

Abstract
DNMT3L is a regulator of imprint establishment of normally methylated maternal genomic sequences. DNMT3L shows high similarity to the de novo DNA methyltransferases, DNMT3A and DNMT3B, however, the amino acid residues needed for DNA cytosine methyltransferase activity have been lost from the DNMT3L protein sequence. Apart from methyltransferase activity, Dnmt3a and Dnmt3b serve as transcriptional repressors associating with histone deacetylase (HDAC) activity. Here we show that DNMT3L can also repress transcription by binding directly to HDAC1 protein. We have identified the PHD-like zinc finger of the ATRX domain as a main repression motif of DNMT3L, through which DNMT3L recruits the HDAC activity needed for transcriptional silencing. Furthermore, we show that DNMT3L protein contains an active nuclear localisation signal at amino acids 156–159. These results describe DNMT3L as a co-repressor protein and suggest that a transcriptionally repressed chromatin organisation through HDAC activity is needed for establishment of genomic imprints.
INTRODUCTION
In genomic imprinting one of the two alleles of an autosomal gene is silenced epigenetically depending on their parental origin. Evidently, imprinting is controlled by several consequent and inter-dependent cis-acting mechanisms that regulate the chromatin structure (1). The critical role in transmitting the imprinting signals is the chromatin methylation pattern, which in mammals is established predominantly at cytosine nucleotides within CpG islands (2). Aberrant changes in DNA methylation profiles are common features of cancer and cause developmental abnormalities (3–6). Examples are imprinting disorders like Prader-Willi and Angelman syndromes, which result from altered methylation on human chromosome 15q11–q13 (7,8).
The molecular basis of methylation-mediated gene regulation is related to changes in chromatin folding. The dynamic changes of chromatin are regulated by histone N-terminal modifications determining the transcriptionally active or repressed chromatin state (reviewed in 9). Direct connection between methylation of DNA and transcriptional repression is mediated via binding of methylated cytosine-binding protein MeCP2 to methyl-CpG dinucleotides and formation of a complex with histone deacetylase (HDAC) (10,11). In this way, the methylation pattern represents epigenetic marking leading to transcriptional silencing through chromatin remodelling.
We have recently isolated a novel DNA-cytosine 5-methyltransferase-like gene (DNMT3L) on chromosome 21q22.3 (12). The DNMT3L protein shows high similarity to human and mouse DNA-cytosine 5-methyltransferase 3 family (DNMT3) members (13,14), proteins involved in de novo methylation of CpG dinucleotide sequences in genome. In addition to DNMT3L, four DNMTs have been characterised altogether, of which three, DNMT1 (15), DNMT3A and DNMT3B (3), have been demonstrated to have cytosine methyltransferase activity. Like all DNMT3 family members, DNMT3L shares similarity with ATRX protein (16,17) in its cysteine-rich zinc finger motif region. Dnmt3a and Dnm3b are essential for de novo methylation and for normal mouse development (3). The two proteins exhibit non-overlapping function in development and produce a methylation pattern that is not random and rather reflects specific DNA targets (3,18). Binding of DNMT proteins to DNA, however, has little dependence on sequence context and, therefore, de novo methylation has been proposed to be under the control of regulatory factors that interact with methyltransferases and mediate the DNA binding specificity (19). As an indication of this, Dnmt3a has been demonstrated to associate, like other co-repressors, with HDAC1 and with transcription factor RP58 (20). These interacting partners identify Dnmt3a as a co-repressor protein carrying deacetylase activity which is targeted to regulatory foci via DNA-binding transcription factors.
Recently, Bourc’his et al. (21) generated a targeted disruption of the Dnmt3l gene in mouse in which male testes had severe hypogonadism and a Sertoli cell only phenotype with azoospermia. The heterozygous progeny of the Dnmt3l-deficient homozygous females failed to develop past 9.5 days due to embryonic defects resulting in biallelic expression of genes that are normally expressed only from the allele of paternal origin. The authors concluded that Dnmt3l is required specifically for the establishment of maternal genomic imprints and it is essential for the de novo methylation of single copy DNA sequences. As the Dnmt3l protein sequence lacks the catalytic motifs needed for methyltransferase activity, it is likely to function as a regulator of methylation at imprinted loci rather than a DNA cytosine methyltransferase.
