


Abstract
Alternative splicing of α-tropomyosin (α-TM) involves mutually exclusive selection of exons 2 and 3. Selection of exon 2 in smooth muscle (SM) cells is due to inhibition of exon 3, which requires both binding sites for polypyrimidine tract-binding protein as well as UGC (or CUG) repeat elements on both sides of exon 3. Point mutations or substitutions of the UGC-containing upstream regulatory element (URE) with other UGC elements disrupted the α-TM splicing pattern in transfected cells. Multimerisation of the URE caused enhanced exon skipping in SM and various non-SM cells. In the presence of multiple UREs the degree of splicing regulation was decreased due to the high levels of exon skipping in non-SM cell lines. These results suggest that the URE is not an intrinsically SM- specific element, but that its functional strength is fine tuned to exploit differences in the activities of regulatory factors between SM and other cell types. Co-transfection of tropomyosin reporters with members of the CUG-binding protein family, which are candidate URE-binding proteins, indicated that these factors do not mediate repression of tropomyosin exon 3.
INTRODUCTION
Alternative pre-mRNA splicing is a key mechanism for regulation of gene expression, allowing generation of different mRNAs encoding distinct protein products from a single gene (1–3). At least a third and probably the majority of human genes are alternatively spliced (reviewed in 4,5) and in some spectacular examples alternative splicing from a single gene can generate hundreds or even thousands of protein isoforms (3,6). Expression of different protein isoforms is usually regulated in a cell type- or developmental stage-specific manner.
Regulation of alternative splicing is mediated by cis-acting pre-mRNA sequence elements and trans-acting regulatory proteins that recognise them. Cis-acting elements fall into two broad classes, enhancer or silencer elements, according to whether they act positively or negatively (2,7,8). Enhancers and silencers can also be subdivided according to their location, intronic or exonic. Binding of trans-acting factors to these elements either promotes or inhibits spliceosome assembly at adjacent splice sites. The trans-acting regulatory factors that interact with the regulatory sequences also fall into two broad classes: proteins that contain arginine/serine-rich (RS) domains (9–12) and those that do not. The latter group includes the hnRNP proteins (13–15). In a few characterised cases, splicing decisions are switched by the presence or absence of a single regulator. This is the case in the regulation of sex determination in Drosophila, which is controlled by the activity of the female cell-specific splicing regulators tra and sxl (7). Examples of this simple type of control have been more scarce among the mammalian model systems of alternative splicing. The more common scenario is of combinatorial regulation, with splicing decisions being determined by combinations of widespread factors (2,16,17).
We have used the mutually exclusive exons 2 and 3 of the rat α-tropomyosin (α-TM) gene as a mammalian model for regulated alternative splicing. Exon 3 is incorporated predominantly in most cells, except for smooth muscle (SM) tissue, where it is replaced by exon 2 (18; Fig. 1A). Exon 2 is selected in SM cells due to inhibition of exon 3 (19). Although exon 2 contains splicing enhancers (20), there is no indication that exon 2 selection is directly regulated. Exon 3 repression is mediated by three negatively acting cis-sequences (19,21,22). The polypyrimidine tract of exon 3 (P3), located ∼120–170 nt upstream of exon 3, contains three UCUU motifs, which are optimal binding sites for polypyrimidine tract-binding protein (PTB, also known as hnRNP-I) (22). The upstream regulatory element (URE) is a 15 nt sequence between P3 and exon 3, which contains three UGC motifs (19). The downstream regulatory element (DRE) lies ∼130–200 nt downstream of exon 3 and comprises a short sequence with four UGC motifs, designated DUGC, followed by a pyrimidine-rich tract referred to as DY (Fig. 1B). The core of the DY tract consists of two overlapping PTB-binding UCUU motifs. Mutation of the PTB-binding sites in P3 and DY impairs negative regulation of splicing in SM cell cultures and this correlates with the ability of PTB to bind to RNAs in vitro (21,22). Moreover, in vitro splicing of α-TM transcripts in HeLa nuclear extracts shows a residual low level of exon 3 skipping that is dependent upon PTB (21,23). PTB is a generally expressed protein and no increase in its level is observed in SM cells relative to non-SM cells (24). However, repression of α-TM exon 3 is largely restricted to SM cells, so additional factors must be involved in α-TM regulation. Obvious candidates would be proteins that bind to the UGC repeat elements, but to date no such factors have been identified.
