

Abstract
Cartilage oligomeric matrix protein (COMP) belongs to the thrombospondin family and is a homopentamer primarily expressed in cartilage. Mutations in the COMP gene result in the autosomal dominant chondrodysplasias pseudoachondroplasia (PSACH) and some types of multiple epiphyseal dysplasia (MED), which are characterized by mild to severe short-limb dwarfism and early-onset osteoarthritis. We have generated COMP-null mice to study the role of COMP in vivo. These mice show no anatomical, histological, or ultrastructural abnormalities and show none of the clinical signs of PSACH or MED. Northern blot analysis and immunohistochemical analysis of cartilage indicate that the lack of COMP is not compensated for by any other member of the thrombospondin family. The results also show that the phenotype in PSACH/MED cartilage disorders is not caused by the reduced amount of COMP.
Cartilage oligomeric matrix protein (COMP) is a member of the thrombospondin family of extracellular matrix proteins (1, 4, 30, 35). The protein consists of five 87-kDa subunits held together by interchain disulfide bonds (19), forming a 435-kDa pentameric protein. COMP is expressed in all types of cartilage (14, 19), vitreous of the eye (33), tendon (12, 38), and vascular smooth muscle cells (36). Immunohistological staining of articular cartilage has revealed a developmentally regulated localization of COMP to the chondrocyte territorial and interterritorial matrix (8). COMP binds in a zinc-dependent manner to collagen type I and type II (37) and also to collagen type IX (20). The thrombospondin family currently includes five members: TSP-1 (9, 25), TSP-2 (5, 23), TSP-3 (42), TSP-4 (26), and COMP (also referred to as TSP-5). These proteins share overall homology, and all contain type 2 (epidermal growth factor-like) and type 3 (calmodulin-like) repeats in their central domains. The homologies are most pronounced in the C-terminal globular domains, whereas the N-terminal amino acid sequences are less conserved. TSP-1 and TSP-2 form trimers containing structurally related N-terminal domains with procollagen-like sequences. TSP-3, TSP-4, and COMP are structurally similar, lacking the N-terminal procollagen homology region, and they form homopentamers (19, 30). Targeted disruptions of TSP-1 or TSP-2 in mice lead to a complex phenotype, affecting numerous organs and connective tissues. Interestingly, TSP-1-deficient mice have a phenotype similar to that of transforming growth factor β-null mice, suggesting that TSP-1 is a major activator of this factor (10). The TSP-2-deficient mice have abnormal collagen fibers, altered bone density, and abnormal bleeding time (22). The COMP gene is located on mouse chromosome 8 and on human chromosome 19 (32). Mutations in the COMP gene are responsible for the human genetic disorders pseudoachondroplasia (PSACH) (6, 7, 18) and some types of multiple epiphyseal dysplasia (MED) (3, 6). The majority of the mutations are missense mutations located in the type 3 calmodulin-like repeat domain. Recombinant COMP carrying PSACH or MED mutations binds fewer divalent cations and show a slightly altered affinity for collagen in the presence of zinc (41). PSACH and MED are autosomal dominant chondrodysplasias causing mild to severe short-limb dwarfism and early-onset osteoarthritis (21). The clinical features are similar, but PSACH is usually more severe and is characterized by dwarfism and joint laxity. MED patients generally show a milder phenotype with short stature and early-onset osteoarthritis. Radiographic analysis of patients show irregular, sometimes small, epiphyses with poor ossification, and patients with PSACH also have irregular metaphyses and delayed ossification of the annular epiphyses of the vertebrae. Ultrastructural analysis of chondrocytes from patients with PSACH and MED show accumulation of material in the rough endoplasmic reticulum (rER) (28, 39), which have a unique lamellar appearance. The accumulated material in rER consists mainly of COMP and type IX collagen (11, 27).
The role of COMP in vivo is unknown. Here we report the targeted disruption of COMP in mice. Mice heterozygous or homozygous for the null mutation are viable and show no detectable skeletal defects.
MATERIALS AND METHODS
Construction of the targeting vector and generation of chimeric mice.
A replacement targeting vector was constructed from cosmid clones isolated from a 129/sv library (kindly provided by J. S. Mudgett, Merck, Rahway, N.J.). A 9-kb genomic EcoRI fragment was subcloned into pBluescript II KS (Stratagene Corp., La Jolla, Calif.). Parts of this fragment were sequenced, and a HindIII site located 3.5 kb from the 5′ end of the genomic fragment was used for the insertion of a phosphoglycerate kinase-neomycin resistance cassette (pGKNeo) (29), interrupting the sequence at a Lys residue of the first type 3 repeat of COMP. The pGKNeo cassette was blunt-end ligated into the HindIII site in reverse transcriptional orientation (Fig. 1).
