

Abstract
Iron regulatory protein 1 (IRP1), a major posttranscriptional regulator of cellular iron and energy metabolism, is controlled by an iron-sulfur cluster switch. Cysteine-437 is critical for coordinating the cluster, and its replacement yields mutants that do not respond to iron perturbations and constitutively bind to cognate mRNA iron-responsive elements (IREs). The expression of IRP1C437S in cells has been associated with aberrations in iron homeostasis and toxicity. We have established clones of human lung (H1299) and breast (MCF7) cancer cells that express high levels of IRP1C437S in a tetracycline-inducible manner. As expected, IRP1C437S stabilizes transferrin receptor mRNA and inhibits translation of ferritin mRNA in both cell types by binding to their respective IREs. However, H1299 transfectants grown at high densities are able to overcome the IRP1C437S-mediated inhibition in ferritin synthesis. The mechanism involves neither alteration in ferritin mRNA levels nor utilization of alternative transcription start sites to eliminate the IRE or relocate it in less inhibitory downstream positions. The derepression of ferritin mRNA translation occurs under conditions where global protein synthesis appears to be impaired, as judged by a significant enrichment in the expression of the underphosphorylated form of the translational regulator 4E-BP1. Collectively, these data document an example where ferritin mRNA translation evades control of the IRE-IRP system. The physiological implications of this response are reflected in protection against iron-mediated toxicity, oxidative stress, and apoptosis.
Iron regulatory protein 1 (IRP1) and IRP2 are involved in the coordinate posttranscriptional regulation of iron metabolism by binding to mRNA iron-responsive elements (IREs). These are hairpin structures in the untranslated regions (UTRs) of a growing family of mRNAs that encode proteins of iron uptake, storage, utilization, and transport as well as energy metabolism (34). IRE-IRP interactions control mRNA translation and stability in response to iron levels and other signals such as nitric oxide, hypoxia, and oxidative stress (2, 10, 28). The best-characterized IRE-containing mRNAs encode the transferrin receptor (TfR), which plays a critical role in cellular iron uptake, and ferritin, a protein for intracellular iron storage. Iron starvation induces IRP binding to multiple IREs in the 3′ UTR of TfR mRNA, resulting in its stabilization, and to a single IRE in ferritin (heavy [H] and light [L] chain) mRNAs, inhibiting their translation.
While IRP2 is regulated at the level of protein stability, IRP1 is controlled by an iron-sulfur cluster switch. In iron-replete cells, IRP1 assembles a cubane 4Fe-4S cluster that converts it to cytosolic aconitase and prevents IRE-binding. Iron starvation, nitric oxide, and extracellular H2O2 trigger dissociation of the cluster, loss of aconitase, and acquisition of IRE-binding activity, presumably facilitated by a conformational change. Mutational analysis has identified three cysteine residues at positions 437, 503, and 506 as indispensable for anchoring the cluster (20, 30). C437 is particularly important as a site for in vitro manipulations of IRP1. When not engaged in the coordination of the cluster, this residue is a target of alkylation by N-ethylmaleimide and, in addition, it forms disulfide bridges with either C503 or C506 upon treatment of apoIRP1 (which does not contain iron) with diamide (20, 30).
As expected, C437S, as well as C503S and C506S, mutants of human IRP1 display maximal IRE-binding activity and do not respond to iron perturbations when transfected transiently into mouse L cells (20) or when expressed in HeLa cells from a glucocorticoid-inducible episomal vector (31). Attempts to obtain stable transfectants of these mutants in L or HeLa cells have failed, indicating that their expression may be associated with a toxic phenotype. Similar data have been obtained in a slightly different experimental setting. Expression of IRP1C437S from the glucocorticoid-inducible system in human rhabdomyosarcoma RD4 cells disrupts iron-dependent regulation of TfR and ferritin and appears to render the cells more sensitive to iron overload (9). However, the inability of RD4 cells to retain the transfected episome has impeded the efforts to study the physiological implications associated with IRP1C437S expression. To this end, we have developed human cancer cell lines expressing IRP1C437S under the tight control of a tetracycline-inducible promoter.
MATERIALS AND METHODS
Plasmid constructions.
Human IRP1 cDNA was excised from pMS-56-hIRF (17) and cloned in two steps (first, a 43-bp EcoRI/BamHI fragment containing the translation start codon and second, the remaining 2.94-kb BamHI/BamHI fragment) into the EcoRI/BamHI sites of the pSG5 vector (18). A C-terminal FLAG epitope was introduced into pSG5-hIRP1 by inserting a pair of annealed oligonucleotides (5′-CCAAGGACTACAAGGACGACGATGACAAGTAGA-3′ and 5′-AGCTTCTACTTGTCATCGTCGTCCTTGTAGTCCTTGG-3′) between its MscI/HindIII sites. This manipulation replaces the authentic stop codon following the MscI site with another one downstream of the FLAG epitope. The tagged IRP1 cDNA was cloned (in two steps) at the EcoRI/BamHI sites of pUHD10-3 (http://www.zmbh.uni-heidelberg.de/Bujard/reporter/pUHD10-3.html), which contains a tetracycline-inducible human cytomegalovirus minimal promoter (16), to yield pUHD10-3-hIRP1. Finally, pUHD10-3-hIRP1C437S was generated by replacing a 1.5-kb PstI/EcoRV fragment in the coding region of IRP1 with the respective fragment from pGEM-IRF-S437 (20). The pIRE.hGH indicator was constructed by excising the ferritin promoter in L5-GH (8) with EcoRI/BamHI and introducing a simian virus 40 promoter, generated by PCR amplification from pSG5-iNOS (27).
Cell culture and transfections.
HIRP1mut and MIRP1mut cells were established by cotransfecting tTA-H1299 (7) or tTA-MCF7 (36) cells with pUHD10-3-hIRP1C437S and the puromycin resistant pBabe by using the calcium phosphate method. Stable transfectants were selected in media containing 2 μg of puromycin/ml, 250 μg of G418/ml, and 2 μg of tetracycline/ml. The cells were maintained in medium supplemented with a tetracycline-free fetal bovine serum (Clontech). Transient transfections with the pIRE.hGH indicator were performed with Lipofectamine Plus (Gibco BRL).
Metabolic labeling and IP.
Cells were metabolically labeled for 2 h with 50 μCi of Tran35S-label/ml, a mixture of 70:30 [35S]methionine-cysteine (ICN). Quantitative immunoprecipitation (IP) from equal amounts of trichloroacetic acid-insoluble radioactivity with antibodies against ferritin (Roche), TfR (Zymed), or human growth hormone (hGH; National Hormone and Pituitary Program) was performed as described in reference 23 and followed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and autoradiography. The protocols for 59Fe-transferrin uptake experiments and analysis of 59Fe-ferritin immunoprecipitates were described in reference 4.
EMSA.
IRE-binding activity was analyzed by electrophoretic mobility shift assay (EMSA) as described in reference 27 and quantified by densitometric scanning.
Western blotting.
Cells were washed twice in phosphate-buffered saline (PBS), and lysates were prepared by direct lysis (unless otherwise indicated) in Laemmli sample buffer (21) (50 μl/106 cells). The lysates were immediately boiled for 5 min, and equal aliquots were resolved by SDS-PAGE on 11% (unless otherwise indicated) gels and transferred onto nitrocellulose filters. The blots were saturated with 10% nonfat milk in PBS and probed with M2-FLAG (Sigma), TfR (Zymed), β-actin (Sigma), ferritin (Roche), eukaryotic initiation factor 4E (eIF-4E; BD Transduction Laboratories), IRP1 (26), or 4E-BP1 (15) antibodies diluted in PBS containing 0.5% Tween 20. Dilutions were 1:500 for IRP1; 1:1,000 for M2-FLAG, TfR, β-actin, and ferritin; and 1:2,000 for eIF-4E and 4E-BP1 antibodies. Following washing with PBS containing 0.5% Tween 20, the blots with monoclonal FLAG, TfR, or eIF-4E antibodies were incubated with peroxidase-coupled rabbit anti-mouse immunoglobulin G (1:4,000 dilution). The blots with all other (polyclonal) antibodies were incubated with peroxidase-coupled goat anti-rabbit immunoglobulin G (1:5,000 dilution). The detection of peroxidase-coupled antibodies was performed with the enhanced chemiluminescence method (Amersham). The blots were quantified by densitometric scanning.