Figure 1.

Mutually exclusive splicing of α-tropomyosin (α-TM) pre-mRNA. (A) The organisation of the 5′ end of the rat α-TM gene is shown schematically. Exons 1–4 are shown as boxes, introns as lines. The broken line reflects the fact that the intron between exons 3 and 4 is ∼12 kb. Splicing patterns are represented as diagonal dashed lines. In most cells the spliced product is 1–3–4, while in SM cells it is 1–2–4. (B) The pTWT construct contains α-TM exons 1, 3 and 4 surrounded by flanking regulatory sequences. Deletion of exon 2 does not interfere with the splicing regulation of this construct (19,21). Expression of pTWT in vivo is driven by the SV40 promoter and enhancer. pTWT contains two point mutations between exon 3 and the URE (C→U, U→G, original wild-type base indicated above sequence) that introduce convenient restriction sites for cloning. The branch point (BP3) and polypyrimidine tract (P3) of exon 3 are shown as a circle and rectangle, respectively. The upstream regulatory element of exon 3 is denoted by a diamond labelled URE. The downstream regulatory element (DRE) consists of polypyrimidine tract DY, denoted by a rectangle, and a UGC-rich region, DUGC, denoted by a diamond. Vertical lines indicate optimal PTB-binding sites (UCUU motifs). The sequence from BP3 to the 3′ end of the DRE is shown below the diagram. The branch point sequence of exon 3, UGC motifs of the DRE and URE and PTB-binding sites (UCUU motifs) are underlined. The sequences of exon 3, URE and DUGC are shown in bold (21). All sequence features (BP3, P3-1, P3-2, etc.) are indicated above the sequence.
In this study, we have carried out a detailed analysis of the URE. Consistent with its central role in control of α-TM splicing, many mutations within the URE reduced exon 3 skipping. Conversely, URE multimerisation caused enhanced exon skipping in SM and various non-SM cells. In the presence of multiple UREs the degree of splicing regulation was decreased due to the high levels of exon skipping in the non-SM cells. These results suggest that the URE is not an intrinsically SM-specific element, but that its functional strength is fine tuned to exploit differences in the activities of regulatory factors between SM and other cell types. Three widely expressed members of the CELF family of proteins (25), which are able to bind to UGC/CUG motifs, were tested for their effects upon TM splicing. None of the proteins promoted exon skipping and under some circumstances they antagonised regulation of splicing, suggesting that they are able to bind the cis-acting elements but that they do not act as repressors of TM exon 3.
MATERIALS AND METHODS
Constructs
The pTWT splicing reporter is a derivative of the construct DREΔ1 (21) with two point mutations upstream of exon 3 (Fig. 1B) which facilitate cloning of further mutants but which do not affect splicing regulation (C.Gooding and C.W.J.Smith, unpublished observations). It contains tropomyosin exons 1, 3 and 4 in the context of flanking regulatory intron sequences. Expression is driven by the SV40 early promoter and enhancer. ΔURE has an EcoRV restriction site introduced upstream of exon 3, which was created by a 15 nt URE deletion, coupled with a single C→T mutation (Fig. 1B). pTWT and all constructs containing mutations within the URE sequence were created by cloning the corresponding oligonucleotides into the EcoRV site of ΔURE. The 1URE/P3-2 and 6URE/P3-2 constructs contained mutation of the UCUU to a CCCC motif within the second PTB-binding site of the exon 3 polypyrimidine tract of the 1URE and 6URE reporters, respectively (22). The 6URE/ΔDRE construct was generated by deletion of the DRE sequence between the BstXI and EcoRI sites of the 6URE reporter. 6URE/ΔDUGC was generated by a 27 base deletion between the BstXI and FspI sites of 6URE. 6URE/ΔDY was created by deletion of the DY sequence between the FspI and EcoRI sites of 6URE. The DUGC/DUGC construct was generated by inserting the DUGC sequence (CTGGATGCCGCCTCTGCTGCTGC; see also Fig. 4A) into the EcoRV site of ΔURE. The URE/URE construct was created by replacing the DUGC sequence between the BstXI and FspI sites of the pTWT reporter by the wild-type URE sequence containing BstXI and FspI compatible ends (ctggatCACGCTGCCTGCTGCa). The DUGC/URE construct was created by replacing the DUGC sequence between the BstXI and FspI sites of the DUGC/DUGC reporter by the wild-type URE sequence containing BstXI and FspI overhangs (ctggatCACGCTGCCTGCTGCa). All the mutants were verified by DNA sequencing. The only construct that contained an unintended mutation was 4URE, which has a single C→T point mutation at the third C in the second repeat of the URE, which does not alter any UGC motifs.