FIG. 1.

Targeted disruption of the COMP gene. (A) Restriction enzyme map showing the relevant sites (B, BamHI; E, EcoRI; H, HindIII; N, NotI) in the COMP gene (top), the structure of the targeting vector (middle), and the structure of the targeted gene (bottom). (B) Southern blot analysis of DNA isolated from a mouse homozygous for the wild-type allele (+/+), a heterozygous mouse (+/−), and a mouse homozygous for the mutated allele (−/−). The wild-type and targeted alleles gave 8.5- and 6.6-kb fragments, respectively, after digestion with BamHI and hybridization with the indicated 600-bp HindIII/EcoRI probe.
R1 embryonic stem (ES) cells (31) were grown on a feeder cell layer as previously described (13). The targeting vector DNA (45 μg) was linearized at a NotI site and used in the electroporation of 3 × 107 ES cells with a Bio-Rad gene pulser set at 800 V and 3 μF. After 24 h without selection, recombinant clones were selected by adding 400 μg of G418 per ml to the culture media. Clones were picked, and isolated DNA was analyzed by BamHI digestion and Southern hybridization to an external 600-bp HindIII/EcoRI probe. The probe hybridizes to an 8.5-kb wild-type BamHI fragment and a 6.6-kb mutant BamHI fragment. Two individually targeted ES cell clones were microinjected into C57/B6 blastocysts to generate chimeric mice as described elsewhere (13). Chimeric males were mated with C57/B6 mice, and males with germ line transmission were further bred with 129/sv females to establish an inbred strain of COMP knockout mice. The use of animals for research complied with national guidelines, and permission was given by the regional ethical board.
RNA analysis.
Total RNA was isolated from the processus xiphoideus of 5-week-old mice using TRIzol (Gibco BRL), according to the manufacturer's recommendations, and subjected to agarose gel electrophoresis in formaldehyde as previously described (35). Filters were hybridized with TSP-1 and TSP-3 probes (gifts from Paul Bornstein, University of Washington), a TSP-4 probe (a gift from Frank Zaucke, University of Cologne, Cologne, Germany), a COMP probe (35), and a glyceraldehyde phosphodehydrogenase probe was used to check loading of RNA.
Reverse transcription-PCR (RT-PCR) was also performed to confirm the absence of truncated COMP mRNA in the COMP-null mice. Poly(A)+ RNA was isolated with an Oligotex direct mRNA minikit (Qiagen) and reverse transcribed with Moloney murine leukemia virus reverse transcriptase (Roche). Single-stranded cDNA was then subjected to PCR amplification using a mouse 5′-COMP 17-mer (GAATGCAGCCCGCACCC) complementary to exon sequence 16 amino acids prior to the start of type 2 repeats and a 3′-COMP 17-mer (GCACAGGAGCCCGTTGC) corresponding to the last amino acids of the fourth type 2 repeat as primers, giving rise to a 578-bp PCR product. As a positive control, cDNAs derived from both wild-type and COMP-null mice were also amplified with primers complementary to mouse fibromodulin (5′-FM 18-mer GACTCTCACTGGTGGATC and 3′-FM 18-mer GTACAGCCTCTCCAAGTG), generating a 417-bp PCR fragment. PCRs were performed in a thermal cycler, with 30 cycles of 95°C for 30 s, 55°C for 45 s, and 72°C for 2 min. PCR products were separated on a 1.2% agarose gel and stained with ethidium bromide.
Protein transfer blot.
Proteins were extracted from the processus xiphoideus of 6-week-old mice by using 4 M guanidine-HCl-50 mM sodium acetate (pH 5.8) containing the protease inhibitor Complete (Boehringer Mannheim GmbH, Mannheim, Germany) as described previously (24). After 48 h at 4°C with continuous mixing, the extracts were cleared by centrifugation and the supernatants were subjected to precipitation with 95% ethanol. The protein precipitates were washed, dissolved in reducing sample buffer, and electrophoresed on a 10% sodium dodecyl sulfate-polyacrylamide gel. Proteins were transferred to nitrocellulose filters by diffusion and detected with a rabbit antiserum against bovine COMP (35). As a control, the filters were incubated with a fibromodulin antiserum (34). Bound antibodies were visualized with 125I-labeled goat anti-rabbit immunoglobulin G (IgG) as previously described (40).
Histology, immunohistochemistry, and electron microscopy.