Northern blotting.
Cells were washed twice in PBS and lysed with the Trizol reagent (Gibco BRL), and RNA was prepared according to the manufacturer's recommendation. Total cellular RNA (10 μg) was electrophoretically resolved on denaturing agarose gels, transferred onto nylon membranes, and hybridized to radiolabeled human TfR, human H-ferritin, rat L-ferritin, or rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA probes. Autoradiograms were quantified by densitometric scanning.
Analysis of IRP1C437S-associated mRNAs by RT-PCR and mapping of H-ferritin transcription start site.
IRP1C437S was immunoprecipitated from extracts of HIRP1mut cells with the FLAG antibody according to a protocol described in reference 6. Reverse transcription-PCRs (RT-PCRs) were performed with the following pairs of primers: TfR, 5′-GAGACTGTCCCTCTGACTGGAAAAC-3′ and 5′-GGCAAAGATAATGCTTCTGCTGG-3′; H-ferritin, 5′-GCCACTGACAAAAATGACCCC-3′ and 5′-ATTCCGCCAAGCCAGATTCG-3′; and GAPDH, 5′-ACCACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′. The transcription start site of H-ferritin was mapped by means of the 5′ rapid amplification of cDNA ends (5′ RACE) technique with the SMART RACE cDNA amplification kit (Clontech), according to the manufacturer's recommendation. The reverse H-ferritin specific primer was 5′-TGGCGGCGACTAAGGAGAGGGCG-3′. The amplified cDNA was transferred to a nylon membrane and probed with 5′-labeled 5′-GCACTGTTGAAGCAGGAAAC-3′.
Analysis of cell viability, cell cycle, apoptosis, and intracellular ROS.
Cell viability was monitored by the MTS colorimetric assay (Promega). Cell cycle analysis with propidium iodide staining was as described in reference 7, and apoptosis was assayed with the Annexin V staining kit (Roche). Levels of intracellular reactive oxygen species (ROS) were monitored by employing the redox-sensitive probe 2′,7′-dichlorodihydrofluorescein diacetate (H2DCF-DA; Molecular Probes) and measuring the fluorescent derivative 2′,7′-dichlorofluorescin (DCF) (13). Fluorescence-activated cell sorter (FACS) analysis was performed in a Beckman Coulter instrument.
RESULTS
Tetracycline-inducible expression of IRP1C437S in H1299 cells.
For conditional expression of IRP1C437S we first utilized human H1299 (lung cancer) cells expressing the transactivator tTA (7). The cells were transfected with an IRP1C437S cDNA driven by a tetracycline-inducible promoter (tet-off system). Several clones were isolated in selective media, a total of 30 were screened for expression of IRP1C437S (tagged with a FLAG epitope at its C terminus), and 5 of them were found to be positive. Western blot analysis of two representative clones (18 and 29) is depicted in Fig. 1A. Probing with a FLAG antibody reveals that neither of the clones expresses IRP1C437S in the presence of tetracycline (Fig. 1A, lanes 1 and 3, top). However, removal of tetracycline for 2 days results in a profound induction of IRP1C437S (Fig. 1A, lanes 2 and 4). Probing the same filter with a β-actin antibody shows that the antibiotic does not affect the steady-state levels of this endogenous protein (Fig. 1A, bottom). While expression of IRP1C437S is very tightly regulated by tetracycline in both clones 18 and 29, the former appears to express slightly higher levels of IRP1C437S. We have utilized clone 18 for further characterization, and the cells are hereafter referred to as HIRP1mut.
FIG. 1.

Tetracycline-inducible expression of IRP1C437S in H1299 cells. (A) Two representative IRP1C437S transfectants (clones 18 and 29) were grown for 48 h with 2 μg of tetracycline (tet)/ml (+) or without tetracycline (−) and analyzed for expression of epitope-tagged IRP1C437S by Western blotting. The filter was probed with FLAG (top) and β-actin (bottom) antibodies. (B) HIRP1mut cells (clone 18) grown as described for panel A were left untreated (lanes 1 and 4) or treated overnight with 100 μM DFO (lanes 2 and 5) or 100 μM hemin (lanes 3 and 6). Cytoplasmic extracts were analyzed by EMSA with a 32P-labeled IRE probe in the absence (top) or presence (bottom) of 2% 2-ME.
To confirm that IRP1C437S is functional and unresponsive to iron manipulations, we evaluated IRE-binding activity by EMSA (Fig. 1B). Preliminary experiments suggested that H1299 cells are relatively poor in IRE-binding activity compared to other human and rodent cell lines (data not shown). An analysis of 50 μg of cytoplasmic extracts from iron-perturbed tetracycline-treated [tet(+)] HIRP1mut cells displays an endogenous IRE-binding activity (Fig. 1B, lanes 1 to 3, top) that is induced approximately 3.5-fold by treatment with the iron chelator desferrioxamine (DFO) (Fig. 1B, lane 2). Incubation of the same protein extracts with 2-mercaptoethanol (2-ME), which is commonly employed to assess levels of activatable IRP1 (28), augments endogenous IRE-binding activity in extracts of control and iron-loaded cells to the levels of DFO-treated cells (Fig. 1B, bottom). Removal of tetracycline leads to a dramatic increase in IRE-binding activity. An analysis of 5 μg of cytoplasmic extracts reveals an ∼100-fold stimulation in IRE-binding that is not affected by iron manipulations (Fig. 1B, lanes 4 to 6, top panel). First, IRE-binding is not further activated in iron-chelated cells (Fig. 1B, lane 5), and second, it is not negatively regulated in iron-loaded cells (Fig. 1B, lane 6). Importantly, the analysis with 2-ME does not show any further activation of IRE-binding (Fig. 1B, lanes 4 to 6). We conclude that the expression of IRP1C437S accounts for the dramatic increase in nonregulated IRE-binding activity.
Induction of IRP1C437S stimulates expression of TfR.
Having established that withdrawal of tetracycline results in overexpression of functional IRP1C437S, we went on to analyze TfR, which is known to be regulated by the IRE-IRP system. HIRP1mut cells were grown with or without tetracycline for 2 days, and TfR mRNA was analyzed by Northern blotting (Fig. 2A, top panel). Removal of the antibiotic results in an ∼3.3-fold increase in TfR mRNA levels (Fig. 2A, lanes 1, 2, 7, and 8). These are sustained even in cells previously treated with hemin for 4 h (Fig. 2A, lane 6) or overnight (Fig. 2A, lane 12), despite the (expected) complete absence of the TfR mRNA signal in iron-loaded tet(+) cells (Fig. 2A, lanes 5 and 11). Thus, the expression of IRP1C437S correlates with an induction of TfR mRNA, suggesting that IRP1C437S promotes its stabilization. This is also evident in cells treated with DFO for 4 h {compare the ∼3.5-fold induction in tet(+) cells with the ∼8-fold induction in non-tetracycline-treated [tet(−)] cells [Fig. 2A, lanes 3 and 4]}. The removal of tetracycline does not affect H-ferritin and GAPDH (control) mRNAs (Fig. 2A, middle and bottom panels). The ∼4-fold increase of H-ferritin mRNA in response to hemin (Fig. 2A, lanes 5, 6, 11, and 12) is probably due to hemin-mediated transcriptional activation (13).
FIG. 2.