The mechanism by which Dnmt3l is involved in establishing genomic imprints in the germline has remained unknown. Here we show that DNMT3L can mediate transcriptional repression and that the repression is mediated through interaction with HDAC protein and its activity. These results indicate that the mechanisms to establish genomic imprints may employ histone deacetylation-mediated repression processes and suggest that other DNA-binding regulatory factors are needed to mediate specific de novo methylation of single copy DNA sequences.
MATERIALS AND METHODS
Plasmids
To create the different DNMT3L constructs used in the CAT assay DNMT3L fragments DNMT3L 1–84 (nucleotides 482–736), DNMT3L 45–84 (nucleotides 617–736) and DNMT3L 88–153 (nucleotides 746–943) were amplified by PCR and cloned into the pM vector (Clontech) EcoRI and SalI sites downstream of the Gal4 DNA-binding domain (Gal4DB). pM-DNMT3L 1–195 (nucleotides 482–1069), pM-DNMT3L (nucleotides 482–1651) and pM-DNMT3L 164–387 (nucleotides 974–1651) were constructed by digesting the pcDNMT3L 1–195, pcDNMT3L and pGEX-DNMT3L vectors, respectively, with EcoRI and HindIII restriction enzymes and subcloning the fragments into pM vector. DNMT3L 88–387 (nucleotides 746–1651) was cloned into pM vector by PCR using primers with EcoRI and HindIII sites. To create plasmids pcDNMT3L 1–195 and pcDNMT3L, which encode N-terminal and full-length myc epitope-tagged DNMT3L, nucleotides 482–1069 (amino acids 1–195) and 482–1651 (amino acids 1–387) were amplified by PCR and cloned between the pcDNA3.1 vector (Invitrogen) EcoRI and HindIII sites. pING14AHDAC1 and GST–HDAC1 were generous gifts from Drs G. Akusjärvi and T. Punga (University of Uppsala). pcHDAC1 with an anti-Flag tag was kindly provided by Dr T. Kouzarides (University of Cambridge). Different GST–DNMT3L fragments (GST–DNMT3L 1–84, GST–DNMT3L 45–84, GST–DNMT3L 88–153, GST–DNMT3L 1–195, GST– DNMT3L 88–387 and GST–DNMT3L) used for GST pull-down assays were digested out from corresponding pM- or pcDNMT3L vectors and subcloned downstream of the glutathione S-transferase (GST) gene into the pGEX-1λT vector between EcoRI and XhoI or HindIII sites. GST–DNMT3L 164–387 was amplified by PCR and cloned into the pGEX-1λT vector between EcoRI and XhoI sites. The pGEX-1λT vector, which contains a modified multiple cloning site, was a gift from Dr K. Saksela (IMT, University of Tampere). To construct plasmids pGFP-NLS and pGFP-DNMT3L for immunofluorescence, DNMT3L nucleotides 906–1005 (amino acids 142–173) and 482–1651 (amino acids 1–387) were amplified by PCR and cloned downstream of green fluorescent protein between the pEGFP-C3 vector (Clontech) EcoRI and SalI sites. All PCR cloned constructs were verified by sequencing.
Cell cultures
Human rhabdomyosarcoma (RD) cells (American Type Culture Collection) and Cos-7 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% foetal calf serum and antibiotics. THP-1 cells were grown in RPMI 1640 supplemented with 10% foetal calf serum, 2 mM l-glutamine and antibiotics. All media and supplements were obtained from Bio Whittaker Europe.
CAT assay
Aliquots of 3 × 105 RD cells were transiently transfected with 500 ng pM, pM-DNMT3L 1–195, pM-DNMT3L 88–387, pM-DNMT3L 164–387, pM-DNMT3L 1–84, pM-DNMT3L 45–84, pM-DNMT3L 88–153 or full-length pM-DNMT3L 1–387 together with 1 µg Gal4TKCAT reporter (kindly provided by Dr Y. Shi, Harvard Medical School) using Lipofectamine (Life Technologies) according to the manufacturer’s instructions. Forty-six hours after transfection cells were washed twice with ice-cold phosphate-buffered saline (PBS) and lysed with the CAT ELISA kit (Roche) lysis buffer for 20 min at 4°C. The CAT activity assay was performed using a CAT ELISA kit (Roche) according to the manufacturer’s protocol. For the dose-dependence study 1 µg Gal4TKCAT reporter gene was co-transfected with 200 ng or 1 µg pM-DNMT3L 1–195 as mentioned above. Trichostatin A (TSA) treatment was performed with RD cells co-transfected with 1 µg Gal4TKCAT reporter and 500 ng pM-DNMT3L 1–195. At 22 h after transfection 100 nM TSA (Sigma) was added for 24 h. All CAT assay transfections were performed as duplicates and normalised against total protein. CAT activity of the pM-vector with a Gal4DB without a fusion partner was set to 100%.