Tissues from mice were fixed in ice-cold 95% ethanol-1% acetic acid for 16 h at 4°C, dehydrated in increasing ethanol concentrations, and embedded in Histowax (Histolab, Gothenburg, Sweden) as previously described (40). Bone-containing tissues were decalcified in 7% EDTA in phosphate-buffered saline (PBS) at 4°C for 5 days prior to dehydration and embedding. Sections of 6 μm were cut, deparaffinized in xylene, rehydrated in ethanol, and stained with Mayer hematoxylin (Histolab) and Chromotrope 2R (Sigma).
For immunohistochemistry, tissue sections were stained as described previously (40) with rabbit antibodies against bovine COMP (34), TSP-1 and TSP-2 (both gifts from D. Mosher, University of Wisconsin), and TSP-4 (a gift from M. Paulsson, University of Cologne). TSP-3 antisera were TSP-3 peptide (RLRGPSRPS and PLQTDRDEDC) antisera (gifts from A. Qabar, Madigan Army Medical Center, Tacoma, Wash.). Prior to incubation with antiserum, the sections were deparaffinized in xylene, rehydrated in ethanol, treated with bovine testicular hyaluronidase (2 mg/ml, type I-S; Sigma) in PBS (pH 5.0) for 30 min at 37°C, blocked for endogenous peroxidase activity with 1% H2O2 in methanol for 20 min, and incubated with normal goat serum (Vector Laboratories, Inc., Burlingame, Calif.) in 1% bovine serum albumin in PBS for 1 h at room temperature. After incubation with the antiserum (diluted 1/200), the sections were incubated with peroxidase-conjugated goat anti-rabbit IgG (Sigma) diluted 1/200 in 1% bovine serum albumin in PBS, washed in PBS, and developed with 3,3′-diaminobenzidine solution.
Ultrastructural analysis using electron microscopy was performed as previously described (2, 40).
For X-ray analysis, mice were anaesthetized with avertin, and X-ray images were taken with a Siemens Polymat 70 using 48 kV and 0.2 mA (Siemens, Langen, Germany).
RESULTS
Generation of COMP-deficient mice.
The mouse COMP gene was inactivated by homologous recombination in ES cells with a targeting vector interrupting the coding sequence in the first type 3 repeat with a neomycin resistance cassette (Fig. 1). The neomycin resistance cassette integration site is located in exon 8 of the COMP gene. After transformation of ES cells and selection with G418, we identified six correctly targeted ES cell clones out of 170 screened. Chimeric mice were generated by blastocyst microinjection, and resulting males were mated with C57/B6 female mice. Males showing germ line transmission were thereafter mated with 129/sv females to produce an inbred strain. COMP-null mice were obtained according to the Mendelian rule and did not reveal any gross anatomical abnormalities. They grew to normal size, had a normal life span, and were fertile.
RNA and protein analysis.
Transfer blot analysis of RNA showed the absence of COMP mRNA in mutated mice (Fig. 2A). The steady-state levels of TSP-1, TSP-3, and TSP-4 mRNAs were not significantly altered in the COMP-null mice. To further assess if the mutant mice were deficient in COMP mRNA, we performed RT-PCR assays (Fig. 2B). We could not detect any PCR product using cDNA reverse transcribed from COMP-null mRNA and primers complementary to sequences located upstream of the pGKNeo cassette. The expected 578-bp PCR product was detected in wild-type mice. Control RT-PCR with fibromodulin primers resulted in a 417-bp PCR product in wild-type and COMP-null mice. Controls without reverse transcriptase demonstrated the absence of genomic DNA in the poly(A)+ RNA preparations.
FIG. 2.

Northern blot and RT-PCR. (A) Northern blot analysis of RNA isolated from the processus xiphoideus of wild-type (wt) and mutant (m) littermates. Filters were hybridized with TSP-1, TSP-3, TSP-4, and a COMP probe. A glyceraldehyde phosphodehydrogenase (GAPDH) probe was used as a control. (B) RT-PCR analysis of COMP mRNA. COMP cDNA was reverse transcribed from processus xiphoideus poly(A)+ RNA by using reverse transcriptase. PCR primers located upstream of the HindIII site used for insertion of the pGKNeo gene were used in the amplification of wild-type (lane 1) and COMP-null (lane 2) cDNAs. As a control, cDNAs were amplified with wild-type (lane 3) and COMP-null (lane 4) fibromodulin primers. Lane 5, control amplification without prior reverse transcription of poly(A)+ RNA from COMP-null mice with fibromodulin primers.