Induction of IRP1C437S regulates expression of TfR. HIRP1mut cells were grown for 48 h with 2 μg of tetracycline (tet)/ml (+) or without tetracycline (−) and treated as indicated. (A) Analysis of TfR (top), H-ferritin (middle), and GAPDH (bottom) mRNA by Northern blotting. Lanes 1, 2, 7, and 8, untreated controls; lanes 3, 4, 9, and 10, treatment with 100 μM DFO for 4 h or overnight (o/n); lanes 5, 6, 11, and 12, treatment with 100 μM hemin for 4 h or overnight. (B) Analysis of TfR (top) and GAPDH (bottom) mRNA by Northern blotting. Lanes 1 and 2, untreated controls; lanes 3 to 6, treatment with 100 μM DFO for 8 h with (+) or without (−) 5 μM actinomycin D (act D). (C) Western blotting with FLAG (top), TfR (middle), and β-actin (bottom) antibodies. Lanes 1 and 2, untreated controls; lanes 3 to 6, overnight treatment with 100 μM DFO or 100 μM hemin.
Interestingly, the protection to TfR mRNA afforded by IRP1C437S is not sufficient to yield TfR mRNA steady-state levels comparable to those observed after prolonged iron chelation. An overnight treatment with DFO stimulates TfR mRNA dramatically (∼30-fold), and this does not appear to further increase with IRP1C437S (Fig. 2A, lanes 9 and 10; the densitometric quantification was performed at a lower exposure). These data suggest that the profound stimulation of TfR mRNA in response to iron chelation may also involve a transcriptional component. To examine this, the cells were exposed for 8 h to DFO in the presence or absence of the transcriptional inhibitor actinomycin D and TfR mRNA was assessed by Northern blotting (Fig. 2B). The treatment with actinomycin D clearly mitigates the response to DFO, strongly suggesting that transcription activation contributes significantly to the increase in TfR mRNA in response to iron chelation.
The IRP1C437S-dependent alterations in TfR mRNA are reflected at the protein level (Fig. 2C). Western blotting shows that removal of tetracycline for 2 days activates IRP1C437S expression (Fig. 2C, top) and correlates with an ∼2-fold increase in TfR (Fig. 2C, middle, lanes 1 and 2). The effect is more prominent in cells pretreated overnight with hemin, where removal of the antibiotic correlates with recovery of TfR from ∼10% to almost control levels (Fig. 2C, lanes 5 and 6). Interestingly, at the protein level, stimulation of TfR expression by IRP1C437S is quantitatively comparable to that observed in iron-starved cells (treated overnight with DFO), no matter whether IRP1C437S is turned on or not (Fig. 2C, lanes 2 to 4). These manipulations leave β-actin (the internal control) largely unaffected (Fig. 2C, bottom). Thus, the induction of IRP1C437S in HIRP1mut cells activates expression of TfR mRNA and protein.
Conditional translational repressor activity of IRP1C437S.
To assess the effect of IRP1C437S in the expression of ferritin, a known target of IRP-mediated translational control, HIRP1mut cells were grown for up to 7 days with or without tetracycline and ferritin expression was analyzed by Western blotting (Fig. 3A). Probing the filter with a polyclonal IRP1 antibody (26) shows a clear induction of IRP1C437S in tet(−) cells over the entire course of the experiment (Fig. 3A, top) (this antibody also cross-reacts with endogenous wild-type IRP1). Interestingly, the steady-state levels of IRP1C437S tend to increase (∼3.5-fold) after the third day of tetracycline withdrawal, very likely reflecting the accumulation of protein that has, at least in the wild type, a relatively long half-life of ∼24 h (26, 33). In agreement with the data shown in Fig. 2, TfR is consistently upregulated in tet(−) cells (Fig. 3A, second panel), correlating well with the expression pattern of IRP1C437S over 7 days. As expected, ferritin diminishes in cells grown for 2 days without tetracycline (Fig. 3A, third panel, lanes 1 to 4). However, surprisingly, ferritin recovers after 3 days and, moreover, its levels even appear to increase (∼2-fold) after 7 days of tetracycline withdrawal (Fig. 3A, lanes 5 to 10). No differences are observed in control β-actin (Fig. 3A, bottom).
FIG. 3.

Time-dependent recovery of ferritin in HIRP1mut cells expressing active IRP1C437S. Cells (2 × 106) were plated in 100-mm-diameter dishes and grown with 2 μg of tetracycline (tet)/ml (+) or without tetracycline (−) for 1 to 7 days. (A) Western blotting with IRP1 (top), TfR (second panel), ferritin (third panel), or β-actin (bottom) antibodies. (B) Analysis of cytoplasmic extracts (5 μg) for IRE-binding by EMSA with a 32P-labeled IRE probe. (C) Analysis of TfR (top), H-ferritin (second panel), L-ferritin (third panel), and GAPDH (bottom) mRNA by Northern blotting. t, time.
To make sure that IRP1C437S remains active over the course of the treatment, we analyzed the same cell extracts for IRE-binding by EMSA (Fig. 3B). The result is consistent with that of the Western blotting, showing a marked activation in tet(−) cells that is further increased (∼3.5-fold) after 5 to 7 days of growth without tetracycline. The possibility remains that the recovery of ferritin expression could be due to a massive increase in ferritin mRNA that may outnumber active IRP molecules. We performed Northern blot analysis to address this issue (Fig. 3C). Hybridizations with TfR (Fig. 3C, top) and GAPDH (Fig. 3C, bottom) probes serve as controls. While there are no appreciable alterations in H- and L-ferritin mRNA levels up to day 3 (Fig. 3C, second and third panels, lanes 1 to 6), the removal of tetracycline stimulates a slight (∼1.5-fold) increase after 5 days and a profound (∼4-fold) increase after 7 days. It is evident that the recovery of ferritin at days 3 to 5 of IRP1C437S induction cannot be attributed to alterations in the ratio between the inhibitor and the mRNA. But even on day 7, the increase in ferritin mRNA is compensated by a concomitant ∼3.5-fold further stimulation in IRP1C437S expression and activity (Fig. 3A and B, compare lanes 1 and 2 with lanes 7 to 10). Thus, the recovery of ferritin expression cannot be justified on the basis of dramatic alterations in the IRP/ferritin mRNA ratio (see Discussion). Taken together, these results suggest that in the early phase (first 2 days) of IRP1C437S induction, the expression of ferritin is coordinately regulated with that of TfR by IRP1C437S but afterwards it appears to be uncoupled from this control.
We wondered whether this unanticipated biphasic regulation of ferritin could be related to growth conditions. To address this important question, we examined ferritin levels in response to variable conditions at the interface of the switch from inhibition to recovery in its expression. The HIRP1mut cells were seeded at three different dilutions in medium supplemented with either 10 or 30% fetal bovine serum and grown for 3 days with or without tetracycline. Following overnight treatment with hemin to increase steady-state levels of ferritin, the cells were harvested and extracts were analyzed by Western blotting. As expected, probing with the FLAG antibody shows tetracycline-dependent induction of IRP1C437S (Fig. 4A, top) that correlates with an ∼2-fold increase in TfR (Fig. 4A, second panel) and has no influence on β-actin (Fig. 4A, bottom). These data suggest that serum concentration and cell density affect neither the expression of IRP1C437S and TfR (and β-actin) nor the regulation of TfR by IRP1C437S. In contrast, these factors have a profound impact on the ability of IRP1C437S to regulate ferritin. Withdrawal of tetracycline correlates with complete inhibition of ferritin expression only in cells seeded at a low density (1 × 106 cells/100-mm-diameter dish) (Fig. 4A, third panel, lanes 1 and 2). The inhibition is compromised by either increasing the serum concentration to 30% (Fig. 4A, lanes 7 and 8) or doubling or tripling the number of seeded cells (Fig. 4A, lanes 3 and 4 or 5 and 6, respectively). The combination of high cell density and serum concentration results in almost complete recovery of ferritin (Fig. 4A, lanes 9 to 12) despite overexpression of highly active IRP1C437S (Fig. 4B). Moreover, this effect is not due to significant tetracycline-dependent alterations in H- or L-ferritin mRNAs (Fig. 4C). The above findings suggest that, under these experimental conditions, constitutive IRE-binding activity fails to block ferritin expression.