HDAC1 GST pull-down assays
In vitro translation of pcDNMT3L 1–195 and pING14AHDAC was performed using the TNT Coupled Reticulocyte Lysate System (Promega) according to the manufacturer’s instructions. GST fusion proteins were purified as described by Frangioni and Neel (22) from 200 ml of 1 mM IPTG-induced Escherichia coli XL-1 Blue culture. Twenty microlitres of purified GST fusion proteins (total volume of purified GST protein/bead slurry was 300 µl) and 2.5 µl in vitro translation products (50 µl) were used for in vitro binding assay that was performed as described elsewhere (23). Bound proteins were separated by SDS–PAGE and subjected to autoradiography. For GST pull-down from RD cell lysate 24 µg Flag epitope-tagged pcHDAC1 was transiently transfected into 2 × 106 RD cells using ExGen500 in vitro transfection reagent (MBI Fermentas) according to the manufacturer’s protocol. After 48 h cells were lysed by freezing and thawing in 1 ml of NET0.2 lysis buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 5 mM EDTA, 2 µg/ml aprotinin, 500 ng/ml leupeptin). Lysates were passed five times through a 19G needle and centrifuged at 14 000 g at 4°C for 20 min. Equivalent amounts of GST, GST–DNMT3L 1–195 and GST–DNMT3L proteins were incubated with pcHDAC1 lysate in blocking buffer (5% milk, 1% BSA in PBS) at 4°C for 1 h. Pull-down products were washed once with PBS, 0.5% NP-40 and twice with PBS. Bound proteins were separated by SDS–PAGE and HDAC1/DNMT3L interaction was detected by western blotting with anti-Flag antibody (M2 clone; Sigma).
Histone deacetylation activity assay
THP-1 monocytes were lysed in NET0.2 buffer for 20 min on ice and the protein concentration of total protein lysate was measured using a Bio-Rad DC Protein Assay Kit. HDAC1 immunoprecipitations were done by incubating 1 µg anti-HDAC1 and anti-GAL4 antibodies (H-51 and RK5C1; Santa Cruz) with 1 mg total THP-1 extract in blocking solution (5% milk, 1% BSA in PBS) at 4°C. After 1–2 h protein A–Sepharose beads (Amersham Pharmacia Biotech) were added to immunoprecipitation samples and incubation was continued for 1–2 h at 4°C. HDAC1 GST pull-downs were performed by incubating different GST–DNMT3L constructs attached to glutathione–Sepharose with 1 mg total THP-1 extract in blocking solution (5% milk, 1% BSA in PBS) for 2–4 h at 4°C. Protein A–Sepharose and GST–Sepharose beads were washed three times with NET0.2 buffer and once with HDAC10 buffer (10 mM Tris–HCl pH 8.0, 10 mM NaCl, 10% glycerol). Finally, Sepharose beads and the attached proteins were resuspended in 40 µl of HDAC10 buffer and used for the histone deacetylation assay, which was performed using a Histone Deacetylation Assay Kit (histone H4 peptide substrate; Upstate Biotechnology). The assay was carried out according to the manufacturer’s protocol with the following exceptions: 50 000 c.p.m. of labelled peptide was used instead of 20 000 c.p.m. and HDAC10 buffer was used instead of the HDAC buffer supplied with the kit.
Immunofluorescence studies
Aliquots of 1 × 105 Cos-7 cells were grown on coverslips in 6-well plates and transfected with 1 µg plasmid pEGFP-C3, pGFP-NLS or pGFP-DNMT3L using ExGen500 transfection reagent (MBI, Fermentas) according to the instructions provided by the manufacturer. After 48 h cells were fixed with 4% paraformaldehyde and immunofluorescence data were acquired using an Olympus IX70 microscope. Images were captured with a digital CCD camera (Wallac) and UltraVIEW v.4.0.15 software for the PC.
