ratios and incubated for 10 min at room temperature. The formed complexes were then characterized by gel filtration chromatography (Superdex-200 column, Pharmacia) coupled with a light scattering detector (Precision Detectors, Franklin, MA) as described previously (Caldentey et al., 2000). The apparent masses of the eluted complexes were calculated using calibration with monomeric actin and the manufacturer's software.
Kinetic Measurements
Kinetics of binding and dissociation of twinfilin or its two ADF-H domains to NBD-G-actin was observed using the MOS250 fluorometer coupled to a μSFM-20 stopped-flow apparatus (BioLogic, France). Contents of two 10-ml syringes containing a solution of twinfilin constructs, NBD-actin, or unlabeled actin were mixed in variable ratios, and changes in NBD-fluorescence were observed in a TC-100/15 cell. The dead time of the flow cell under the conditions used was determined to be ≤5 ms using a standard method of DCIP reduction. Because of mechano-optical instability, however, reliable data could be collected at 15 ms. NBD fluorescence was exited at 482 nm and recorded at 535 nm using 10-nm excitation and 20-nm emission bandwidth. In addition to this, an external photomultiplier with a cutoff filter of 500 nm was used. Time-resolved data were collected using 0.1-ms intervals and an optimal A/D conversion range to improve signal-to-noise ratio. Apparent first-order rate constants were obtained by fitting the experimental data to single exponentials using Biokine software and the simplex method. In addition, second-order kinetics and off-rate competition were simulated using the KINSIM package (Frieden, 1994). To obtain an off-rate corrected for the on-rate contribution, each experimental curve was fitted with a single exponential to obtain the apparent first-order rate constant. The reaction was then simulated using the known on-rate under the conditions of each experiment while the off-rate was systematically varied. For each simulation, a single exponential was used to approximate the simulated curve, and the fitted rate was compared with the apparent experimental first-order rate until agreement within an experimental error was achieved.
Miscellaneous
PAGE was carried out by using the buffer system described by Laemmli (1970). Native PAGEs to study protein-protein interactions were performed as described by Safer (1989) and modified by Weeds and Maciver (1993). Protein concentrations were determined with a Hewlett Packard 8452A diode array spectrophotometer by using calculated extinction coefficients: ε = 36.4 mM−1 cm−1 (at 280 nm) for mouse wild-type twinfilin; ε = 26.1 mM−1 cm−1 for Twf1–142 and Twf1–174; ε = 10.3 mM−1 cm−1 for Twf141–350, Twf169–350, Twf141–322, and Twf169–322; ε = 13.5 mM−1 cm−1 for human nonmuscle cofilin; and ε = 26.6 mM−1 cm−1 (at 290–400 nm) for rabbit muscle actin. The extent of NBD labeling of actin was determined at 482–400 nm, where the absorption coefficient used was 26 mM−1 cm−1. Protein distribution in SDS-PAGE gels was quantified by Fluor-S MultiImager with Quantity One software version 4.1.0 (Bio-Rad). The rate of nucleotide exchange of actin in the absence and presence of mouse twinfilin was measured as described previously (Hawkins et al., 1993).
RESULTS
Twinfilin Binds ADP-G-Actin with a High Affinity
The fluorescence of NBD-G-actin is modulated on binding to ADF/cofilin, thereby providing a means of determining the affinity of interaction with ADF/cofilins (Carlier et al., 1997). Because twinfilin is composed of two ADF/cofilin-like (ADF-H) domains, we examined whether twinfilin would also affect the fluorescence of NBD-G-actin.
Binding of wild-type mouse twinfilin results in maximally 35–50% enhancement in fluorescence of NBD-G-actin (Figure 1). KD values, assuming a 1:1 complex, were 0.16 and 0.02 μM for ATP-G-actin and ADP-G-actin in low ionic strength, respectively, whereas corresponding values under physiological ionic conditions (0.1 M KCl, 1 mM MgCl2, pH 7.5) were 0.47 and 0.05 μM, respectively. These data show that twinfilin interacts with ADP-G-actin with ∼10-fold higher affinity than with ATP-G-actin both under physiological ionic conditions and at low ionic strength. The higher affinity observed at low ionic strength suggests that the interaction between twinfilin and G-actin is largely electrostatic in nature.
Figure 1.

Interaction of mouse twinfilin with G-actin. The increase in the fluorescence of NBD-labeled MgATP-G-actin (open circles) or MgADP-G-actin (closed circles) was measured at different concentrations of mouse twinfilin. The experiment was carried out with 0.2 μM actin under physiological ionic conditions (A) and with 0.5 μM actin at low-ionic-strength conditions (B). Symbols are data averaged from three independent experiments, and the solid lines are fitted binding curves for a complex with 1:1 stoichiometry.
Previous studies have shown that yeast twinfilin decreases the rate of nucleotide exchange on binding to G-actin (Goode et al., 1998). The ability of mouse twinfilin to inhibit the exchange of ε-ATP to ATP was measured by using 1 μM G-actin and 0–5 μM twinfilin, confirming that the mouse protein also inhibits the spontaneous nucleotide exchange of G-actin in a manner similar to yeast twinfilin (our unpublished data).