FIG. 4.

Cell density- and serum-dependent recovery of ferritin in HIRP1mut cells expressing active IRP1C437S. Cells (1 × 106, 2 × 106, or 3 × 106) were plated in 100-mm-diameter dishes and grown for 3 days with 2 μg of tetracycline (tet)/ml (+) or without tetracycline (−) in the presence of 10 or 30% fetal bovine serum. Prior to harvesting, the cells were treated overnight (o/n) with hemin. (A) Western blotting with FLAG (top), TfR (second panel), ferritin (third panel), or β-actin (bottom) antibodies. (B) Analysis of cytoplasmic extracts (5 μg) for IRE-binding by EMSA with a 32P-labeled IRE probe. (C) Analysis of TfR (top), H-ferritin (second panel), L-ferritin (third panel), and GAPDH (bottom) mRNA by Northern blotting.
To directly investigate the effects of IRP1C437S on ferritin mRNA translation, HIRP1mut cells were plated at various densities and grown for 3 days with or without tetracycline in the presence of 10 or 30% serum. Following pretreatment with hemin to augment ferritin synthesis or not, the cells were metabolically labeled with [35S]methionine-cysteine and TfR and ferritin were analyzed by simultaneous IP (Fig. 5A). Removal of tetracycline correlates with a notable increase in TfR synthesis, regardless of whether the cells have been previously loaded with hemin or not, and this effect is completely independent from growth conditions. In contrast, inhibition in ferritin synthesis is only observed in sparse tet(−) cells (Fig. 5A, lanes 1 to 4 and 9 to 12). Cells that have reached four-times-higher densities are able to overcome IRP1C437S-mediated inhibition in ferritin synthesis, either partially (when supplemented with 10% serum) (Fig. 5A, lanes 5 to 8) or completely (in the presence of 30% serum) (Fig. 5A, lanes 13 to 16). A shorter exposure of the film with a better resolution of the ferritin H and L chains and a longer exposure to compare ferritin levels in cells not pretreated with hemin are shown at the bottom of Fig. 5A. Taken together, our data suggest that, in HIRP1mut cells, the function of IRP1C437S as a translational repressor of ferritin mRNAs is conditional.
FIG. 5.

Cell density- and serum-dependent derepression of ferritin mRNA translation in HIRP1mut cells expressing active IRP1C437S. (A) Cells (1 × 106 or 4 × 106) were plated in 100-mm-diameter dishes and grown for 3 days with 2 μg of tetracycline (tet)/ml (+) or without tetracycline (−) in the presence of 10 or 30% fetal bovine serum. Following treatment with (+) or without (−) 100 μM hemin for 4 h, the cells were metabolically labeled with [35S]methionine-cysteine for 2 h and cell extracts were subjected to simultaneous quantitative IP with 2 μl of TfR and 5 μl of ferritin antibodies. Immunoprecipitated material was analyzed by SDS-PAGE on an 11% gel, and TfR and ferritin H and L chains (arrows) were visualized by autoradiography. The positions of molecular mass standards (in kilodaltons) are indicated on the right. Shorter and longer exposures of ferritin are also shown. (B) Low (∼120 cells/mm2) and high (∼500 cells/mm2) density cells grown with (+) or without (−) tetracycline for 24 h were transfected with pIRE.hGH and incubated for another 2 days to allow expression of the indicator. Following pretreatment with (+) or without (−) 100 μM hemin, the cells were metabolically labeled with [35S]methionine-cysteine for 2 h and cell lysates were subjected to quantitative IP with TfR (2 μl) and ferritin (5 μl) antibodies (one half) or with an hGH (5 μl) antibody (the other half). Immunoprecipitated material was analyzed as described for panel A. The positions of TfR, ferritin (H and L chains), and hGH are shown by arrows.
To exclude the possibility that this unprecedented response is mediated by elements other than IREs that could be unique to ferritin mRNAs, we utilized an indicator encoding an hGH mRNA under the control of a functional ferritin IRE in its 5′ UTR. It was previously shown that expression of this construct in transfected mouse B6 fibroblasts is iron regulated via the IRE-IRP system (8, 13). HIRP1mut cells were plated at either low (∼1 × 106 cells/100-mm-diameter dish) or high (∼4 × 106 cells/100-mm-diameter dish) density, transiently transfected with pIRE.hGH, and grown with or without tetracycline. Following metabolic labeling with [35S]methionine-cysteine, the synthesis of endogenous TfR and ferritin and transfected hGH were assessed by IP. In agreement with the data shown in Fig. 5A, the expression of IRP1C437S in sparse (control and iron loaded) cells results in coordinated regulation in the synthesis of TfR (Fig. 5B, top), ferritin (Fig. 5B, middle), and, importantly, hGH (Fig. 5B, bottom) (Fig. 5B, lanes 1 to 4). However, in confluent cultures, IRP1C437S is not sufficient to inhibit ferritin and hGH synthesis (Fig. 5B, lanes 5 to 8). We conclude that the function of IRP1C437S as a translational repressor of IRE-containing mRNAs depends on cell density and is also affected by factors present in serum.
The HIRP1mut cells define the first tissue culture model in which growth conditions promote ferritin mRNA translation by evading the control of the IRE-IRP system. To investigate whether this is a cell-specific or a more general phenomenon, we developed a similar system for conditional expression of IRP1C437S in human MCF7 (breast cancer) cells. A representative clone (referred to as MIRP1mut) was analyzed for IRP1C437S expression and function in a time course experiment as described for Fig. 3. Western blot analysis (Fig. 6A) shows a clear tetracycline-dependent induction of IRP1C437S (Fig. 6A, top) that is accompanied by stimulation of TfR (Fig. 6A, middle) and does not affect β-actin (Fig. 6A, bottom). As expected, IRP1C437S is highly active in IRE-binding (Fig. 6B). To examine the effect of IRP1C437S in ferritin synthesis, MIRP1mut cells grown at the different time points with or without tetracycline were metabolically labeled with [35S]methionine-cysteine and ferritin was analyzed by IP (Fig. 6C). In contrast to results obtained with HIRP1mut cells, the ability of IRP1C437S (Fig. 6C, top) to inhibit ferritin synthesis (Fig. 6C, bottom) in MIRP1mut cells is not compromised at the high densities reached at late time points (it should be noted that MCF7 cells grow at a relatively slower rate). Increasing the serum concentration to 30% in a similar time course experiment also failed to derepress ferritin mRNA translation (data not shown). Thus, the mechanism(s) to overcome IRP-mediated inhibition in ferritin synthesis appears to be cell specific.
FIG. 6.

The function of IRP1C437S as a translational inhibitor of ferritin is not compromised in dense MIRP1mut cells. Cells (2 × 106) were plated in 100-mm-diameter dishes and grown with 2 μg of tetracycline (tet)/ml (+) or without tetracycline (−) for 1 to 7 days. (A) Western blotting with FLAG (top), TfR (second panel), ferritin (third panel), or β-actin (bottom) antibodies. (B) Analysis of cytoplasmic extracts (5 μg) for IRE-binding by EMSA with a 32P-labeled IRE probe. (C) Metabolic labeling with [35S]methionine-cysteine for 2 h and quantitative IP with FLAG and ferritin antibodies. t, time.
Overcoming the IRE-IRP blockade: mechanistic investigations.