RESULTS
DNMT3L can repress transcription through the PHD-like zinc finger of the ATRX domain
Dnmt3a and Dnmt3b have been shown to have the ability to repress transcription (20,24), so we wanted to test whether DNMT3L could act as a transcriptional co-repressor as well. In our system a DNMT3L construct was fused to the Gal4DB of the pM vector (pM-DNMT3L) and co-transfected into RD cells together with a CAT reporter gene that contained Gal4-binding sites upstream of a strong thymidine kinase promoter. Co-transfection repressed the reporter activity by ∼30% when compared with empty pM vector, indicating that DNMT3L indeed has co-repressor activity. We further constructed several DNMT3L truncation constructs in order to identify the protein domain responsible for the repressional effect (Fig. 1A). Transfection of the reporter constructs pM-DNMT3L 1–195, pM-DNMT3L 88–387, pM-DNMT3L 1–84 and pM-DNMT3L 164–387 showed that the repressional activity was largely mediated through the PHD-like zinc finger of the ATRX domain, as pM-DNMT3L 1–195 inhibited transcriptional activity by up to 80% and the strongest repression (90%) was observed with the fragment pM-DNMT3L 88–387, which contains the C-terminal region of the protein with the PHD-like zinc finger but not the C2C2 part of the ATRX domain. Less transcriptional repression (60%) than with PHD-containing fragments was also found with the pM-DNMT3L 164–387 fragment, a C-terminal region without the PHD finger, suggesting that the C-terminal part may contain another repressional region (Fig. 1B).
Figure 1.

DNMT3L represses transcription in a TSA-sensitive manner. (A) Schematic representation of the DNMT3L fragments used for transfection experiments. Numbers indicate DNMT3L amino acids. (B) DNMT3L represses transcription when fused to the Gal4DB. RD cells were co-transfected with five different pMDNMT3L constructs fused to Gal4DB and a CAT repoter gene that contained five Gal4-binding sites upstream from a thymidine kinase promoter. CAT activity from cells transfected only with Gal4DB (pM), without a fusion partner, was set at 100%. (C) DNMT3L repression is dose-dependent. RD cells were transfected with the CAT reporter together with increasing amounts (200 ng or 1 µg) of the DNMT3L 1–195 construct. (D) DNMT3L repression is inhibited by TSA, a specific inhibitor of HDAC activity. RD cells were co-transfected with a CAT reporter gene and the N-terminal pM-DNMT3L 1–195 construct. After 22 h, cells were treated or left untreated with 100 nM TSA for 24 h.
To further dissect the role of the ATRX domain in repression we used a construct with the N-terminal region of DNMT3L with the C2C2 domain but lacking the PHD-like finger (pM-DNMT3L 1–84). Interestingly enough, this fragment did not have repressional activity and was even able to slightly activate the Gal4 system (Fig. 1B). Experiments with only the C2C2 (pM-DNMT3L 45–84) and PHD (pM-DNMT3L 88–153) fragments, however, gave similar results to empty pM vector, which might be due to the fact that these shorter fragments covered only a minimal domain and did not fold correctly.
Taken together these results demonstrate that DNMT3L has a repressional activity, which is predominantly mediated by the PHD-like zinc finger of ATRX as a strong repressional domain.
DNMT3L repression is dose-dependent and inhibited by TSA
After finding that the PHD-like zinc finger of the ATRX domain was responsible for transcriptional repression by DNMT3L, we next wanted to determine whether the repression was dependent on dosage of the DNMT3L protein fragment containing the repression domain. Increasing amounts of pM-DNMT3L 1–195 repressed the CAT activity in a dose-dependent manner, as increasing the DNA construct concentration from 200 ng to 1 µg decreased the activity 4-fold (Fig. 1C). Since HDAC activity is related to transcriptional repression and as the repression of many proteins has been shown to be mediated by HDAC activity, we further tested whether the repression activity in our system was dependent on a HDAC-specific inhibitor, TSA. Addition of 100 nM TSA to pM-DNMT3L 1–195-transfected cells increased the CAT activity to a comparable level with empty pM vector, suggesting that the DNMT3L repression is HDAC-dependent (Fig. 1D).
