

I am by training a clinician-scientist in the fields of cardiology, vascular biology and hematology. In regard to being on “one side of the fence” or the “other” of the COX-2 inhibitor controversy, I'll let the data speak for themselves, as I focus my attention on several mechanistic insights that have emerged over the past several years.
I will begin with the recent Advisory Panel's summary of recommendations to the FDA, and discuss the supporting evidence. The FDA Advisory Panel concluded that 1) COX-2 inhibitors increase the risk for cardiovascular events; 2) the risk appears to differ across individual drugs within the COX-2 inhibitor class; and 3) the cardiovascular risk may be dose related, possibly duration related; and there is no evidence, albeit with scarce data, that aspirin prevents or attenuates the observed risk. As a result, 4) the COX-2 inhibitors should be restricted to use as 2nd-line, possibly as 3rd-line, agents for carefully chosen patients; and 5) randomized trials versus placebo or an accepted comparator must be performed, to better define the overall safety profile in diverse patient populations (Table I).
TABLE I. FDA Advisory Committee on COX-2 Inhibitors Summary

The Biology
Cyclooxygenase (COX) is found in many tissues and organ systems: platelets, mast cells, gastric mucosa, renal medulla, and the vascular endothelium (both microvascular and macrovascular) (Fig. 1). Not only does inhibition of COX cause reduced platelet aggregation, but—because both COX-1 and COX-2 reside within the vascular endothelium—changes in vasoreactivity and thromboresistance mechanisms governed by prostacyclin and nitric oxide are also anticipated. McAdam and colleagues1 have shown that the COX-1 antagonist ibuprofen causes a marked reduction in thromboxane A2 synthesis (as represented by the urinary metabolite 11-dehydrothromboxane B2). COX-2 inhibitors have a more modest effect on thromboxane A2 production. In contrast, they exhibit varying inhibitory effects on synthesis.1 When the biological effects are considered collectively, COX-2 inhibitors shift the balance toward preserved thromboxane A2 effects (platelet aggregation/vasoconstriction) and away from the protective effects attributable to prostacyclin (which include platelet inhibition, vasodilation, and resistance to inflammation).
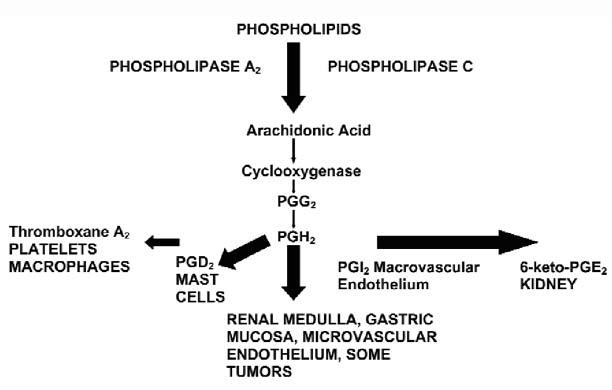
Fig. 1 The anatomy and biochemistry of COX pathways.
Biology-Physiology Interface
The semi-selective COX-2 inhibitor celecoxib, in 100-mg, 400-mg, or even 800-mg doses, exerts a modest inhibitory effect on arachidonic acid-induced platelet aggregation. As mentioned previously, this observation falls in distinct contrast to most COX-1 inhibitors.2
Sowers3 studied patients treated with varying doses of celecoxib, rofecoxib (selective COX-2 inhibitor), or naproxen (COX-1 inhibitor). Patients received treatment for 6 weeks and underwent 24-hour ambulatory blood pressure monitoring. Individuals receiving rofecoxib were more likely to have elevations in blood pressure than those given celecoxib. What does this mean clinically? Given the well-known relationship between systemic hypertension and cardiovascular events, this effect may be particularly relevant. What about individuals with previously diagnosed systemic hypertension whose blood pressure was controlled adequately with drug therapy? After 6 weeks, there was a much larger proportion of individuals treated with rofecoxib (compared with those receiving celecoxib or naproxen) who no longer had acceptable blood pressure control (systolic blood pressure consistently >135 mmHg). Elevated blood pressures during activity were also more likely in the presence of rofecoxib. Accordingly, the previously summarized imbalance between thromboxane A2 and prostacyclin, at the vascular level, may translate into a well-recognized risk factor for cardiovascular events—systemic hypertension.
Cellular Proliferation and Inflammation
Cheng and colleagues4 investigated the effect of thromboxane A2 on vascular responses following carotid artery injury in rats. Animals in which the thromboxane A2 receptor is over-expressed have a marked increase in smooth muscle proliferative and inflammatory responses. In contrast, animals in which thromboxane A2 is over-expressed—but is then inhibited with a thromboxane A2 antagonist—have attenuated responses. Similarly, in animals in which the prostacyclin receptors have been genetically “knocked out,” one observes the same features as with over-expression of thromboxane A2, which strongly suggests that prostacyclin may attenuate the response to injury—mediated, at least in part, by thromboxane A2.