We focused on HIRP1mut cells to study ferritin mRNA translation. As an adaptive response to prolonged expression of IRP1C437S, the cells may develop mechanisms to overcome ferritin translational inhibition by utilizing cryptic start sites for ferritin mRNA transcription to either completely eliminate its IRE or relocate it to a less inhibitory cap-distal position. We directly addressed this hypothesis by mapping the 5′ end of H-ferritin mRNA with the 5′ RACE technique. A control RT-PCR with RNA from HIRP1mut cells and a combination of universal and gene-specific primers yields the predicted 253-bp fragment (Fig. 7A). Importantly, similar analysis of RNA from cells grown with or without tetracycline over 6 days yields the same single fragment. This is unambiguously assigned to H-ferritin because it hybridizes to a radiolabeled ferritin-specific oligonucleotide (corresponding to a fragment within the IRE) on a Southern blot (Fig. 7B). A similar picture was obtained with RNAs from dense cells grown with 30% serum (data not shown). These data demonstrate that the bypass of the translational block of the IRE-IRP complex does not involve alterations in the transcription start site of H-ferritin.
FIG. 7.

Mapping of the 5′ end in H-ferritin mRNA by 5′ RACE. (A) RNA from HIRP1mut cells was subjected to RT-PCR analysis with the universal primer A mix (Clontech) and a ferritin-specific primer. The 253-bp product (arrow) was resolved on a 1% agarose gel and visualized by ethidium bromide staining. (B) HIRP1mut cells (2 × 106) were plated in 100-mm-diameter dishes and grown with 2 μg of tetracycline (tet)/ml (+) or without tetracycline (−) for 1 to 6 days. RNA was extracted and analyzed as described for panel A. The reaction products were transferred onto a nylon membrane and probed with a 5′ end-labeled ferritin-specific oligonucleotide (corresponding to a fragment within the IRE). The hybridizing band (arrow) corresponds to the 253-bp fragment in panel A. The stronger signal in lane 6 possibly correlates to the increased H-ferritin mRNA levels observed in Fig. 3C. t, time.
Does IRP1C437S remain bound to ferritin IRE during translation or does it get displaced from the translational apparatus? To investigate this critical question, HIRP1mut cells were grown under various conditions, IRP1C437S was immunoprecipitated, and the association of ferritin mRNA was examined by an RT-PCR assay (6). The expression and recovery of IRP1C437S in the immunoprecipitate are shown in Fig. 8A. It is expected that the input and the IP material from both low- and high-density cells contain TfR mRNA bound to IRP1C437S. This is confirmed with the detection of a 302-bp fragment in the RT-PCR assay (Fig. 8B, lanes 2 to 6) with a pair of TfR-specific primers. Conversely, the specificity of this assay is demonstrated by the absence of a GAPDH signal (negative control) in the immunoprecipitate (Fig. 8B, lanes 14 to 17) while a 451-bp band corresponding to GAPDH is readily detected in the input (Fig. 8B, lane 13). Employment of H-ferritin-specific primers results in the detection of a 142-bp fragment in the immunoprecipitate from both sparse and dense tet(+) and tet(−) cells (and the input). Thus, the interaction of IRP1C437S with ferritin IRE is detectable even under conditions in which ferritin mRNA translation is relieved. While a negative outcome would provide a strong argument for a displacement of IRP1C437S from ferritin IRE in dense cultures, the semiquantitative nature of this assay does not allow us to draw reliable conclusions about whether IRE-IRP complexes are dissociated or simply ignored by the translational machinery. Nevertheless, this experiment provides another piece of evidence that IRP1C437S is competent in binding to IRE-containing mRNAs in vivo.
FIG. 8.

Detection of ferritin mRNA in IRP1C437S immunoprecipitates of sparse and dense HIRP1mut cells. Sparse (∼170 cells/mm2) and dense (∼550 cells/mm2) cells were grown for 3 days with 2 μg of tetracycline (tet)/ml (+) or without tetracycline (−) in the presence of 10 or 30% fetal bovine serum. Cell extracts (4.5 mg of protein) were subjected to quantitative IP with 15 μl of FLAG antibody. (A) Western blotting with FLAG (top) and β-actin (bottom) antibodies in cell lysates (30 μg) prior to IP (input, lanes 1 to 4) and in 1/20 of the FLAG IP material (lanes 5 to 8). (B) RT-PCR with gene-specific primers for human TfR, H-ferritin, and GAPDH in the input of low-density cells (lanes 2, 7, and 13) and the IP eluate of low-density (lanes 3, 4, 8, 9, 14, and 15) and high-density (lanes 5, 6, 10, 11, 16, and 17) cells. The products were resolved on a 1% agarose gel and visualized by ethidium bromide staining. The positions of bands corresponding to TfR (302 bp), H-ferritin (142 bp), and GAPDH (451 bp) are indicated by arrows.
We examined the possibility that the bypass of the IRE-IRP translational blockade in dense cultures might be associated with a global stimulation of translation. To this end, we analyzed the effect of cell density on the expression of eIF-4E and of its regulator 4E-BP1. The Western blot shown in Fig. 9 reveals that the levels of eIF-4E are not affected by cell density (Fig. 9, second panel). However, dense cells express a substantially enriched fraction of underphosphorylated 4E-BP1 (Fig. 9, third panel), whereas both eIF-4E and 4E-BP1 do not appear to be significantly affected by the expression of IRP1C437S (Fig. 9, upper panel) or differences in serum concentration. The enrichment of underphosphorylated 4E-BP1 is known to negatively regulate translation because this form of 4E-BP1 binds to eIF-4E and inhibits its association with the mRNA cap (14). Therefore, the derepression of ferritin synthesis in IRP1C437S-expressing cells is unlikely to be a result of a global stimulation of cap-dependent translation.
FIG. 9.

Cell density-dependent increase in the expression of underphosphorylated 4E-BP1. HIRP1mut cells (1 × 106 or 3 × 106) were plated in 100-mm-diameter dishes and grown for 3 days with 2 μg of tetracycline (tet)/ml (+) or without tetracycline (−) in the presence of 10 or 30% fetal bovine serum. Cell extracts were prepared and analyzed by SDS-PAGE. Two equal aliquots were resolved on 11 or 15% gels, followed by Western blotting. The filter corresponding to the 11% gel was probed with FLAG (top), eIF-4E (second panel), and β-actin (bottom) antibodies. The other filter was probed with a 4E-BP1 antibody (third panel). Three bands corresponding to different phosphorylation states of 4E-BP1 (arrows) were detected.  , phosphorylation.
, phosphorylation.
Derepression of ferritin mRNA translation confers protection against iron toxicity.
Previous studies suggested that overexpression of IRP1C437S in mammalian cells is associated with toxicity, especially in iron overload (9, 20, 31). A possible explanation for this is offered by a predicted IRP1C437S-mediated increase in iron uptake and concomitant inhibition of iron storage. In light of these findings and of the data described above, we designed experiments to study the physiological responses linked to the expression of IRP1C437S in HIRP1mut cells. First, we studied the effect of IRP1C437S in iron uptake and storage. Cells grown for 3 days at low or high densities with or without tetracycline were incubated with 59Fe-transferrin, and cell-associated radioactivity was measured in a gamma counter (Fig. 10). Removal of tetracycline stimulates iron uptake by ∼66 and ∼24% in sparse and dense cultures, respectively (Fig. 10). Quantitative ferritin IP followed by counting of the immunoprecipitate reveals that sparse cells expressing IRP1C437S display a reduced (by ∼44%) capacity to store 59Fe in ferritin (Fig. 10A), which is in line with the inhibitory effects in ferritin synthesis. In contrast, IRP1C437S does not affect storage of 59Fe in ferritin in dense cells (Fig. 10B), which is in agreement with its failure to modulate ferritin mRNA translation.
FIG. 10.