The C-Terminal ADF-H Domain Binds Actin with Higher Affinity than the N-Terminal ADF-H Domain and Is Responsible for the Ability of Twinfilin to Increase the Amount of Monomeric Actin
To examine the roles of the two ADF-H domains of twinfilin as well as the conserved linker and the C-terminal tail region for actin interactions, we designed a series of deletion mutants. The domain structure as well as the N- and C-terminal sequences of the six deletion mutants used in this study are shown in Table 1. Proteins were purified as GST-fusion proteins with a glutathione agarose affinity column, followed by a thrombin cleavage and anion-exchange and gel filtration chromatographies. All purified deletion proteins were soluble and appeared to be monomeric, as judged by size-exclusion chromatography.
Table 1.
Mouse twinfilin constructs used in this study. Twinfilin is composed of two ADF-H domains (N and C), separated by a short linker region and followed by a COOH-terminal tail region. The recombinant twinfilins used in this study were produced in E. coli as GST-fusion proteins. The thrombin-cleavage of the fusion proteins leaves a 2–4 amino-acid extension at the NH2-terminus of the proteins (underlined). Furthermore, in wild-type twinfilin, Twf1–142 and Twf1–174 constructs the serine residue following the NH2-terminal methionine is replaced by alanine (indicated in italics).
 |
We examined the ability of the six twinfilin deletion proteins to prevent actin filament assembly under physiological ionic conditions (100 mM NaCl, 1 mM MgCl2, 1 mM ATP) to assess the G-actin sequestering activity of different parts of the protein. Wild-type twinfilin and all constructs containing the C-terminal ADF-H domain (Twf141–322, Twf141–350, Twf169–322, Twf169–350) efficiently increased the amount of monomeric actin in filament sedimentation assays (Figure 2). Similar results were also obtained when the twinfilin was mixed with G-actin before assembly. In contrast, the constructs lacking the C-terminal ADF-H domain (Twf1–142, Twf1–174) were significantly less efficient in increasing the amount of monomeric actin in sedimentation assays. Interestingly, Twf141–322 and Twf141–350, which contain the C-terminal ADF-H domain as well as the linker region, were slightly less efficient in shifting actin to the monomeric fraction than the wild-type twinfilin and the deletion proteins containing the C-terminal ADF-H domain without the linker region (Figure 2). This indicates that in the absence of the N-terminal ADF-H domain, the linker region slightly inhibits the actin monomer sequestering activity of the C-terminal ADF-H domain. It is also important to note that the deletion proteins examined in this assay did not display any interaction with actin filaments in cosedimentation assays (our unpublished data).
Figure 2.

The ability of wild-type twinfilin and deletion constructs to decrease the amount of filamentous actin in solution. Actin filaments (3 μM) were mixed with 0, 2, 4, 6, and 12 μM wild-type twinfilin or with twinfilin deletion proteins. Actin filaments were sedimented, and actin monomer concentrations were quantified in three independent experiments. Wild-type twinfilin and all constucts containing the C-terminal ADF-H domain (A, D, E, F, and G) efficiently increase the amount of monomeric actin, whereas the constructs containing only the N-terminal ADF-H domain (B and C) are significantly less efficient in increasing the amount of monomeric actin. (H) Comparison of actin monomer sequestering activities of wild-type twinfilin (closed circles), Twf169-322 (closed triangles), and Twf1-142 (open circles).
The affinities of the twinfilin deletion constructs for ADP-G-actin were determined by use of the fluorescence assay with NBD-G-actin. All deletion proteins containing the C-terminal ADF-H domain of twinfilin resulted in a 35–60% increase in the fluorescence of NBD-G-actin under saturating conditions. In contrast, the protein containing the N-terminal ADF-H domain and the linker region (Twf1–174) resulted in ∼15% quenching of the NBD-G-actin fluorescence, whereas the Twf1–142 deletion protein did not result in a significant change in the NBD fluorescence. Under physiological ionic conditions, the KD values for ADP-G-actin and proteins containing the C-terminal ADF-H domain of twinfilin were 0.03 μM (Twf169–322), 0.08 μM (Twf141–322), and 0.06 μM (Twf169–350). These affinities are similar to that of wild-type twinfilin (KD = 0.05 μM). The deletion protein containing only the N-terminal ADF-H domain and the linker region (Twf1–174) binds ADP-G-actin with ∼10-fold lower affinity (KD = 0.7 μM) than wild-type twinfilin and the C-terminal domains (Figure 3).
Figure 3.

Interaction of twinfilin deletion constructs with ADP-G-actin. The change in the fluorescence of NBD-labeled MgADP-G-actin was measured at different concentrations of wild-type twinfilin and twinfilin deletion proteins. These experiments were carried out at physiological ionic conditions, and the actin concentration was 0.2 μM. (A) Wild-type twinfilin, (B)Twf169–322, (C) Twf141–322, and (D) Twf169–350 increase the NBD fluorescence, whereas (E) the binding of Twf1–174 to G-actin quenches the NBD fluorescence. Symbols are data averaged from three independent experiments, and the solid lines are fitted binding curves for complexes with 1:1 stoichiometry.