A recent study by Egan5 examined the effects of COX-2 inhibition, thromboxane A2 receptor inhibition, and combined COX-2/thromboxane A2 inhibition on atherosclerotic plaques in an animal atherosclerosis model. Over the years, it has become clear that the fibrous cap affords an element of protection from future thrombotic events. Animals given COX-2 inhibitors were less likely to develop a fibrous cap on their atherosclerotic lesions, despite a large necrotic core. Similarly, with combined COX-2 and thromboxane A2 inhibition, fibrous caps did not develop. These findings suggest strongly that prostacyclin is not only an important part of the response to vascular injury, but also a determining factor for atheromatous plaque architecture. Thus, one begins to understand the biology of the linkage between cardiovascular events and COX-2 inhibition.
Inflammatory diseases such as rheumatoid arthritis and some types of carcinoma are “angiogenesis dependent.” COX-2 is highly expressed in many solid tumors. In this setting, inhibiting angiogenesis may provide therapeutic benefit. In contrast, angiogenesis (the development of collateral circulation) is an important protective mechanism among individuals with severe, obstructive atherosclerotic coronary artery disease.
A study published recently in Arteriosclerosis, Thrombosis and Vascular Biology6 investigated the angiogenic response to CYP2C9 and the effect of celecoxib on this response. Normally, with CYP2C9 activation, endothelial tubes are formed—the angiogenic response; but with COX-2 inhibition, angiogenesis is attenuated.
Caspase-3 and caspase-97 contribute to plaque instability by degrading a portion of the plaque's extracellular matrix. When certain chemotherapeutic agents and a COX-2 selective inhibitor are combined, there is evidence of increased Caspase-3 concentrations and cellular apoptosis. This is a desirable effect for treating malignancies, but not what one would welcome in the setting of vulnerable atherosclerotic plaques.
As an agent becomes less COX-2 specific (semi-selective) and more COX-1 specific, thromboxane A2 inhibition increases, but not linearly. As a result, the medical community would be remiss to conclude that the degree of COX-2 selectivity is the primary consideration in assessing drug-specific cardiovascular safety.
Emerging Clinical Experience with COX Inhibitors and Their Cardiovascular Risk
A recent issue of the New England Journal of Medicine8 included data from the APC study (Adenoma Prevention with Celecoxib) in individuals with known colorectal adenomas who underwent endoscopic resection and were randomized to either 200 mg or 400 mg of celecoxib twice daily, or a placebo. Patients were followed for at least 3 years. There were significant differences between groups in the hard cardiovascular endpoints of cardiovascular death, myocardial infarction, stroke, and heart failure. In a 2nd study, the APPROVe (Adenomatous Polyp Prevention on Vioxx) study, similar increases in cardiovascular events were reported with rofecoxib. A question of considerable importance is, “Exactly where do the curves begin to diverge significantly in these studies?” Is there an early risk associated with the use of these agents? Based on the experiences of roughly 4,500-patients in these 2 trials, I don't believe that we have the statistical power to state with confidence that either drug (at the doses used) is safe in all patients for even short-term administration.9
How do these data translate in terms of daily clinical practice? The patients were, in general, at moderate risk for cardiovascular events. When going from 400 mg twice daily to 800 mg twice daily, there appeared to be a “dose–response” relationship for the composite of cardiovascular death, angina, stroke, heart failure, and revascularization; the overall risk was greatest for a combined endpoint of death and myocardial infarction. Considering myocardial infarction specifically, one could argue that the absolute event rate is low; however, there is anywhere from a 2- to 4-fold relative increase in events. In common clinical conditions such as osteoarthritis or back pain, COX-2 inhibitors could be used by millions of patients, and even relatively low event rates would involve significant numbers of individuals.
A placebo-controlled trial of COX-2 inhibitors has been published in the New England Journal of Medicine.10 Patients were randomized to valdecoxib and parecoxib postoperatively—intravenously for the first 3 days and then orally for 7 days, with outcomes assessed at 30 days. There was an early separation in events, including sudden cardiac death, stroke, and thromboembolic events between treatments, highlighting the potential risk of both arterial and venous thrombotic events with COX-2 inhibition.
David Graham recently published a paper in Lancet11 that used the large Kaiser-Permanente database, with over a million patients. Individuals who had a remote exposure to any COX inhibitor, whether COX-1 or COX-2, were compared (for cardiovascular events) with those exposed recently. Celecoxib appeared to be associated with a lower risk of events (compared with rofecoxib); ibuprofen had an increased risk (compared with naproxen, which itself was associated with increased risk). There also appeared to be a dose-related risk for rofecoxib, at least when doses greater than 25 mg daily were compared with those less than 25 mg daily. Does the cardiovascular risk manifest itself only after 18 months of daily COX-2 inhibition as suggested by the APC and APPROVe trial results? Further investigation is needed to answer this important and highly relevant question.