IRP1C437S-mediated stimulation of 59Fe uptake and cell density- and serum-dependent sequestration of 59Fe in ferritin. Sparse (∼120 cells/mm2) and dense (∼380 cells/mm2) HIRP1mut cells were grown for 3 days with 2 μg of tetracycline/ml [tet(+)] or without tetracycline [tet(−)] in the presence of 10 or 30% fetal bovine serum. The cells were incubated for 6 h with 10 μM human 59Fe-transferrin (16.27 cpm/pmol) and washed 3 times with ice-cold PBS, and radioactivity was monitored on a gamma counter. Subsequently, cell lysates were subjected to quantitative IP with a ferritin antibody (5 μl) and ferritin-associated 59Fe was counted. Values correspond to triplicate samples and are expressed in picomoles of 59Fe/106 cells/6 h. White bars, total cellular 59Fe; black bars, ferritin-associated 59Fe; ∗∗, P < 0.01 versus control (Student's t test)
How does IRP1C437S affect growth of HIRP1mut cells? To address this question, the cells were seeded at low or high densities and incubated for up to 7 days with or without tetracycline. Cell growth was monitored at different time points by a colorimetric proliferation-toxicity assay. In contrast to what would be expected on the basis of the previous experiments (9, 20, 31), expression of IRP1C437S has virtually no effect on cell growth either in sparse or in dense cultures (Fig. 11A and D). However, the addition of 50 μM ferric ammonium citrate (FAC) moderately (∼25% on day 7) inhibits growth of sparse IRP1C437S-expressing cells (Fig. 11B) while it does not affect dense cultures (Fig. 11E). Moreover, the addition of 25 μM hemin dramatically (∼75% on day 7) impairs growth of sparse IRP1C437S-expressing cells (Fig. 11C), leaving dense cultures unaffected (Fig. 11F).
FIG. 11.

Sensitivity of sparse IRP1C437S-expressing cells to iron. Sparse (∼50 cells/mm2) (A to C) and dense (∼200 cells/mm2) (D to F) HIRP1mut cells were grown for 7 days either without any added iron source (A and D) or in the presence of 50 μM FAC (B and E) or 25 μM hemin (C and F). The growth rate was monitored at different time points by the MTS assay. Formazan is plotted against time. Open squares and diamonds correspond to values from cells grown with 2 μg of tetracyline/ml [tet(+)] or without tetracycline [tet(−)], respectively. Data are means ± standard errors of the means (n = 5) from a representative of three independent experiments. OD490, optical density at 490 nm.
To investigate the mechanism for the iron-mediated inhibition of growth of HIRP1mut cells, we analyzed the effects of IRP1C437S on the cell cycle under inhibitory conditions, e.g., in low-density cultures grown for 4 days with 25 μM hemin. Propidium iodide staining and analysis by FACS reveals a slight (2%) but significant (P < 0.01) increase in the number of tet(−) cells arrested in the G2-M phase and a 7% increase (P < 0.0001) in apoptotic cells in the sub-G1 phase (Fig. 12A and B). The latter is confirmed upon staining with annexin V, which shows a 9% increase in tet(−) apoptotic cells (Fig. 12C and D). To elucidate whether this phenomenon is related to iron-induced oxidative stress, we analyzed the levels of ROS with the redox-sensitive dye H2DCF-DA. Expression of IRP1C437S correlates with an ∼2.4-fold increase in DCF fluorescence, which reflects intracellular ROS (Fig. 12E and F). Experiments with tet(+) and tet(−) sparse and dense cultures grown without extra iron, or with dense cultures supplemented with FAC or hemin, did not show any significant IRP1C437S-dependent alterations in cell cycle or redox status (data not shown). Taken together, we conclude that expression of IRP1C437S in HIRP1mut cells is not toxic per se but clearly aggravates iron toxicity and impairs the capacity of cells to cope with iron overload when it inhibits ferritin synthesis.
FIG. 12.

IRP1C437S-mediated apoptosis and oxidative stress in iron-loaded sparse cells. HIRP1mut cells (∼50 cells/mm2) were grown for 4 days with 2 μg of tetracycline/ml [tet(+)] or without tetracycline [tet(−)] in the presence of 25 μM hemin. DNA content was quantified by propidium iodide staining; the percentage of cells in various phases of the cell cycle is shown in the insets (A and B). Apoptotic cells were analyzed by propidium iodide-annexin V double staining (C and D). The generation of ROS was monitored by probing cells with the redox-sensitive dye H2DCF-DA (E and F). All values are the means of triplicate samples. 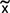 , median value of DCF fluorescence intensity; **, P < 0.01 versus control (Fisher's exact test); ∗∗∗, P < 0.0001 versus control (Fisher's exact test).
, median value of DCF fluorescence intensity; **, P < 0.01 versus control (Fisher's exact test); ∗∗∗, P < 0.0001 versus control (Fisher's exact test).
DISCUSSION
IRP1 and IRP2 are important regulators of cellular iron metabolism. Disruption of their activity by means of gene targeting or, conversely, expression of constitutive mutants, are valuable approaches to better understand their function at the cellular and systemic levels. Along these lines, it was recently shown that ablation of the IRP2 gene in Ireb2−/− mice is associated with aberrant iron metabolism in the intestinal mucosa and the central nervous system and leads to severe neurodegeneration (22). In contrast, targeted disruption of the IRP1 gene produced viable mice that do not exhibit a detectable phenotype (32). While no studies involving the expression of constitutive IRP mutants in animals are thus far available, earlier work has provided evidence of iron-dependent toxicity associated with the expression of the constitutive mutant IRP1C437S in mammalian cell lines (9, 20, 31).
To mitigate these obstacles and establish conditions for studying the physiological implications associated with the expression of IRP1C437S, we have employed a tetracycline-inducible expression system in H1299 and MCF7 cells. Clonal selection of IRP1C437S transfectants yielded the HIRP1mut and MIRP1mut cell lines that express high levels of functional IRP1C437S under the tight control of a tetracycline-responsive promoter (Fig. 1A and 6A). The removal of tetracycline results in the supraphysiological increase of an IRE-binding activity unresponsive to iron perturbations (Fig. 1B and 6B). These properties render HIRP1mut and MIRP1mut cells suitable models to challenge the IRE-IRP system and study the function of IRP1 in cellular iron homeostasis, especially in the context of iron toxicity. Our findings are discussed below.
Conditional uncoupling of coordinated regulation of TfR and ferritin expression by the IRE-IRP system.
As expected, induction of IRP1C437S stimulates TfR mRNA and protein levels (Fig. 2 to 6). However, the ∼3.3-fold stimulation of TfR mRNA in tet(−) HIRP1mut cells lags almost an order of magnitude behind the ∼30-fold increase achieved in tet(+) DFO-treated cells (Fig. 2A), implying that IRE-IRP1C437S interactions do not fully account for its dramatic upregulation in response to the chelator. In fact, the analysis with actinomycin D (Fig. 2B) suggests that, in these cells, transcription contributes significantly to the increase in TfR mRNA expression following treatment with DFO. A similar conclusion was reached earlier in experiments with B6 fibroblasts (5). However, in L fibroblasts, iron regulation of TfR was almost exclusively posttranscriptional (25), suggesting that the relative contribution of the TfR promoter and the IRE-IRP system may vary in different cell types. Nevertheless, it is noteworthy that the modest (∼3.3-fold) and the dramatic (∼30-fold) upregulation of TfR mRNA in response to the expression of IRP1C437S or treatment with DFO, respectively (Fig. 2A), are reflected in a mere ∼2-fold stimulation of TfR protein levels (Fig. 2C). An analogous discrepancy was also recently observed in control and DFO-treated B6 cells (4), arguing that the expression of TfR may be subjected to regulation by additional posttranscriptional and/or posttranslational mechanisms.