Kinetics of Association and Dissociation of Twinfilin–ADP-G-Actin Complex
The kinetics of dissociation of the twinfilin–ADP-G-actin complex were monitored by fluorescence using a stopped-flow apparatus. In these experiments, a 0.5 μM twinfilin–0.5 μM NBD-G-actin complex was mixed with 0.5–1.5 μM unlabeled ADP-G-actin, and the kinetics of dissociation were followed by the decrease in fluorescence in a stopped-flow apparatus. Because an excess of unlabeled actin could not be used because of its tendency to form filaments, the apparent off-rates were affected by the reverse association reaction. This was taken into account by simulating the course of the dissociation competition using KINSIM (Frieden, 1994) and correcting the off-rates as described in “MATERIALS AND METHODS.” The dissociation rate constant for wild-type twinfilin–ADP-G-actin was 1.8 ± 0.2 s−1 at room temperature and physiological ionic strength (Figure 4A). Similar assays for twinfilin deletion proteins containing the C-terminal ADF-H domain gave dissociation rate constants of 2.2 ± 0.4 s−1 for Twf141–322, 2.3 ± 0.3 s−1 for Twf169–322, and 2.2 ± 0.3 s−1 for Twf169–350 (Figure 4, B–D). Therefore, the kinetic stability of the complex is primarily a result of slow dissociation from the C-terminal ADF-H domain. Similar measurements carried out with the N-terminal ADF domain of twinfilin (Twf1–174) suggest that it releases actin monomer by an order of magnitude faster (koff = 19 ± 2 s−1) than wild-type twinfilin (koff = 1.8 s−1) (Figure 4E). However, because of the smaller amplitude in the change of NBD fluorescence and lower affinity of the N-terminal ADF-H domain for G-actin, the signal-to-noise ratio and consequently the experimental error were larger in the measurements carried out for the N-terminal ADF-H domain (Twf1–174)– ADP-G-actin complex.
Figure 4.

Dissociation rates of the twinfilin–actin monomer complex. NBD-labeled ADP-actin (0.5 μM) equilibrated with twinfilin (0.5 μM) was mixed in a stopped-flow fluorometer with unlabeled actin (0.5 μM). Noisy curves represent the data coming from at least seven averaged traces. Solid lines represent kinetic models fitted by the Simplex method within a first-order reaction scheme for dissociation of the complexes. The data were then simulated by KINSIM software with known kon rates to yield dissociation rate constants between ADP-G-actin and wild-type twinfilin (A, koff = 1.8 ± 0.2 s−1), Twf141–322 (B, koff = 2.2 ± 0.4 s−1), Twf169–322 (C, koff = 2.3 ± 0.3 s−1), Twf169–350 (D, koff = 2.2 ± 0.3 s−1), and N-terminal ADF-H domain Twf1–174 (E, koff = 19 ± 2 s−1). Experiments were carried out at room temperature under physiological ionic conditions (100 mM NaCl, 1 mM MgCl2, 0.5 mg/ml BSA) at 20°C and pH 8.0.
To close the thermodynamic cycle and verify the dissociation constants, we next examined the association kinetics of the twinfilin–ADP-G-actin complex and complexes containing the C-terminal (Twf169–322) or the N-terminal (Twf1–174) ADF-H domain of mouse twinfilin. The pseudo first-order rates for association of ADP-G-actin–Twf169–322 complex showed a linear concentration dependence that corresponds to a second-order association rate of 23.6 ± 0.6 μM−1 s−1 (Figure 5, A and D). The equilibrium dissociation constant calculated from these rate constants (2.3 s−1/23.6 μM−1 s−1 = 0.09 ± 0.02 μM) is comparable to that derived from the equilibrium measurements (∼0.05 μM).
Figure 5.

Kinetics of binding of twinfilin to ADP-G-actin. NBD-labeled ADP-actin (0.25 μM for Twf1–174 and Twf169–322 and 0.1 μM for wild-type twinfilin) was mixed in a stopped-flow apparatus with different concentrations of wild-type twinfilin or deletion proteins, and the associations of the complexes were followed by a change in the NBD fluorescence. Noisy curves represent typical time courses of the fluorescence changes in the reaction between ADP-G-actin and Twf169–322 (A), Twf1–174 (B), and wild-type twinfilin (C). The associations of the isolated C-terminal ADF-H domain (Twf169–322, D) and N-terminal ADF-H domain (Twf1–174, E) follow linear concentration dependence consistent with association rates of 23.6 μM−1 s−1 and 40 μM−1 s−1, respectively. The rate of NBD fluorescence increase resulting from the interaction of wild-type twinfilin with ADP-G-actin shows a linear increase only with small protein concentrations and reaches a maximum rate of 10.6 s−1 at 4 μM twinfilin. The apparent association rate was fitted with a simple Michaelis-Menten model reflecting a fast binding followed by a slow conformational change. The apparent KD obtained was 0.8 μM. Experiments were carried out at room temperature under physiological ionic conditions (100 mM NaCl, 1 mM MgCl2, 0.2 mM ADP, 0.5 mg/ml BSA) at 20°C and pH 8.0.
Similar experiments were carried out with the N-terminal ADF-H domain (Twf1–174) and ADP-G-actin. The second-order association rate constant was 40 ± 8 μM−1 s−1 (Figure 5, B and E). Hence, the equilibrium dissociation constant calculated from kinetic measurements (0.48 ± 0.14 μM) is similar to that measured in the equilibrium assay (0.7 μM).
Experiments using wild-type twinfilin did not show a linear relationship between association rate constant and twinfilin concentration: rather, the apparent association rate showed saturation behavior, with a plateau at 10.6 ± 0.3 s−1, which is governed by apparent dissociation constant of 0.8 ± 0.1 μM (Figure 5F) and estimated off-rate k−2 = 1.3 ± 0.3 s−1. This provides clear evidence that the rate of the fluorescence change is governed by a fast equilibrium with a KD of 0.8 μM followed by a slower first-order process (conformational change) with a limiting forward rate of k2 = 9.3 ± 0.6 s−1 (see DISCUSSION for the equation).