Summary
The totality of data—the vascular biology and clinical trial data available to date—support the following conclusions: if COX-2 inhibitors are to be used, they should be considered as 2nd- or 3rd-line agents. They should be used for brief periods of time among patients who are at low risk for cardiovascular events. It is worth pointing out that COX-2 inhibitors were designed initially for use in patients at risk for bleeding ulcers, to minimize gastrointestinal toxicity. Large healthcare databases show, however, that the largest growth of COX-2-inhibitor use has occurred among individuals at low risk for GI side effects. COX-2 inhibitors increase the risk for cardiovascular events. The risk differs, to some degree, across agents, and does appear to be dose related. The relationship between cardiovascular risk and duration of therapy is an important question that requires further consideration. Early risk, from the perspective of pathobiology, may differ from long-term risk. The mechanism of cardiovascular risk is multifactorial and relates to sites of COX-2 synthesis, expression within the vasculature, and related local consequences of an imbalance between thromboxane A2 and prostacyclin. Considered collectively, increased platelet aggregation, hypertension, endothelial cell dysfunction, impaired angiogenesis, and destabilization of the atherosclerotic plaque matrix are important contributors to the “prothrombotic environment.” Randomized clinical trials are required to better understand the hazards among individuals at low and high risk for cardiovascular events.
Footnotes
Address for reprints: Richard C. Becker, MD, Duke Cardiovascular Thrombosis Center, Duke Clinical Research Institute, Room 0311 Terrace Level, 2400 Pratt St., Durham, NC 27705
E-mail: beck021@dcri.duke.edu
Presented at the Texas Heart Institute's symposium “Current Issues in Cardiology;” held at the Sheraton World Resort; 5 March 2005; Orlando
References
- 1.McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2 [published erratum appears in Proc Natl Acad Sci U S A 1999;96:5890]. Proc Natl Acad Sci U S A 1999;96:272–7. [DOI] [PMC free article] [PubMed]
- 2.Van Hecken A, Schwartz JI, Depre M, De Lepeleire I, Dallob A, Tanaka W, et al. Comparative inhibitory activity of rofecoxib, meloxicam, diclofenac, ibuprofen, and naproxen on COX-2 versus COX-1 in healthy volunteers. J Clin Pharmacol 2000;40:1109–20. [PubMed]
- 3.Sowers JR, White WB, Pitt B, Whelton A, Simon LS, Winer N, et al. The effects of cyclooxygenase-2 inhibitors and nonsteroidal anti-inflammatory therapy on 24-hour blood pressure in patients with hypertension, osteoarthritis, and type 2 diabetes mellitus [published erratum appears in Arch Intern Med 2005;165:551]. Arch Intern Med 2005; 165:161–8. [DOI] [PubMed]
- 4.Cheng Y, Austin SC, Rocca B, Koller BH, Coffman TM, Grosser T, et al. Role of prostacyclin in the cardiovascular response to thromboxane A2. Science 2002;296:539–41. [DOI] [PubMed]
- 5.Egan KM, Wang M, Fries S, Lucitt MB, Zukas AM, Pure E, et al. Cyclooxygenases, thromboxane, and atherosclerosis: plaque destabilization by cyclooxygenase-2 inhibition combined with thromboxane receptor antagonism [published erratum appears in Circulation 2005;111:2412]. Circulation 2005;111:334–42. [DOI] [PubMed]
- 6.Michaelis UR, Falck JR, Schmidt R, Busse R, Fleming I. Cytochrome P4502C9-derived epoxyeicosatrienoic acids induce the expression of cyclooxygenase-2 in endothelial cells. Arterioscler Thromb Vasc Biol 2005;25:321–6. [DOI] [PubMed]
- 7.Dandekar DS, Lopez M, Carey RI, Lokeshwar BL. Cyclooxygenase-2 inhibitor celecoxib augments chemotherapeutic drug-induced apoptosis by enhancing activation of caspase-3 and -9 in prostate cancer cells. Int J Cancer 2005;115:484–92. [DOI] [PubMed]
- 8.Solomon SD, McMurray JJ, Pfeffer MA, Wittes J, Fowler R, Finn P, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med 2005;352:1071–80. [DOI] [PubMed]
- 9.Psaty BM, Furberg CD. COX-2 inhibitors—lessons in drug safety. N Engl J Med 2005;352;1133–5. [DOI] [PubMed]
- 10.Nussmeier NA, Whelton AA, Brown MT, Langford RM, Hoeft A, Parlow JL, et al. Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. N Engl J Med 2005;352:1081–91. [DOI] [PubMed]
- 11.Graham DJ, Campen D, Hui R, Spence M, Cheetham C, Levy G, et al. Risk of acute myocardial infarction and sudden cardiac death in patients treated with cyclo-oxygenase 2 selective and non-selective non-steroidal anti-inflammatory drugs: nested case-control study. Lancet 2005;365:475–81. [DOI] [PubMed]