Transfer blot analysis of proteins extracted from processus xiphoideus showed the absence of COMP in mutated mice (Fig. 3). COMP subunits with an apparent molecular mass of 100 kDa were detected in extracts from wild type animals but were absent in the COMP-null mice. Similar results were obtained with an extract from femoral heads (not shown). Analysis of tail and Achilles tendon extracts derived from mice of different ages showed the absence of COMP also in wild-type mice. This indicates a species difference, since human and bovine tendons have been reported to contain COMP (12, 38).
FIG. 3.

Western blot analysis of proteins extracted from the processus xiphoideus of a wild-type mouse (lanes 1 and 3) and a COMP-null mouse (lanes 2 and 4). Proteins were separated on a sodium dodecyl sulfate-10% polyacrylamide gel and transferred to nitrocellulose filters. COMP (lanes 1 and 2) and fibromodulin (lanes 3 and 4), as controls, were detected with antisera followed by 125I-labeled goat anti-rabbit IgG.
Anatomy, histology, and immunohistochemistry.
X-ray analysis of wild-type and COMP-null mice did not show any skeletal abnormalities (Fig. 4). The epiphysis and metaphysis of long bones were similar in size and morphology, indicating normal ossification in the COMP-null mice. Histological investigations of tibias from newborn (Fig. 5A and B) and 4-month-old (Fig. 5C and D) animals did not reveal any obvious defects. The structure of the growth plates and the ossification were identical in COMP-null and wild-type littermates. Histological examination of knee articular cartilage did not show any signs of osteoarthritis in 7-month-old COMP-null mice (Fig. 5E and F). Furthermore, no osteoarthritis was detected in COMP-null mice up to 14 months of age (data not shown). No morphological abnormalities in the COMP-null mice were detected in sternum, vertebra, or tendons in the tail. The morphology of the inner organs (heart, liver, lung, kidney, spleen, duodenum, and testis) was normal (data not shown).
FIG. 4.

X-ray skeletal analysis of 11-week-old wild-type (left) and COMP-null (right) littermates. Lower panels show side views of hind legs.
FIG. 5.
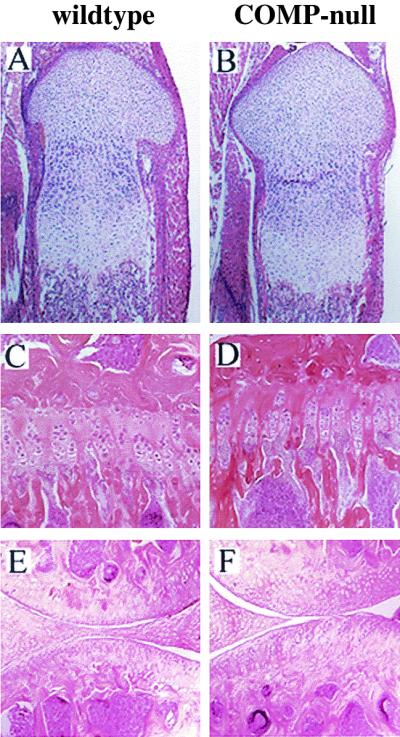
Histology. Hematoxylin-eosin staining was carried out on tibias from newborn wild-type (A) and COMP-null (B) mice, tibias from 4-month-old wild-type (C) and COMP-null (D) littermates, and knee articular cartilage from 7-month-old wild-type (E) and COMP-null (F) littermates.
Immunostaining of tibias from newborn mice with COMP antiserum shows the absence of COMP in mutant cartilage (Fig. 6A and B). No change in staining patterns or increase in the amounts of TSP-1, TSP-2, TSP-3, and TSP-4 could be detected by immunohistochemistry. TSP-1 antiserum stained the cartilage in both wild-type and COMP-null tibias (Fib. 6C and D). The TSP-2 antiserum stained cartilage and tendon tissue where the patellar tendon contacts cartilage (Fig. 6E and F). TSP-3 antisera stained cartilage, tendon, ligament, and muscle tissue (Fig. 6G and H). No staining was observed with a TSP-4 antiserum (Fig. 6I and J).
FIG. 6.

Immunohistochemistry. Tibias from newborn wild-type (A, C, E, G, and I) and COMP-null (B, D, F, H, and J) mice were stained with antibodies against COMP (A and B), TSP-1 (C and D), TSP-2 (E and F), TSP-3 (G and H), and TSP-4 (I and J). Sections were counterstained with hematoxylin.