The Two ADF-H Domains of Twinfilin Compete with Each Other in Binding to G-Actin
Experiments with yeast and mouse twinfilins suggest that despite two actin monomer–binding ADF-H domains, these proteins form a 1:1 molar ratio complex with G-actin (Goode et al., 1998; Vartiainen et al., 2000). To elucidate whether the two ADF-H domains interact with overlapping or different interfaces on actin monomer, we carried out competition experiments with the N-terminal and C-terminal ADF-H domains of twinfilin in the G-actin–binding assay (Figure 6A). The results showed that the two ADF-H domains of twinfilin compete with each other in binding to G-actin and most likely interact with actin through partially or completely overlapping interfaces. The data were fitted as described in “MATERIALS AND METHODS” by using the KD value of 0.03 μM for Twf169–322 (from Figure 3B) and gave equilibrium dissociation constants of 0.5–0.6 μM for the Twf1–174–ADP-G-actin complex. These values are very similar to the ones obtained for Twf1–174 from steady-state (Figure 3) and kinetic-binding assays (Figures 4 and 5) (KD = 0.48–0.7 μM). It is important to note that the Twf1–142 protein, whose affinity to G-actin could not be determined because of the lack of fluorescence signal on binding to NBD-actin, also competes with Twf169–322 in binding for ADP-G-actin in a manner similar to Twf1–174 (our unpublished data). Thus, both constructs containing the N-terminal ADF-H domain (Twf1–142 and Twf1–174) bind ADP-G-actin with similar affinities, indicating that the presence of the linker region in the longer construct does not affect the binding affinity.
Figure 6.

Cofilin and the two ADF-H domains of twinfilin compete with each other for the same binding site on G-actin. (A) The competition of the N-terminal (Twf1–174) and C-terminal (Twf169–322) ADF-H domains of twinfilin for actin binding was examined by adding different concentrations (0.5–32 μM) of Twf1–174 to a mixture of 0.2 μM ADP-G-actin and 0.4, 0.8, and 2 μM Twf169–322. Twf1–174, which alone results in a 15% decrease in the NBD fluorescence of ADP-G-actin, results in >35% quenching of the NBD fluorescence of the Twf169–322–ADP-G-actin complex. Filled circles, triangles, and squares are data with 0.4, 0.8, and 2 μM Twf169–322, and the solid lines are the fitted binding curves with KD values of 0.03 μM for Twf169–322 and 0.5, 0.6, and 0.6 μM for Twf1–174 for ADP-G-actin, respectively. (B) Competition between cofilin (Cof) and twinfilin for ADP-actin (Act) monomer binding was analyzed on native gels at a pH of 9. Lane 1, 10 μM actin; lane 2, 10 μM twinfilin; lane 3, 10 μM cofilin; lane 4, 10 μM actin + 10 μM twinfilin; lane 5, 10 μM actin + 10 μM cofilin; lane 6, 10 μM actin + 10 μM twinfilin + 10 μM cofilin. Cofilin dissociates the twinfilin–ADP-G-actin complex and shifts twinfilin to its original migration position. (C) The competition of cofilin and twinfilin for binding to G-actin under physiological conditions was examined by adding different amounts (0–50 μM) of human cofilin to a mixture of 1 μM NBD-ADP-G-actin and 1 μM twinfilin. The complexes were monitored by size-exclusion chromatography coupled with light-scattering detector, light absorption detector, and refractive index detector. Inset shows deconvolved mass abundance distribution across the included peaks analyzed on the basis of 90° light scattering and absorbance at 482 nm. The apparent higher mass of free actin is caused by scattering of free twinfilin, which elutes at a similar position but does not absorb at 482 nm. Thin solid line, 1 μM actin, 1 μM twinfilin; bold solid line, 1 μM actin, 1 μM twinfilin, 1 μM cofilin; dotted line, 1 μM actin, 1 μM twinfilin, 10 μM cofilin; dashed line, 1 μM twinfilin, 50 μM cofilin. The arrows indicate apparent masses of G-actin and its complexes as follows: A, actin alone; AC, actin/cofilin; AT, actin-twinfilin; ATC, the putative actin-twinfilin-cofilin complex.
Twinfilin Competes With ADF/Cofilin for G-Actin Binding
Because each of the two ADF-H domains of twinfilin shows ∼20% sequence identity to ADF/cofilins, we examined, using human cofilin-1, whether twinfilin and ADF/cofilin compete with each other in binding to ADP-G-actin. As shown in Figure 6B, lane 4, a complex between twinfilin and ADP-actin monomers is detected on native polyacrylamide gels run at a pH of 9. An addition of cofilin-1 to the reaction mixture before loading on gel dissociates the twinfilin–actin monomer complex (Figure 6B, lane 6), suggesting that twinfilin and cofilin compete for the same high-affinity binding site on actin monomers.