While our data are in perfect agreement with the regulatory model for IRP-mediated stabilization of TfR mRNA, the effects of IRP1C437S on ferritin expression are more complex. At a first glance, IRP1C437S appears to be sufficient to inhibit ferritin mRNA translation in both HIRP1mut and MIRP1mut cells (Fig. 3 and 6). However, tet(−) HIRP1mut cells grown at high densities are able to synthesize ferritin despite the expression of highly active and functional (with regard to TfR regulation) IRP1C437S. Increased serum concentrations stimulate this response (Fig. 4 and 5). We have estimated a density of ∼600 cells/mm2 as the cutoff at which IRP1C437S ceases to function as a translational inhibitor. A similar uncoupling of the coordinated regulation of TfR and ferritin mRNAs has previously been observed in a different experimental system involving physiological levels of IRE-binding activity. The administration of carbon tetrachloride to rats triggered regeneration of the liver associated with enhanced TfR and ferritin synthesis in a background of increased IRP2 activity (3). It remains to be shown whether there is a common denominator in the relief of IRP-mediated translational inhibition of ferritin synthesis in rapidly proliferating cells like cancer or nontransformed cells of a regenerating tissue, neither of which are constrained by density-dependent growth arrest (contact inhibition). However, the predicted (at least under the conditions tested) phenotype of MIRP1mut cells, as opposed to that of HIRP1mut cells, indicates that the mechanism(s) for derepression of ferritin mRNA translation may operate in a cell- and tissue-specific context. Our data may help to identify physiologically relevant situations in which ferritin mRNA translation evades the control of the IRE-IRP system.
Mechanistic aspects of ferritin mRNA translation in IRP1C437S-expressing cells.
From the mechanistic point of view, the failure of IRP1C437S to perform as an inhibitor of ferritin (and IRE.hGH) mRNA translation in dense HIRP1mut cell cultures is striking, considering that IRP binding to translation-type IREs sterically abolishes binding of the 43S ribosome (24). However, IRE-IRP complexes in cap-distal positions are not inhibitory to translation (29). Plausible scenarios based on alternative ferritin mRNA transcription from cryptic sites could explain the overcome in the IRE-IRP blockade. Thus, utilization of putative downstream site(s) would bypass the IRE while upstream initiation would shift it away from the cap. The experiments described for Fig. 7 clearly rule out these possibilities and demonstrate that the recovery of ferritin mRNA translation in dense IRP1C437S-expressing cells does not involve elimination or relocation of the IRE.
The prolonged growth of HIRP1mut cells in the absence of tetracycline seems to stimulate H- and L-ferritin mRNA expression on day 7 (Fig. 3C). Conceivably, this may be related to oxidative stress-mediated transcriptional activation via an antioxidant response element located in the H- and L-ferritin promoter (35). Since IRP1C437S expression and activity, which is already upregulated ∼100-fold upon removal of tetracycline (Fig. 1 and 3), is further induced after 5 days of growth (Fig. 3A and B), it seems unlikely that the concentration of ferritin mRNA exceeds that of the inhibitor, even on day 7. Nonetheless, this increase in ferritin mRNA may well account for the ∼2-fold stimulation in ferritin levels (Fig. 3A, lanes 9 and 10). Taken together, our data do not support a model for ferritin mRNA translational derepression based on alterations in the IRE/IRP ratio.
It is not clear whether IRP1C437S gets displaced from the IREs during translation or not. To address this, we performed the experiment described for Fig. 8, which is based on an RT-PCR assay that showed association of FMRP, a fragile X-related protein, with its own mRNA in vivo (6). This assay revealed the presence of ferritin mRNA in FLAG immunoprecipitates from both sparse and dense cells, implying that in the latter, IRP1C437S remains bound to ferritin IRE under conditions in which the mRNA is translated. However, we cannot rule out the possibility that the assay has resulted in the amplification of a translationally silent fraction of ferritin mRNA bound to the inhibitor. Employment of quantitative methods (1) or analysis of IRP1C437S in polysome-associated ferritin mRNA would give more-definite answers.
The stimulatory effect of serum in the derepression of ferritin mRNA translation prompted us to analyze the levels of eIF-4E and of its regulator 4E-BP1. The data in Fig. 9 demonstrate that HRP1C437S cells grown at high densities express a significantly enriched fraction of underphosphorylated 4E-BP1. This is known to negatively affect cap-dependent translation (14). Interestingly, a similar response was observed in nontransformed human skin fibroblasts, but this was accompanied by a decrease in eIF-4E expression (12). We conclude that cell density-dependent bypass of the IRE-IRP block is not a result of a global stimulation of cap-dependent translation but rather an mRNA-specific phenomenon.
Effects of IRP1C437S expression on iron toxicity and cell cycle.
IRP1C437S stimulates the uptake of 59Fe from 59Fe-transferrin (Fig. 10). This effect is more prominent in sparse cultures. In dense cells the basal 59Fe uptake is higher, possibly because of a cell density-dependent increase in the internalization efficiency of TfR (19). Nevertheless, the increase in iron uptake has no obvious effects on the growth of both sparse and dense cells, as judged by the proliferation assay (Fig. 11), despite the fact that the formers have a reduced capacity to store iron in ferritin (Fig. 10). However, loading cells with extra iron, either in the form of FAC or hemin, has a profound negative effect on the growth of sparse cells while dense cultures remain largely unaffected. This is, at least in part, due to iron-induced apoptosis that correlates with intracellular oxidative stress (Fig. 12). Remarkably, these responses are only observed under conditions where IRP1C437S inhibits ferritin synthesis. When the inhibition is bypassed, growth is normal and neither apoptosis nor oxidative stress is observed. These results underlie the paramount significance of ferritin in iron detoxification and the control of cell growth and are in line with the early embryonic lethal phenotype of Fth−/− (H-ferritin knockout) mice (11).
In conclusion, the HIRP1mut (and MIRP1mut) cells can be utilized as useful models not only to study the effects of iron on cell growth and the cell cycle but also to explore the overall physiology of the IRE-IRP system and the mechanism for ferritin mRNA translation. Along these lines, and in light of the data from the IRP1 (32) and IRP2 (22) knockout mice, it would also be of interest to examine whether the expression of IRP2 constitutive mutants elicits responses similar to the ones described here.
Acknowledgments
We thank Xinbin Chen (University of Alabama—Birmingham, Birmingham) for providing us with the tTA-H1299 and tTA-MCF7 cells, Anne-Claude Gingras and Nahum Sonenberg for the eIF-4E and 4E-BP1 antibodies, Franca Sicilia for technical assistance with FACS, and Joan Buss, Prem Ponka, and Antonis Koromilas for critical readings of the manuscript.
This work was supported by a grant from the Canadian Institutes of Health Research (CIHR). J.W. is a recipient of a fellowship from CIHR; K.P. is a scholar of CIHR and a researcher of the Canada Foundation for Innovation.