Ultrastructural analysis of growth plate cartilage, articular cartilage, and Achilles tendons showed normal cell morphology and a normal collagen fibrillar network in the extracellular matrix (Fig. 7). The Achilles tendon fibrils had average diameters of 122 nm (n = 400) and 128 nm (n = 400) in the wild-type and COMP-null mice, respectively. Morphometric analysis of tail tendon fibrils show an average fibril diameter of 121 nm (n = 300) and 119 nm (n = 300) in wild-type and COMP-null animals, respectively.
FIG. 7.

Ultrastructure. Shown are chondrocytes in the proliferating zone of tibias from 3-month-old wild-type (A) and COMP-null (B) littermates, interterritorial matrix in the proliferative zone from newborn wild-type (C) and COMP-null (D) mice, and transverse sections of fibrils from 1-month-old wild-type (E) and COMP-null (F) Achilles tendons (bar = 500 nm).
DISCUSSION
It has been proposed that COMP plays an important role in cartilage by binding to type II and type IX collagen (20, 37). Such interactions bridging collagen fibrils would be of great importance for maintaining the structure and mechanical properties of the tissue. In addition to the putative structural functions of COMP, it has been postulated that the protein may have a role in storage and delivery of hydrophobic hormones. COMP binds retinol, retinoic acid, and vitamin D with high affinity in the N-terminal axial pore of the five-stranded coiled-coil domain (15). This binding may have implications in the morphogenesis and repair of cartilage and for the mineralization of bone. The absence of an apparent phenotype in the COMP-null mice indicates that there may be functionally complementary proteins taking over the roles of COMP, possibly a closely related thrombospondin. However, RNA transfer blot analysis did not reveal any changes in the steady-state levels of TSP-1, TSP-3, and TSP-4 mRNA in the COMP-null mice. In addition, immunostainings of cartilage with TSP-1, TSP-2, TSP-3, and TSP-4 antisera did not reveal any differences between wild-type and COMP-null mice. Generation of double-deficient mice, lacking e.g., COMP and TSP-3, should resolve the question of redundancy among thrombospondins. These results suggest that development and normal homeostasis of cartilage does not require COMP.
Some 50 COMP mutations have been documented in patients with PSACH and MED. The clinical manifestations can be mild to severe. PSACH generally shows the more severe clinical features, with dwarfism, joint laxity, and characteristic vertebral, epiphyseal, and metaphyseal abnormalities. The PSACH cartilage matrix contains dramatically reduced amounts of COMP, and the chondrocytes show large rER cisternae with a unique lamellar appearance (17). These cisternae contain COMP, decorin, fibromodulin, and type IX, type XI, and type XII collagen (16, 43). On the other hand, type II collagen and aggrecan appear to be secreted normally from PSACH chondrocytes (11, 43). The mutations in COMP apparently cause severe alterations in the secondary structure and result in misfolding and retention in the chondrocyte rER. Mutated and misfolded COMP molecules may bind to other cartilage components in the rER and also reduce their secretion. This reduced secretion of many extracellular matrix components may lead to the pathology of PSACH. The accumulation of extracellular matrix components in rER may also seriously impair the activity of chondrocytes, leading to deficient production of the components required for a functional cartilage matrix. In some PSACH patients COMP can be detected at low levels in the cartilage matrix, indicating that some mutated COMP is secreted (43). It has been hypothesized that mutations in the C-terminal domain of COMP disrupt the interaction with type IX collagen and prevent the assembly of a functional cartilage matrix in PSACH/MED (20). These extracellular assembly abnormalities could result from mutations in the binding sites or from reduced amounts of COMP or type IX collagen in the cartilage matrix. Mice lacking COMP do not show a phenotype resembling the clinical manifestations of PSACH/MED in humans, strongly suggesting that COMP-type IX collagen interactions are not essential for a functional cartilage matrix. However, it cannot be ruled out that the phenotype in PSACH/MED cartilage results from the presence of mutated COMP in the extracellular matrix, preventing key events in matrix assembly.
Our results show that the phenotype in PSACH/MED cartilage disorders is caused not by the reduced amount of COMP but by some other mechanism, such as folding defects or extracellular assembly abnormalities due to dysfunctional mutated COMP.
Acknowledgments
We thank Mathias Mörgelin for providing tail tendon electron microscopy data.
This work was supported by the Swedish Medical Research Council, Anna-Greta Craafoord's Stiftelse, Greta och Johan Kock's Stiftelse, Gustav V 80-års Fond, and Alfred Österlunds Stiftelse.