The competition between twinfilin and cofilin for G-actin binding under physiological conditions is more difficult to follow. An NBD-fluorescence–binding assay similar to the one described in Figure 6A for the isolated ADF-H domains of twinfilin suggested that the equilibrium dissociation constant of human cofilin for ADP-G-actin is ∼0.2 μM. In the presence of 2 μM twinfilin, the apparent KD increases to 1.5 μM, suggesting that twinfilin disturbs the binding of cofilin to actin monomers under physiological conditions as well. However, the lack of linear concentration dependency and inconsistency in the apparent KD value obtained by this assay indicate that cofilin, twinfilin, and actin may also form a ternary complex at higher protein concentrations (our unpublished data). To further evaluate the possible competition of cofilin and twinfilin for actin binding under physiological conditions, we analyzed size distributions and masses of actin-containing complexes in the presence of twinfilin and increasing amounts of cofilin (Figure 6C). In the absence of cofilin, 85% of actin was found in the complex with twinfilin with an apparent mass of 85 kDa, e.g., 1:1 complex. The remaining actin (∼15%) eluted in the void volume in filaments with apparent mass >2 MDa. Apparently, at the micromolar protein concentrations used in this assay, residual ADP-NBD-actin polymerization is difficult to prevent and twinfilin cannot effectively dissociate such assemblies. However, in the presence of a small amount of cofilin, these assemblies disappeared and actin was distributed in several species: free actin (apparent mass 50 kDa, 30%), actin-cofilin (apparent mass 65 kDa, 15%), and actin-twinfilin (apparent mass 85 kDa, 55%). Although 15% of the free actin was probably released from the filaments, ≥30% of the total actin was released from the actin-twinfilin complex by cofilin competition. At excess cofilin, the free actin is sequestered in the actin-cofilin complex. In the presence of a great excess of cofilin (10 and 50 μM), the actin-twinfilin complex gradually disappeared and another actin-containing species with an apparent mass of 95 kDa was populated. Because of overlap between chromatography bands, the exact composition could not be determined. Thus, this complex could be either ternary actin-twinfilin-cofilin or actin-cofilin2 (based on the apparent mass). Control runs of actin in the presence of a great excess of cofilin failed to detect the latter (actin-cofilin2) species, suggesting that a weaker ternary complex, actin-twinfilin-cofilin, is formed at higher cofilin concentrations and coexists with the actin-cofilin complex. This complex is most likely a result of a low-affinity cofilin-binding site on actin monomer that is different from the high-affinity twinfilin-cofilin site. Recent electron microscopy studies suggested that ADF-cofilin proteins indeed have two different binding sites at least on F-actin (Galkin et al., 2001).
DISCUSSION
A number of actin-binding proteins show preference in their binding to the nucleotide present on the actin, and recent evidence has shown structural differences between ADP-actin and ATP-actin (Otterbein et al., 2001). Here, we show that under physiological ionic conditions, mouse twinfilin binds ADP-G-actin with ∼10-fold higher affinity than ATP-G-actin. Previous studies have demonstrated that other ADF-H domain proteins, ADF/cofilins, also bind ADP-G-actin with ∼50-fold higher affinity than ATP-G-actin (Maciver and Weeds, 1994; Carlier et al., 1997). Hence, it appears that the ADF-H domain has evolved as a specific ADP-actin–binding protein motif. It is also important to note that mouse twinfilin binds ADP-G-actin with a very high affinity (KD = 0.05 μM) under physiological ionic conditions. This affinity is significantly higher than that of profilin (KD = 0.5–3 μM) and thymosin-β-4 (KD ∼50 μM) for ADP-G-actin (Carlier et al., 1993; Pantaloni and Carlier, 1993; Perelroizen et al., 1996; Vinson et al., 1998), suggesting that the majority of the cytoplasmic ADP-actin monomers are associated with either twinfilin or ADF/cofilin. In contrast, twinfilin binds ATP-G-actin with a lower affinity (KD = 0.47 μM) than profilin (KD = 0.1–0.15 μM), suggesting that twinfilin does not significantly affect the dynamics of the cytoplasmic ATP-G-actin pool.
Our results further suggest that the affinity of twinfilin for ADP-G-actin is somewhat higher than that of ADF/cofilins for ADP-G-actin, because the mean value of measurements for a variety of isoforms of ADF and cofilin is ∼0.15 μM (Blanchoin and Pollard, 1998; Ressad et al., 1998; Vartiainen et al., 2002; Yeoh et al., 2002). This, together with a relatively high cellular concentration of twinfilin, suggests that twinfilin might displace ADF/cofilin from ADP-actin (Palmgren et al., 2001). Although the difference in KD is not large, any sequestration of ADP-G-actin by twinfilin will reduce the concentration of ADF/cofilin–ADP-G-actin complexes and thereby reduce both the addition of complexes to filament ends and the spontaneous nucleation of these complexes, which is a highly cooperative process in its concentration dependence (Yeoh et al., 2002). Moreover, the high dissociation rate constant of the ADF/cofilin–ADP-G-actin complex will facilitate rapid exchange of ADP-G-actin between ADF/cofilin and twinfilin. (The off-rate for Arabidopsis ADF1–ADP-G-actin complex was 12–16 s−1 at 4°C and would be significantly higher at higher temperatures [Ressad et al., 1998].) The relatively slow koff rates of twinfilin–actin monomer complexes, together with fast koff rates of ADF/cofilin–actin monomer complexes, provides kinetic evidence to support the argument that ADP-G-actin is recycled from ADF/cofilin to twinfilin. These stable complexes also show much slower nucleotide exchange than actin itself.
It is important to note that twinfilin does not prevent actin filament assembly as efficiently as would be expected for a sequestering protein with submicromolar affinity for actin monomers (Figure 2). However, because similar results were obtained whether twinfilin was mixed with F-actin or G-actin, it appears that the incomplete depolymerization at high concentrations cannot be explained on the basis of the slow kinetics of disassembly. Further experiments will be needed to establish whether twinfilin has other effects on actin dynamics than monomer sequestration.