DNMT3L interacts with HDAC1
Previously, it has been shown that DNMT1 can repress transcription through directly interacting with HDAC1 (23). More recently, it was shown that by using its zinc finger domain for association with HDAC1, Dnmt3a can also act as a transcriptional repressor (20). With this data, we asked whether DNMT3L could use its ATRX region for physical interaction with HDAC1. We performed in vitro GST pull-down assays using GST–DNMT3L 1–195 and in vitro translated full-length HDAC1. Figure 2A shows that a GST–ATRX domain fusion interacted with HDAC1 whereas GST alone did not. To test the same interaction in the opposite way, we used a GST–HDAC1 protein in pull-down assays with in vitro translated ATRX domain. As again seen in Figure 2B, interaction of the ATRX domain was present with HDAC1 but not with GST protein alone. To prove even further that DNMT3L associates with HDAC1, we performed GST pull-down assays with GST–DNMT3L 1–195 and GST–DNMT3L 1–387 using a lysate of RD cells transfected with pcHDAC1. Figure 2C shows that both full-length GST–DNMT3L and GST–DNMT3L 1–195 interact with HDAC1 as detected by western blotting against HDAC1 protein. These results indicate that DNMT3L, similarly to Dnmt3a, can physically interact with HDAC1 protein using its zinc finger ATRX region for interaction.
Figure 2.
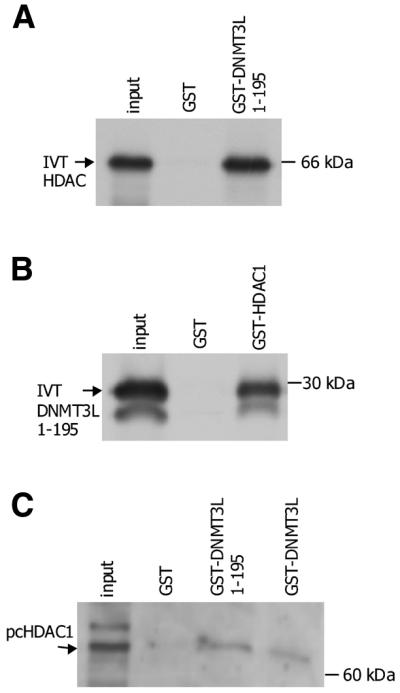
DNMT3L interacts with the histone deacetylase HDAC1. (A) GST pull-down assay using in vitro translated (IVT) full-length HDAC1 and GST-DNMT3L 1–195. (B) GST pull-down assay using IVT pcDNMT3L 1–195 and full-length GST–HDAC1. (C) GST pull-down from RD cell lysate. Total lysate of RD cells transfected with pcHDAC1 was used for a GST pull-down assay performed with GST–DNMT3L 1–195, full-length GST–DNMT3L and GST alone. Input proteins are indicated by arrows on the left and molecular weights are indicated on the right.
DNMT3L associates with HDAC activity
After showing an interaction between DNMT3L and HDAC1, we next studied whether DNMT3L binding to HDAC1 is associated with the enzymatically active deacetylase. To test this, we used THP-1 total protein lysate as a HDAC1 source and performed GST pull-down assays with seven different DNMT3L fragments (Fig. 1A) fused to GST protein. As positive and negative controls, anti-HDAC1 and anti-GAL4 antibodies, respectively, were used in immunoprecipitations. The pull-down products and precipitates were incubated with 3H-acetylated peptide substrate and deacetylation was measured as released [3H]acetate using a scintillation counter. Positive control immunoprecipitations with anti-HDAC1 antibody efficiently precipitated HDAC activity and negative control antibody to GAL4 gave background activity (Fig. 3). Of the seven different GST–DNMT3L fusion proteins used in the assay, all those fragments that contained the PHD-like zinc finger of the ATRX domain were able to pull down the HDAC activity from THP-1 lysate. GST–DNMT3L 1–195, 1–387, 88–153 and 88–387 fusion proteins consistently associated with HDAC activity in three separate experiments, whereas GST–DNMT3L 164–387, 1–64, and 45–64 (without the PHD finger) and GST alone did not show deacetylase activity in the assay (Fig. 3). The highest HDAC activity was repeatedly seen with the DNMT3L 88–387 fusion protein containing the C-terminal part of the protein in addition to the PHD-like zinc finger domain of the ATRX region. To further verify the enzymatic reaction of HDAC and to show that a deacetylase inhibitor could relieve HDAC activity, we used 100 mM sodium butyrate, similar to TSA, which was added to inhibit the deacetylase reaction. As seen in Figure 3, deacetylase activity associated with all four DNMT3L fragments containing the PHD domain was inhibited by sodium butyrate treatment. These results demonstrate that DNMT3L associated with HDAC1 has functional deacetylase activity and that the PHD-like zinc finger of the ATRX domain is needed for enzymatic reactivity.
Figure 3.