REFERENCES
- 1.Adams, J. C., and J. Lawler. 1993. The thrombospondin family. Curr. Biol. 3:188-190. [DOI] [PubMed] [Google Scholar]
- 2.Aszo'di, A., E. Chan, E. Hunziker, J. F. Bateman, and R. Fässler. 1998. Collagen II is essential for the removal of the notochord and the formation of intervertebral discs. J. Cell Biol. 143:1399-1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ballo, R., M. D. Briggs, D. H. Cohn, R. G. Knowlton, P. H. Beighton, and R. S. Ramesar. 1997. Multiple epiphyseal dysplasia, Ribbing type; a novel point mutation in the COMP gene in a South African family. Am. J. Med. Genet. 68:396-400. [PubMed] [Google Scholar]
- 4.Bornstein, P. 1992. Thrombospondins: structure and regulation of expression. FASEB J. 6:3290-3299. [DOI] [PubMed] [Google Scholar]
- 5.Bornstein, P., K. O'Rourke, K. Wikstrom, F. W. Wolf, R. Katz, P. Li, and V. M. Dixit. 1991. A second, expressed thrombospondin gene (Thbs2) exists in the mouse genome. J. Biol. Chem. 266:12821-12824. [PubMed] [Google Scholar]
- 6.Briggs, M. D., S. M. G. Hoffman, L. M. King, A. S. Olsen, H. Morhenweiser, J. G. Leroy, G. R. Mortier, D. L. Rimoin, R. S. Lachman, E. S. Gaines, L. A. Cekleniak, R. G. Knowlton, and D. H. Cohn. 1995. Pseudoachondroplasia and multiple dysplasia due to mutations in the cartilage oligomeric matrix protein gene. Nat. Genet. 10:330-336. [DOI] [PubMed] [Google Scholar]
- 7.Briggs, M. D., G. R. Mortier, W. G. Cole, L. M. King, S. S. Golik, J. Bonaventure, L. Nuytinck, A. De Paepe, J. G. Leroy, L. Biesecker, M. Lipson, W. R. Wilcox, R. S. Lachman, D. L. Rimoin, R. G. Knowlton, and D. H. Cohn. 1998. Diverse mutations in the gene for cartilage oligomeric matrix protein in the pseudoachondroplasia-multiple epiphyseal dysplasia disease spectrum. Am. J. Hum. Genet. 62:311-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, Z., D. Heinegård, and Y. Sommarin. 1994. Distribution and expression of cartilage oligomeric matrix protein and bone matrix protein shows marked changes during rat femoral head development. Matrix Biol. 14:773-781. [DOI] [PubMed] [Google Scholar]
- 9.Coligan, J. E., and H. S. Slayter. 1984. Structure of thrombospondin. J. Biol. Chem. 259:3944-3948. [PubMed] [Google Scholar]
- 10.Crawford, S. E., V. Stellmach, J. E. Murphy-Ullrich, S. Ribeiro, J. Lawler, R. O. Hynes, G. P. Boivin, and N. Bouck. 1998. Thrombospondin-1 is a major activator of TGF-β1 in vivo. Cell 93:1159-1170. [DOI] [PubMed] [Google Scholar]
- 11.Délot, E., S. G. Brodie, L. M. King, W. R. Wilcox, and D. H. Cohn. 1998. Physiological and pathological secretion of cartilage oligomeric matrix protein by cells in culture. J. Biol. Chem. 273:26692-26697. [DOI] [PubMed] [Google Scholar]
- 12.DiCesare, P., N. Hauser, D. Lehman, S. Pasumarti, and M. Paulsson. 1994. Cartilage oligomeric matrix protein (COMP) is an abundant component of tendon. FEBS Lett. 354:237-240. [DOI] [PubMed] [Google Scholar]
- 13.Fässler, R., P. N. J. Schnegelsberg, J. Dausman, T. Shinya, Y. Muragaki, M. T. McCarthy, B. R. Olsen, and R. Jaenisch. 1994. Mice lacking α 1 (IX) collagen develop noninflammatory degenerative joint disease. Proc. Natl. Acad. Sci. USA 91:5070-5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fife, R. S. 1988. Identification of cartilage matrix glycoprotein in synovial fluid in human osteoarthritis. Arthritis Rheum. 31:553-556. [DOI] [PubMed] [Google Scholar]