Previous studies showed that twinfilin, although composed of two potential actin-binding motifs (ADF-H domains), forms a 1:1 molar ratio complex with G-actin (Goode et al., 1998; Vartiainen et al., 2000). The experiments reported here show that both ADF-H domains of twinfilin interact independently with G-actin, but the C-terminal domain has a 10-fold higher affinity for ADP-G-actin than the N-terminal domain and sequesters actin monomers with an affinity similar to wild-type twinfilin (Figures 2 and 3). The two ADF-H domains compete with each other in binding G-actin, suggesting interaction through overlapping interfaces (Figure 6A). Kinetic analysis shows significant differences between the interaction of actin with N- and C-terminal domains. Thus, the dissociation rate constant for the C-terminal domain is approximately 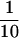 that of the N-terminal domain (Figure 4). The association rates of actin with both N- and C-terminal domains are very rapid, but the second-order rate constant for the N-terminal domain is approximately two times that of the C-terminal (Figure 5). There is therefore very rapid equilibration between the individual domains and actin.
that of the N-terminal domain (Figure 4). The association rates of actin with both N- and C-terminal domains are very rapid, but the second-order rate constant for the N-terminal domain is approximately two times that of the C-terminal (Figure 5). There is therefore very rapid equilibration between the individual domains and actin.
In contrast to the isolated ADF-H domains, the association of wild-type twinfilin with ADP-G-actin follows biphasic kinetics, and at higher twinfilin concentrations, there is saturation behavior with a limiting first-order rate constant of 9.3 s−1. These experiments suggest that the fluorescence enhancement arises as a result of an isomerization process after the binding process, analogous to the fluorescence enhancement of myosin when it binds ATPγS (Bagshaw et al., 1974). This model is described by the following scheme:
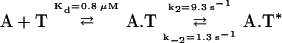 |
where A.T represents the nonfluorescent association intermediate and A.T* represents the fluorescence-promoting, fully assembled complex. Interestingly, the apparent KD of 0.8 μM is close to the equilibrium dissociation constant of the N-terminal ADF-H domain, suggesting that the fast equilibrium step is a result of binding of the N-terminal domain to G-actin.
Similar two-step binding was not observed for the isolated twinfilin ADF-H domains; therefore, this phenomenon probably arises from a conformational change of the twinfilin molecule after association with G-actin. Both wild-type twinfilin and the isolated C-terminal ADF-H domain promote similar enhancement in the NBD-fluorescence on binding to actin; this suggests that the second step during wild-type twinfilin binding to G-actin represents the slow exchange between the N- and C-terminal ADF-H domain on the surface of actin. This is consistent with the fact that the N-terminal ADF-H domain does not promote an increase in the NBD fluorescence. In the future, it will be important to establish whether the physiological function of the N-terminal ADF-H domain is to deliver the ADP-actin monomer from ADF/cofilin to the C-terminal ADF-H domain of twinfilin or whether it plays some other more complex role for twinfilin, such as interaction with capping protein.
Previous studies suggested that yeast twinfilin may function as a protein that localizes G-actin to the sites of rapid actin filament assembly. This appears to be mediated through direct interactions between twinfilin and capping protein (Palmgren et al., 2001). Figure 7 shows a kinetic model of how twinfilin, along with ADF/cofilin and profilin, may contribute to actin dynamics in cells. ADF/cofilin promotes the dissociation of ADP-G-actin at the minus end of the filament with a rate in excess of 9 s−1 (Carlier et al., 1997). Because ADF/cofilins dissociate rapidly from G-actin (Ressad et al., 1998) and compete in G-actin binding with twinfilin, the actin monomer may be delivered to twinfilin, which leaves ADF/cofilin free to associate with actin filaments and promote a new round of depolymerization. Twinfilin may first associate with an actin monomer through its N-terminal ADF-H domain, which promotes a conformational change and a consequent delivery of the monomer to the C-terminal ADF-H domain. Thereafter, twinfilin efficiently keeps actin monomers in ADP-bound form until they are needed for reassociation with the plus ends of filament. Evidence from mutation studies has shown that both fully active twinfilin and capping protein are needed for correct localization of twinfilin in yeast and that there is direct interaction between capping protein and twinfilin (Palmgren et al., 2001). The mechanisms whereby capping proteins modulate the interaction of twinfilin and actin have yet to be investigated. Profilin may promote actin assembly by catalyzing nucleotide exchange within twinfilin-actin complex or on actin monomers after they have dissociated from twinfilin, thus providing ATP-G-actin for filament assembly.
Figure 7.