DNMT3L associates with HDAC activity. Seven different GST–DNMT3L fusion proteins were used to pull-down the HDAC activity from THP-1 monocyte total protein lysates. α-HDAC1 and α-GAL4 antibodies were used as positive and negative controls, respectively. Deacetylase activity was detected as radioactivity released from 3H-labelled acetylated histone peptide substrate. To show that the repressional effect was mediated through HDAC1, 100 mM sodium butyrate (NaBu) was added to the reactions with PHD finger-containing constructs to inhibit the deacetylation reaction.
DNMT3L contains an active nuclear localisation signal (NLS)
We have previously observed that in transiently transfected Cos-1 and RD cells both human and mouse DNMT3L proteins subcellularly localise in the cytoplasm and nucleus. Thus, examination of the DNMT3L sequence revealed a possible NLS, RRRK, at amino acids 156–159. We cloned DNMT3L nucleotides 906–1005 (amino acids 142–173) downstream of green fluorescent protein into pEGFP-C3 vector and transiently transfected the construct into Cos-7 cells. The GFP–NLS 142–173 protein fragment localised to the nucleus (Fig. 4A), demonstrating that DNMT3L has a functional NLS, while GFP–DNMT3L and GFP alone were distributed in the cytoplasm and nucleus (Fig. 4B and C).
Figure 4.

DNMT3L has a functional NLS. (A) DNMT3L has a functional NLS, RRRK, at amino acids 156–159. DNMT3L amino acids 142–173 were cloned into GFP vector pEGFP-C3 and transiently transfected into Cos-7 cells. (Upper) GFP fluorescence; (lower) DAPI staining of the same cell. (B) Full-length GFP–DNMT3L and (C) GFP vector alone are distributed in both the cytoplasm and nucleus.
DISCUSSION
In this study we report experiments identifying DNMT3L protein as a transcriptional co-repressor. Previously it has been shown that the function of methyltransferase proteins includes actions as repressors. The major enzyme responsible for maintenance of DNA methylation, DNMT1, establishes a repressive transcription complex by interacting with HDAC1 (23) and HDAC2 (25). A further connection between methyltransferases and gene silencing comes from experiments showing that the de novo methyltransferases Dnmt3a and Dnmt3b repress transcription in association with HDAC activity (20,24). Interestingly, in this context, Dnmt3a and Dnmt3b have been shown to repress in a manner that does not require its de novo methyltransferase activity. Previously we isolated the human and mouse DNMT3L genes (12,26) and sequence analysis of the two homologous proteins pointed to a lack of amino acid residues needed for methyltransferase activity. Although sharing significant similarity with the conserved motifs I, IV and VI found in the catabolic domain of other DNMT family proteins (27–29), the absence of the critical residues implied that DNMT3L/Dnmt3l protein has no direct methyltransferase activity and functions through other mechanisms. Our current results, showing a transcriptional repression effect of DNMT3L mediated through interaction with HDAC and its activity, support similar findings observed with Dnmt3a.
The majority of the repression effect can be attributed to the ATRX domain of DNMT3L protein. The ATRX domain connected with the N-terminal part of DNMT3L protein was able to more actively repress transcription than full-length DNMT3L protein, decreasing CAT activity to 20% of that in the presence of the pM vector, and in some experiments a reduction was observed to as low as 11%. These results are consistent with recent observations by Fuks et al. (20) and Bachman et al. (24) on other DNMT3 family members, which demonstrated that the repressional activity of Dnmt3a and Dnmt3b was mediated through their ATRX-like regions. To further scrutinise the repression activity of the ATRX-like domain we analysed the C2C2 region in the context of the N-terminal part and the PHD-like finger in the context of the C-terminal region of DNMT3L separately. Repression was still observed with the fragment with the PHD-like finger whereas the N-terminal part even exhibited a slight activation effect. This result may explain the discrepancy in repression activity found between the ATRX domain-containing fragment and full-length protein. In addition, we have demonstrated that the ATRX domain was able to directly interact with HDAC1 and, in concordance with this, associates with HDAC activity. Again, of seven tested constructs the four fragments with the PHD-like finger of the ATRX region gave consistent effects on deacetylase enzymatic activity, thus highlighting the role of this domain in functional repression. Moreover, in both repression and deacetylation assays the PHD-like zinc finger showed the most remarkable effect in the context of the C-terminal region of DNMT3L.