REFERENCES
- 1.Brown, V., P. Jin, S. Ceman, J. C. Darnell, W. T. O'Donnell, S. A. Tenenbaum, X. Jin, Y. Feng, K. D. Wilkinson, J. D. Keene, R. B. Darnell, and S. T. Warren. 2001. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107:477-487. [DOI] [PubMed] [Google Scholar]
- 2.Cairo, G., and A. Pietrangelo. 2000. Iron regulatory proteins in pathobiology. Biochem. J. 352:241-250. [PMC free article] [PubMed] [Google Scholar]
- 3.Cairo, G., L. Tacchini, and A. Pietrangelo. 1998. Lack of coordinate control of ferritin and transferrin receptor expression during rat liver regeneration. Hepatology 28:173-178. [DOI] [PubMed] [Google Scholar]
- 4.Caltagirone, A., G. Weiss, and K. Pantopoulos. 2001. Modulation of cellular iron metabolism by hydrogen peroxide. Effects of H2O2 on the expression and function of iron-responsive element-containing mRNAs in B6 fibroblasts. J. Biol. Chem. 276:19738-19745. [DOI] [PubMed] [Google Scholar]
- 5.Casey, J. L., B. Di Jeso, K. Rao, R. D. Klausner, and J. B. Harford. 1988. Two genetic loci participate in the regulation by iron of the gene for the human transferrin receptor. Proc. Natl. Acad. Sci. USA 85:1787-1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ceman, S., V. Brown, and S. T. Warren. 1999. Isolation of an FMRP-associated messenger ribonucleoprotein particle and identification of nucleolin and the fragile X-related proteins as components of the complex. Mol. Cell. Biol. 19:7925-7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, X., L. J. Ko, L. Jayaraman, and C. Prives. 1996. p53 levels, functional domains, and DNA damage determine the extent of the apoptotic response of tumor cells. Genes Dev. 10:2438-2451. [DOI] [PubMed] [Google Scholar]
- 8.Dandekar, T., R. Stripecke, N. K. Gray, B. Goossen, A. Constable, H. E. Johansson, and M. W. Hentze. 1991. Identification of a novel iron-responsive element in murine and human erythroid δ-aminolevulinic acid synthase mRNA. EMBO J. 10:1903-1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeRusso, P. A., C. C. Philpott, K. Iwai, H. S. Mostowski, R. D. Klausner, and T. A. Rouault. 1995. Expression of a constitutive mutant of iron regulatory protein 1 abolishes iron homeostasis in mammalian cells. J. Biol. Chem. 270:15451-15454. [DOI] [PubMed] [Google Scholar]
- 10.Eisenstein, R. S. 2000. Iron regulatory proteins and the molecular control of mammalian iron metabolism. Annu. Rev. Nutr. 20:627-662. [DOI] [PubMed] [Google Scholar]
- 11.Ferreira, C., D. Bucchini, M. E. Martin, S. Levi, P. Arosio, B. Grandchamp, and C. Beaumont. 2000. Early embryonic lethality of H ferritin gene deletion in mice. J. Biol. Chem. 275:3021-3024. [DOI] [PubMed] [Google Scholar]
- 12.Galy, B., A. Maret, A. C. Prats, and H. Prats. 1999. Cell transformation results in the loss of the density-dependent translational regulation of the expression of fibroblast growth factor 2 isoforms. Cancer Res. 59:165-171. [PubMed] [Google Scholar]
- 13.Gehring, N., M. W. Hentze, and K. Pantopoulos. 1999. Menadione-induced oxidative stress leads to inactivation of both RNA-binding and aconitase activities of iron regulatory protein-1. J. Biol. Chem. 274:6219-6225. [DOI] [PubMed] [Google Scholar]
- 14.Gingras, A. C., B. Raught, and N. Sonenberg. 2001. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 15:807-826. [DOI] [PubMed] [Google Scholar]
- 15.Gingras, A. C., Y. Svitkin, G. J. Belsham, A. Pause, and N. Sonenberg. 1996. Activation of the translational suppressor 4E-BP1 following infection with encephalomyocarditis virus and poliovirus. Proc. Natl. Acad. Sci. USA 93:5578-5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gossen, M., and H. Bujard. 1992. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA 89:5547-5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gray, N. K., S. Quick, B. Goossen, A. Constable, H. Hirling, L. C. Kühn, and M. W. Hentze. 1993. Recombinant iron regulatory factor functions as an iron-responsive element-binding protein, a translational repressor and an aconitase. A functional assay for translational repression and direct demonstration of the iron switch. Eur. J. Biochem. 218:657-667. [DOI] [PubMed] [Google Scholar]
- 18.Green, S., I. Issemann, and E. Sheer. 1988. A versatile in vivo and in vitro eukaryotic expression vector for protein engineering. Nucleic Acids Res. 16:369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hansen, S. H., K. Sandvig, and B. van Deurs. 1992. Internalization efficiency of the transferrin receptor. Exp. Cell Res. 199:19-28. [DOI] [PubMed] [Google Scholar]
- 20.Hirling, H., B. R. Henderson, and L. C. Kühn. 1994. Mutational analysis of the [4Fe-4S]-cluster converting iron regulatory factor from its RNA-binding form to cytoplasmic aconitase. EMBO J. 13:453-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 22.LaVaute, T., S. Smith, S. Cooperman, K. Iwai, W. Land, E. Meyron-Holtz, S. K. Drake, G. Miller, M. Abu-Asab, M. Tsokos, R. Switzer III, A. Grinberg, P. Love, N. Tresser, and T. A. Rouault. 2001. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat. Genet. 27:209-214. [DOI] [PubMed] [Google Scholar]
- 23.Melefors, Ö., B. Goossen, H. E. Johansson, R. Stripecke, N. K. Gray, and M. W. Hentze. 1993. Translational control of 5-aminolevulinate synthase mRNA by iron-responsive elements in erythroid cells. J. Biol. Chem. 268:5974-5978. [PubMed] [Google Scholar]
- 24.Muckenthaler, M., N. K. Gray, and M. W. Hentze. 1998. IRP-1 binding to ferritin mRNA prevents the recruitment of the small ribosomal subunit by the cap-binding complex eIF4F. Mol. Cell 2:383-388. [DOI] [PubMed] [Google Scholar]
- 25.Owen, D., and L. C. Kühn. 1987. Noncoding 3′ sequences of the transferrin receptor gene are required for mRNA regulation by iron. EMBO J. 6:1287-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pantopoulos, K., N. Gray, and M. W. Hentze. 1995. Differential regulation of two related RNA-binding proteins, iron regulatory protein (IRP) and IRPB. RNA 1:155-163. [PMC free article] [PubMed] [Google Scholar]
- 27.Pantopoulos, K., and M. W. Hentze. 1995. Nitric oxide signaling to iron-regulatory protein (IRP): direct control of ferritin mRNA translation and transferrin receptor mRNA stability in transfected fibroblasts. Proc. Natl. Acad. Sci. USA 92:1267-1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pantopoulos, K., and M. W. Hentze. 2000. Nitric oxide, oxygen radicals and iron metabolism, p. 293-313. In L. Ignarro (ed.), Nitric oxide. Academic Press, San Diego, Calif.
- 29.Paraskeva, E., N. K. Gray, B. Schläger, K. Wehr, and M. W. Hentze. 1999. Ribosomal pausing and scanning arrest as mechanisms of translational regulation from cap-distal iron-responsive elements. Mol. Cell. Biol. 19:807-816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Philpott, C. C., D. Haile, T. A. Rouault, and R. D. Klausner. 1993. Modification of a free Fe-S cluster cysteine residue in the active iron-responsive element-binding protein prevents RNA binding. J. Biol. Chem. 268:17655-17658. [PubMed] [Google Scholar]
- 31.Philpott, C. C., R. D. Klausner, and T. A. Rouault. 1994. The bifunctional iron-responsive element binding protein/cytosolic aconitase: the role of active-site residues in ligand binding and regulation. Proc. Natl. Acad. Sci. USA 91:7321-7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rouault, T. A. 2000. The role of iron regulatory proteins in mammalian iron homeostasis, p. 133-144. In D. G. Badman, R. J. Bergeron, and G. M. Brittenham (ed.), Iron chelators: new development strategies. The Saratoga Group, Ponte Vedra Beach, Fla.
- 33.Tang, C. K., J. Chin, J. B. Harford, R. D. Klausner, and T. A. Rouault. 1992. Iron regulates the activity of the iron-responsive element binding protein without changing its rate of synthesis or degradation. J. Biol. Chem. 267:24466-24470. [PubMed] [Google Scholar]
- 34.Theil, E. C., and R. S. Eisenstein. 2000. Combinatorial mRNA regulation: iron regulatory proteins and iso-iron responsive elements (iso-IREs). J. Biol. Chem. 275:40659-40662. [DOI] [PubMed] [Google Scholar]
- 35.Tsuji, Y., H. Ayaki, S. P. Whitman, C. S. Morrow, S. V. Torti, and F. M. Torti. 2000. Coordinate transcriptional and translational regulation of ferritin in response to oxidative stress. Mol. Cell. Biol. 20:5818-5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu, J., and X. Chen. 2000. MCG10, a novel p53 target gene that encodes a KH domain RNA-binding protein, is capable of inducing apoptosis and cell cycle arrest in G2-M. Mol. Cell. Biol. 20:5602-5618. [DOI] [PMC free article] [PubMed] [Google Scholar]