- 15.Guo, Y., D. Bozic, V. N. Malashkeviich, R. A. Kammerer, T. Schulthess, and J. Engel. 1998. All-trans retinol, vitamin D and other hydrophobic compounds bind in the axial pore of the five-stranded coiled-coil domain of cartilage oligomeric matrix protein. EMBO J. 17:5265-5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hecht, J. T., E. Hayes, M. Snuggs, G. Decker, D. Montufar-Solis, K. Doege, F. Mwalle, R. Poole, J. Stevens, and P. J. Duke. 2001. Calreticulin, PDI, Grp94 and Bip chaperone proteins are associated with COMP in pseudoachondroplasia chondrocytes. Matrix Biol. 20:251-262. [DOI] [PubMed] [Google Scholar]
- 17.Hecht, J. T., D. Montufar-Solis, G. Decker, J. Lawler, K. Daniels, and J. Duke. 1998. Retention of cartilage oligomeric matrix protein (COMP) and cell death in redifferentiated pseudoachondroplasia chondrocytes. Matrix Biol. 17:625-633. [DOI] [PubMed] [Google Scholar]
- 18.Hecht, J. T., L. D. Nelson, E. Crowder, Y. Wang, F. F. B. Elder, W. R. Harrison, C. A. Francomano, C. K. Prange, G. G. Lennon, M. Deere, and J. Lawler. 1995. Mutations in exon 17B of cartilage oligomeric matrix protein (COMP) cause pseudoachondroplasia. Nat. Genet. 10:325-329. [DOI] [PubMed] [Google Scholar]
- 19.Hedbom, E., P. Antonsson, A. Hjerpe, D. Aeschlimann, M. Paulsson, E. Rosa-Pimentel, Y. Sommarin, M. Wendel, Å. Oldberg, and D. Heinegård. 1992. Cartilage matrix proteins. An acidic oligomeric protein (COMP) detected only in cartilage. J. Biol. Chem. 267:6132-6136. [PubMed] [Google Scholar]
- 20.Holden, P., R. S. Meadows, K. L. Chapman, M. E. Grant, K. E. Kadler, and M. D. Briggs. 2001. Cartilage oligomeric matrix protein interacts with type IX collagen, and disruptions to these interactions identify a pathogenetic mechanism in a bone dysplasia family. J. Biol. Chem. 276:6046-6055. [DOI] [PubMed] [Google Scholar]
- 21.International Working Group on Constitutional Diseases of Bone. 1992. International classification of osteochondrodysplasias. Am. J. Med. Genet. 44:223-229. [DOI] [PubMed] [Google Scholar]
- 22.Kyriakides, T. R., Y.-H. Zhu, L. T. Smith, S. T. Bain, Z. Yang, M. T. Lin., K. G. Danielsson, R. V. Iozzo, M. LaMarca, C. E. McKinney, E. I. Ginns, and P. Bornstein. 1998. Mice that lack TSP-2 display connective tissue abnormalities that are associated with disordered collagen fibrillogenesis, an increased vascular density and a bleeding diathesis. J. Cell Biol. 140:419-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laherty, C. D., K. O'Rourke, F. W. Wolf, R. Katz, M. F. Selding, and V. M. Dixit. 1992. Characterization of mouse thrombospondin 2 sequence and expression during cell growth and development. J. Biol. Chem. 267:3274-3281. [PubMed] [Google Scholar]
- 24.Larsson, T., Y. Sommarin, M. Paulsson, P. Antonsson, E. Hedbom, and D. Heinegård. 1991. Cartilage matrix proteins; a basic 36 kDa protein with a restricted distribution to cartilage and bone. J. Biol. Chem. 266:20428-20433. [PubMed] [Google Scholar]
- 25.Lawler, J., L. H. Derick, J. E. Connolly, J.-H. Chen, and F. C. Chao. 1985. The structure of human platelet thrombospondin. J. Biol. Chem. 260:3762-3772. [PubMed] [Google Scholar]
- 26.Lawler, J., M. Duquette, C. A. Whittaker, J. C. Adams, K. McHenry, and D. W. DeSimone. 1993. Identification and characterization of thrombospondin-4, a new member of the thrombospondin gene family. J. Cell Biol. 120:1059-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maddox, B. K., D. R. Keene, L. Y. Sakai, N. L. Charbonneau, N. P. Morris, C. C. Ridgway, B. A. Boswell, M. D. Sussman, W. A. Horton, H. P. Bächinger, and J. T. Hecht. 1997. The fate of cartilage oligomeric matrix protein is determined by the cell type in the case of a novel mutation in pseudoachondroplasia. J. Biol. Chem. 272:30993-30997. [DOI] [PubMed] [Google Scholar]
- 28.Maynard, J. A., R. R. Cooper, and I. V. Ponseti 1972. A unique rough surfaced endoplasmic reticulum inclusion in pseudoachondroplasia. Lab. Investig. 26:40-44. [PubMed] [Google Scholar]