Our previous studies showed that DNMT3L subcellularly localises in the nucleus and cytoplasm. In this study we identified a functional NLS, RRRK, at DNMT3L amino acids 156–159. Bachmann et al. (24) showed that in transient transfections of NIH 3T3 cells, Dnmt3a is localised to discrete nuclear foci representing pericentric heterochromatin and that the ATRX-like domain of Dnmt3a was not needed for targeting of the protein. However, alignment of all three DNMT3 family members suggests that the same NLS is also present in DNMT3A and DNMT3B. Whether the NLS identified here in DNMT3L is also functional in DNMT3A and DNMT3B remains to be studied.
In conclusion, our major findings provide information that despite the lack of methyltransferase activity DNMT3L can act as a co-repressor by interacting with HDAC1 through its ATRX-like domain and associates with HDAC activity. These results also indirectly suggest that to regulate the establishment of genomic imprints in sperm and oocytes, DNMT3L may make use of nucleosome deacetylase-linked processes or recruit, through an HDAC complex, active de novo methyltransferases such as DNMT3A or DNMT3B. Lack of obvious DNA-binding domains in the DNMT3L sequence also points to the possibility that DNMT3L, like Dnmt3a, may interact with specific DNA-binding transcription factors.
Acknowledgments
ACKNOWLEDGEMENTS
We thank U. Kiiskinen for excellent technical assistance. This work was supported by the Academy of Finland, Sigrid Juselius Foundation and Medical Research Fund of Tampere University Hospital.
REFERENCES
- 1.Ferguson-Smith A.C. and Surani,M.A. (2001) Imprinting and the epigenetic asymmetry between parental genomes. Science, 293, 1086–1089. [DOI] [PubMed] [Google Scholar]
- 2.Paulsen M. and Ferguson-Smith,A.C. (2001) DNA methylation in genomic imprinting, development and disease. J. Pathol., 95, 97–110. [DOI] [PubMed] [Google Scholar]
- 3.Okano M., Bell,D.W., Haber,D.A. and Li,E. (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell, 99, 247–257. [DOI] [PubMed] [Google Scholar]
- 4.Xu G.L., Bestor,T.H., Bourc’his,D., Hsieh,C.L., Tommerup,N., Bugge,M., Hulten,M., Qu,X., Russo,J.J. and Viegas-Pequignot,E. (1999) Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature, 402, 187–191. [DOI] [PubMed] [Google Scholar]
- 5.Amir R.E., Van den Veyver,I.B., Wan,M., Tran,C.Q., Francke,U. and Zoghbi,H.Y. (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature Genet., 23, 185–188. [DOI] [PubMed] [Google Scholar]
- 6.Rountree M.R, Bachman,K.E, Herman,J.G. and Baylin,S.B. (2001) DNA methylation, chromatin inheritance and cancer. Oncogene, 20, 3156–3165. [DOI] [PubMed] [Google Scholar]
- 7.Knoll J.H., Nicholls,R.D., Magenis,R.E., Graham,J.M., Lalande,M. and Latt,S.A. (1989) Angelman and Prader-Willi syndromes share common chromosome 15 deletion but differ in parental origin of the deletion. Am. J. Med. Genet., 32, 285–290. [DOI] [PubMed] [Google Scholar]
- 8.Nicholls R.D. and Knepper,J.L. (2001) Genome organization, function and imprinting in Prader-Willi and Angelman syndromes. Annu. Rev. Genomics Hum. Genet., 2, 153–175. [DOI] [PubMed] [Google Scholar]
- 9.Jenuwein T. and Allis,D.C. (2001) Translating the histone code. Science, 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- 10.Nan X., Ng,H.H., Johnson,C.A., Laherty,C.D., Turner,B.M., Eisenman,R.N. and Bird,A. (1998) Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature, 393, 386–389. [DOI] [PubMed] [Google Scholar]
- 11.Ng H.H. and Bird,A. (1999) DNA methylation and chromatin modification. Curr. Opin. Genet. Dev., 9, 158–163. [DOI] [PubMed] [Google Scholar]
- 12.Aapola U., Shibuya,K., Scott,H.S., Ollila,J., Vihinen,M., Heino,M., Shintani,A., Kawasaki,K., Minoshima,S., Krohn,K., Antonarakis,S.E., Shimizu,N., Kudoh,J. and Peterson,P. (2000) Isolation and initial characterization of a novel zinc finger gene, DNMT3L, on 21q22.3, related to the cytosine-5-methyltransferase 3 gene family. Genomics, 65, 293–298. [DOI] [PubMed] [Google Scholar]