A kinetic model for the role of twinfilin in actin filament turnover. ADF/cofilin proteins increase the treadmilling of actin filaments by depolymerizing ADP-G-actin from the pointed end of filaments with a rate of 9 s−1. Because ADF/cofilin dissociates from ADP-G-actin with rapid kinetics (koff = 16 s−1) and because twinfilin and ADF/cofilin compete in binding to ADP-G-actin, twinfilin can replace ADF/cofilin on the ADP-actin monomer. The association of twinfilin with an actin monomer probably takes place through the N-terminal, low-affinity (KD = 0.7 μM) ADF-H domain. This promotes a conformational change with a rate of 9.3 s−1 and is followed by a delivery of ADP-G-actin to the C-terminal, high-affinity (KD = 0.05 μM) ADF-H domain. Because twinfilin forms a kinetically more stable complex with ADP-G-actin (koff = 1.8 s−1) than cofilin (koff = 16 s−1), it may be involved in recycling G-actin, in their inactive ADP-form, to the sites of rapid actin filament assembly in cells. The localization of twinfilin to the cortical actin cytoskeleton is mediated by interactions with capping protein (Palmgren et al., 2001). After release from twinfilin, the actin monomer undergoes nucleotide exchange and assembly into the plus-end of the filament. These reactions may be catalyzed by profilin. The references for the kinetic and equilibrium parameters for the ADF/cofilin-actin and profilin-actin complexes are from Carlier et al., 1997; Blanchoin and Pollard, 1998; Vartiainen at al., 2002; Ressad et al., 1998; Perelroizen et al., 1996; and Gutsche-Perelroizen et al., 1999.
In conclusion, we show that twinfilin forms a high-affinity and relatively stable complex with ADP-G-actin. It competes in actin binding with ADF/cofilin, and although the strong actin monomer–binding affinity of twinfilin resides in its C-terminal ADF-H domain, the N-terminal ADF-H domain seems to play an important part in the binding process, because there appears to be a conformational change detected in the fluorescence assays. In the future, it will be important to gain structural information about the twinfilin–actin monomer complex and to elucidate the molecular nature of the conformational change involved in the association of twinfilin with G-actin. The role of capping protein in regulating interactions must also be elucidated.
ACKNOWLEDGMENTS
This work was supported by grants from the Academy of Finland and Biocentrum Helsinki (to P.L.). P.J.O. is supported by a fellowship from the Viikki Graduate School for Biosciences.
Footnotes
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E02–03–0157. Article and publication date are at www.molbiolcell.org/cgi/doi/10.1091/mbc.E02–03–0157.
REFERENCES
- Bagshaw CR, Eccleston JF, Eckstein F, Goody RS, Gutfreund H, Trentham DR. The magnesium ion-dependent adenosine triphosphatase of myosin: two-step processes of adenosine triphosphate association and adenosine diphosphate dissociation. Biochem J. 1974;141:351–364. doi: 10.1042/bj1410351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamburg JR, McGough A, Ono S. Putting a new twist on actin: ADF/cofilins modulate actin dynamics. Trends Cell Biol. 1999;9:364–370. doi: 10.1016/s0962-8924(99)01619-0. [DOI] [PubMed] [Google Scholar]
- Blanchoin L, Pollard TD. Interaction of G-actin with Acanthamoeba actophorin (ADF/cofilin) and profilin. J Biol Chem. 1998;273:25106–25111. doi: 10.1074/jbc.273.39.25106. [DOI] [PubMed] [Google Scholar]
- Caldentey J, Tuma R, Bamford DH. Assembly of bacteriophage PRD1 spike complex: role of the multidomain protein P5. Biochemistry. 2000;39:10566–10573. doi: 10.1021/bi000711+. [DOI] [PubMed] [Google Scholar]
- Carlier M-F, Jean C, Rieger KJ, Lenfant M, Pantaloni D. Modulation of the interaction between G-actin and thymosin β4 by the ATP/ADP ratio: possible implication in the regulation of actin dynamics. Proc Natl Acad Sci USA. 1993;90:5034–5038. doi: 10.1073/pnas.90.11.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlier M-F, Laurent V, Santolini J, Melki R, Didry D, Xia GX, Hong Y, Chua N-H, Pantaloni D. Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J Cell Biol. 1997;136:1307–1322. doi: 10.1083/jcb.136.6.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detmers P, Weber A, Elzinga M, Stephens RE. 7-Chloro-4-nitrobenzeno-2-oxa-1,3-diazole actin as a probe for actin polymerization. J Biol Chem. 1981;256:99–105. [PubMed] [Google Scholar]
- Frieden C. Analysis of kinetic data: practical applications of computer simulation and fitting programs. Methods Enzymol. 1994;240:311–322. doi: 10.1016/s0076-6879(94)40053-9. [DOI] [PubMed] [Google Scholar]
- Galkin VE, Orlova A, Lukoyanova N, Wriggers W, Egelman EH. Actin depolymerizing factor stabilizes an existing state of F-actin and can change the tilt of F-actin subunits. J Cell Biol. 2001;153:75–86. doi: 10.1083/jcb.153.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode BL, Drubin DG, Lappalainen P. Regulation of the cortical actin cytoskeleton in budding yeast by twinfilin, a ubiquitous actin monomer-binding protein. J Cell Biol. 1998;142:723–733. doi: 10.1083/jcb.142.3.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutsche-Perelroizen I, Lepault J, Ott A, Carlier M-F. Filament assembly from profilin-actin. J Biol Chem. 1999;247:6234–6243. doi: 10.1074/jbc.274.10.6234. [DOI] [PubMed] [Google Scholar]