- 29.McBurney, M. W., L. C. Sutherland, C. N. Adra, B. Leclair, M. A. Rudnicki, and K. Jardine. 1991. The mouse Pgk-1 gene promoter contains an upstream activator sequence. Nucleic Acids Res. 20:5755-5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mörgelin, M., D. Heinegård, J. Engel, and M. Paulsson. 1992. Electron microscopy of native cartilage oligomeric matrix protein purified from the Swarm rat chondrosarcoma reveals a five-armed structure. J. Biol. Chem. 267:6137-6141. [PubMed] [Google Scholar]
- 31.Nagy, A. J., R. Rossant, W. Nage, W. Abramow-Newerly, and J. C. Roder. 1993. Derivation of completely cell culture-derived mice from early passage embryonic stem cells. Proc. Natl. Acad. Sci. USA 90:8424-8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Newton, G., S. Weremowics, C. C. Morton, N. G. Copeland, D. J. Gilbert, N. A. Jenkins, and J. Lawler. 1994. Characterization of human and mouse cartilage oligomeric matrix protein. Genomics 24:435-439. [DOI] [PubMed] [Google Scholar]
- 33.Nguyen, B. Q., and R. S. Fife. 1986. Vitreous contains a cartilage-related protein. Exp. Eye Res. 43:375-382. [DOI] [PubMed] [Google Scholar]
- 34.Oldberg, Å., P. Antonsson, K. Lindblom, and D. Heinegård. 1989. A collagen-binding 59 kDa protein (fibromodulin) is structurally related to the small proteoglycans PG-S1 and PG-S2 (decorin). EMBO J. 8:2601-2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oldberg, Å., P. Antonsson, K. Lindblom, and D. Heinegård. 1992. COMP (cartilage oligomeric matrix protein) is structurally related to the thrombospondins. J. Biol. Chem. 267:22346-22350. [PubMed] [Google Scholar]
- 36.Riessen, R., M. Fenchel, H. Chen, D. Axel, K. Karch, and J. Lawler. 2000. Cartilage oligomeric matrix protein (thrombospondin-5) is expressed by vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 21:47-54. [DOI] [PubMed] [Google Scholar]
- 37.Rosenberg. K., H. Olsson, M. Morgelin, and D. Heinegard. 1998. COMP shows high affinity Zn-dependent interaction with triple helical collagen. J. Biol. Chem. 273:20397-20403. [DOI] [PubMed] [Google Scholar]
- 38.Smith, R. K. W., L. Zunino, P. M. Webbon, and D. Heinegård. 1997. The distribution of cartilage oligomeric matrix protein (COMP) in tendon and its variation with tendon site, age and load. Matrix Biol. 16:255-271. [DOI] [PubMed] [Google Scholar]
- 39.Stanescu, R., V. Stanescu, M. P. Muriel, and P. Maroteaux. 1993. Multiple epiphyseal dysplasia, Fairbank type: morphologic and biochemical study of cartilage. Am. J. Med. Genet. 45:501-507. [DOI] [PubMed] [Google Scholar]
- 40.Svensson, L., A. Aszódi, F. P. Reinholt, R. Fässler, D. Heinegård, and Å. Oldberg. 1999. Fibromodulin-null mice have abnormal collagen fibrils, tissue organization, and altered lumican deposition in tendon. J. Biol. Chem. 274:9636-9647. [DOI] [PubMed] [Google Scholar]
- 41.Thur, J., K. Rosenberg, D. P. Nitche, T. Pihlajamaa, L. Ala-Kokko, D. Heinegard, M. Paulsson, and P. Maurer. 2001. Mutations in cartilage oligomeric matrix protein causing pseudoachondroplasia and multiple epiphyseal dysplasia affect binding of calcium and collagen I, II, and IX. J. Biol. Chem. 276:6083-6092. [DOI] [PubMed] [Google Scholar]
- 42.Vos, H. L., S. Devarayalu, Y. De Vries, and P. Bornstein. 1992. Thrombospondin 3 (Thbs3), a new member of the thrombospondin gene family. J. Biol. Chem. 267:12192-12196. [PubMed] [Google Scholar]
- 43.Vranka, J., A. Mokashi, D. R. Keene, S. Tufa, G. Corson, M. Sussman, W. A. Horton, K. Maddox, L. Sakai, and H. P. Bächinger. 2001. Selective intracellular retention of extracellular matrix proteins and chaperones associated with pseudoachondroplasia. Matrix Biol. 20:439-450. [DOI] [PubMed] [Google Scholar]