- 13.Okano M., Xie,S. and Li,E. (1998) Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nature Genet., 19, 219–220. [DOI] [PubMed] [Google Scholar]
- 14.Xie S., Wang,Z., Okano,M., Nogami,M., Li,Y., He,W.W., Okumura,K. and Li,E. (1999) Cloning, expression and chromosome locations of the human DNMT3 gene family. Gene, 236, 87–95. [DOI] [PubMed] [Google Scholar]
- 15.Bestor T., Laudano,A., Mattaliano,R. and Ingram,V. (1988) Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzyme is related to bacterial restriction methyltransferases. J. Mol. Biol., 203, 971–983. [DOI] [PubMed] [Google Scholar]
- 16.Gibbons R.J., Picketts,D.J., Villard,L. and Higgs,D.R. (1995) Mutations in a putative global transcriptional regulator cause X-linked mental retardation with alpha-thalassemia (ATR-X syndrome). Cell, 80, 837–845. [DOI] [PubMed] [Google Scholar]
- 17.Gibbons R.J., Bachoo,S., Picketts,D.J., Aftimos,S., Asenbauer,B., Bergoffen,J., Berry,S.A., Dahl,N., Fryer,A., Keppler,K., Kurosawa,K., Levin,M.L., Masuno,M., Neri,G., Peirpoint,M., Slaney,S.F. and Higgs,D.R. (1997) Mutations in transcriptional regulator ATRX establish the functional significance of a PHD-like domain. Nature Genet., 17, 146–148. [DOI] [PubMed] [Google Scholar]
- 18.Hsieh C.-L. (1999) In vivo activity of murine de novo methyltransferases Dnmt3a and Dnmt3b. Mol. Cell. Biol., 19, 8211–8218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoder J.A., Soman,N.S., Verdine,G.L. and Bestor,T. (1997) DNA (cytosine-5) methyltransferases in mouse cells and tissues. Studies with a mechanism-based probe. J. Mol. Biol., 270, 385–395. [DOI] [PubMed] [Google Scholar]
- 20.Fuks F., Burgers,W.A., Godin,N., Kasai,M. and Kouzarides,T. (2001) Dnmt3a binds deacetylases and is recruited by a sequence-specific repressor to silence transcription. EMBO J., 20, 2536–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bourc’his D., Xu,G.L., Lin,C.S., Bollman,B. and Bestor,T.H. (2001) Dnmt3l and the establishment of maternal genomic imprints. Science, 294, 2536–2539. [DOI] [PubMed] [Google Scholar]
- 22.Fragnioni J.V. and Neel,B.G. (1993) Solibilization and purification of enzymatically active glutathione S-transferase (pGEX) fusion proteins. Anal. Biochem., 210, 179–187. [DOI] [PubMed] [Google Scholar]
- 23.Fuks F., Burgers,W., Brehm,A., Hughes-Davies,L. and Kouzarides,T. (2000) DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nature Genet., 24, 88–91. [DOI] [PubMed] [Google Scholar]
- 24.Bachman K.E., Rountree,M.R. and Baylin,S.B. (2001) Dnmt3a and Dnmt3b are transcriptional repressors that exhibit unique localization properties to heterochromatin. J. Biol. Chem., 276, 32282–32287. [DOI] [PubMed] [Google Scholar]
- 25.Rountree M.R., Bachman,K.E. and Baylin,S.B. (2000) DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nature Genet., 25, 269–277. [DOI] [PubMed] [Google Scholar]
- 26.Aapola U., Lyle,R., Krohn,K., Antonarakis,S.E. and Peterson,P. (2001) Isolation and initial characterisation of the mouse Dnmt3l gene. Cytogenet. Cell Genet., 92, 122–126. [DOI] [PubMed] [Google Scholar]
- 27.Lauster R., Trautner,T.A. and Noyer-Weidner,M. (1989) Cytosine-specific type II DNA methyltransferases. A conserved enzyme core with variable target-recognizing domains. J. Mol. Biol., 206, 305–312. [DOI] [PubMed] [Google Scholar]
- 28.Kumar S., Cheng,X., Klimasauskas,S., Mi,S., Posfai,J., Roberts,R.J. and Wilson,G.G. (1994) The DNA (cytosine-5) methyltransferases. Nucleic Acids Res., 22, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klimasauskas S., Kumar,S., Roberts,R.J. and Cheng,X. (1994) HhaI methyltransferase flips its target base out of the DNA helix. Cell, 76, 357–369. [DOI] [PubMed] [Google Scholar]