- Hawkins M, Pope B, Maciver SK, Weeds AG. Human actin depolymerizing factor mediates a pH-sensitive destruction of actin filaments. Biochemistry. 1993;32:9985–9993. doi: 10.1021/bi00089a014. [DOI] [PubMed] [Google Scholar]
- Hayden SK, Miller PS, Brauweiler A, Bamburg JR. Analysis of the interactions of actin depolymerizing factor with G- and F-actin. Biochemistry. 1993;32:9994–10004. doi: 10.1021/bi00089a015. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lappalainen P, Kessels MM, Cope MJTV, Drubin DG. The ADF-homology (ADF-H) domain, a highly exploited actin-binding module. Mol Biol Cell. 1998;9:1951–1959. doi: 10.1091/mbc.9.8.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciver SK, Weeds AG. Actophorin preferentially binds monomeric ADP-actin over ATP-bound actin: consequences for cell locomotion. FEBS Lett. 1994;347:251–256. doi: 10.1016/0014-5793(94)00552-4. [DOI] [PubMed] [Google Scholar]
- Otterbein LR, Graceffa P, Dominguez R. The crystal structure of uncomplexed actin in the ADP state. Science. 2001;293:708–711. doi: 10.1126/science.1059700. [DOI] [PubMed] [Google Scholar]
- Palmgren S, Ojala PJ, Wear MA, Cooper JA, Lappalainen P. Interactions with PIP2, ADP-G-actin, and capping protein regulate the activity and localization of yeast twinfilin. J Cell Biol. 2001;155:251–260. doi: 10.1083/jcb.200106157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmgren S, Vartiainen MK, Lappalainen P. Twinfilin, a molecular mailman for actin monomers. J Cell Sci. 2002;115:881–886. doi: 10.1242/jcs.115.5.881. [DOI] [PubMed] [Google Scholar]
- Pantaloni D, Carlier M-F. How profilin promotes actin filament assembly in the presence of thymosin β4. Cell. 1993;75:1007–1014. doi: 10.1016/0092-8674(93)90544-z. [DOI] [PubMed] [Google Scholar]
- Peränen J, Rikkonen M, Hyvönen M, Kääriäinen L. T7 vectors with a modified T7lac promoter for expression of proteins in Escherichia coli. Anal Biochem. 1996;236:371–373. doi: 10.1006/abio.1996.0187. [DOI] [PubMed] [Google Scholar]
- Perelroizen I, Didry D, Christensen H, Chua N-H, Carlier M-F. Role of nucleotide exchange and hydrolysis in the function of profilin in actin assembly. J Biol Chem. 1996;271:12302–12309. doi: 10.1074/jbc.271.21.12302. [DOI] [PubMed] [Google Scholar]
- Pollard TD. Rate constants for the reaction of ATP- and ADP-actin with the ends of actin filaments. J Cell Biol. 1986;103:2747–2754. doi: 10.1083/jcb.103.6.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD, Blanchoin L, Mullins DR. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu Rev Biophys Biomol Struct. 2000;29:545–576. doi: 10.1146/annurev.biophys.29.1.545. [DOI] [PubMed] [Google Scholar]
- Ressad F, Didry D, Xia G-X, Hong Y, Chua N-H, Pantaloni D, Carlier M-F. Kinetic analysis of the interaction of actin-depolymerizing factor (ADF/cofilin) with G- and F-actin. J Biol Chem. 1998;273:20894–20902. doi: 10.1074/jbc.273.33.20894. [DOI] [PubMed] [Google Scholar]
- Safer D. An electrophoretic procedure for detecting proteins that bind G-actin. Anal Biochem. 1989;178:32–37. doi: 10.1016/0003-2697(89)90351-5. [DOI] [PubMed] [Google Scholar]
- Spudich JA, Watt S. The regulation of rabbit skeletal muscle contraction. J Biol Chem. 1971;246:4866–4871. [PubMed] [Google Scholar]
- Vartiainen M, Ojala PJ, Auvinen P, Peränen J, Lappalainen P. Mouse A6/twinfilin is an actin monomer-binding protein that localizes to the regions of rapid actin dynamics. Mol Cell Biol. 2000;20:1772–1783. doi: 10.1128/mcb.20.5.1772-1783.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartiainen MK, Mustonen T, Mattila PK, Ojala PJ, Thesleff I, Partanen J, Lappalainen P. The three mouse actin-depolymerizing factor/cofilins evolved to fulfill cell-type-specific requirements for actin dynamics. Mol Biol Cell. 2002;13:183–194. doi: 10.1091/mbc.01-07-0331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinson VK, De La Cruz EM, Higgs HN, Pollard TD. Interactions of Acanthamoeba profilin with actin and nucleotides bound to actin. Biochemistry. 1998;37:10871–10880. doi: 10.1021/bi980093l. [DOI] [PubMed] [Google Scholar]
- Wahlström G, Vartiainen M, Yamamoto L, Mattila PK, Lappalainen P, Heino TI. Twinfilin is required for actin-dependent developmental processes in Drosophila. J Cell Biol. 2001;155:787–795. doi: 10.1083/jcb.200108022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeds AG, Harris H, Gratser W, Gooch J. Interactions of pig plasma gelsolin with G-actin. Eur J Biochem. 1986;161:77–84. doi: 10.1111/j.1432-1033.1986.tb10126.x. [DOI] [PubMed] [Google Scholar]
- Wolven AK, Belmont LD, Mahoney NM, Almo SD, Drubin DG. In vivo importance of actin nucleotide exchange catalyzed by profilin. J Cell Biol. 2000;150:895–904. doi: 10.1083/jcb.150.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeoh S, Pope B, Mannherz HG, Weeds A. Determining the differences in actin binding by human ADF and cofilin. J Mol Biol. 2002;315:911–925. doi: 10.1006/jmbi.2001.5280. [DOI] [PubMed] [Google Scholar]