Figure 4.

URE multimerisation increases exon 3 skipping. Constructs containing 0, 1, 2, 3, 4, 6 and 8 copies of the URE were transfected into PAC-1 SM cells, L cells, HeLa cells and C2C12 myoblasts (A–D) and analysed as in Figure 2. (E) Histogram representation of the data from (A) to (D). (F) Histogram representation of splicing regulation. The degree of regulation for each construct was calculated as the ratio of exon skipping in SM cells compared with the other non-muscle (L, HeLa and C2C12) cell types. URE multimerisation increases exon 3 skipping in all cells but decreases splicing regulation in SM/non-SM cells.
Cell culture and transfections
HeLa cells, PAC-1 rat pulmonary artery vascular smooth muscle cells (26), L mouse fibroblasts and C2C12 mouse myoblasts (27) were grown under standard conditions in DMEM (Gibco BRL) supplemented with 10% (v/v) foetal calf serum (Gibco BRL) and 1% (v/v) l-glutamine (Gibco BRL). Transient transfections were performed in 35 mm plates using the calcium phosphate method (19,21,28) or with LipofectAMINE™ reagent (Gibco BRL). Aliquots of 2–5 µl of LipofectAMINE™ reagent were preincubated with 1 µg DNA in the presence of 0.1 ml of Opti-MEM I Reduced Serum Medium (Gibco BRL) at room temperature for 30 min to form DNA–liposome complexes. Complexes were diluted by addition of 0.8 ml of Opti-MEM and applied to the cells. After incubation for 5–6 h at 37°C, the precipitate was removed and replaced by complete medium. Cells were usually harvested 48 h following the start of transfection.
Detection of expressed RNA
Transiently expressed cytoplasmic RNA was harvested using TRI reagent (Sigma). Total RNA was separated from chromosomal DNA and protein by addition of 0.2 ml of chloroform followed by isopropanol precipitation and a 75% ethanol wash as described (23). The resulting RNA was usually resuspended in 15–20 µl of H2O. For RT–PCR analysis, typically 2 µl of total RNA was added to 5 µl of 2× RT buffer (100 mM Tris pH 8.3, 80 mM KCl, 16 mM MgCl2, 4 mM DTT) and 10 pmol SV3′RT primer (5′-GCA AACTCAGCCACAGGT). Samples were incubated at 55°C for 15 min and then 1 mM dNTPs and 9 U AMV reverse transcriptase (Promega) were added and the reactions were incubated for a further 60 min at 42°C. An aliquot of 2 µl of the RT reaction was used as a template in 25 µl PCRs.
PCRs were carried out by a hot-start procedure in which the reaction was assembled wth 2 µl of the cDNA, 25 pmol SV5′2 primer (5′-GGAGGCCTAGGCTTTTGCAAAAAG), 2.5 µl 10× PCR buffer [0.5 M KCl, 0.1 M Tris pH 8.3, 10–25 mM MgCl2, 0.01% (w/v) gelatine], 0.2 mM dNTPs and H2O. The reactions were incubated at 92°C for 3 min and then cooled down to 80°C for the addition of 1.25 U Taq DNA polymerase (Amersham Pharmacia Biotech) and 2 pmol 32P-labelled SV3′1 primer (5′-ACTCACTGCGTTCCAGGCAATGCT). The PCR consisted of 30 cycles of 94°C for 30 s, 62°C for 30 s and 72°C for 1 min, followed by a final 2 min extension at 72°C. For quantitation of the splicing products, 1 µl of the reaction was diluted into 20 µl of formamide dyes and 10 µl of this mixture was loaded onto a 4–6% denaturing polyacrylamide gel. Using SV5′2 and SV3′1 primers for the PCR the sizes of the expected splicing products were 434 (1–3–4 spliced product) and 308 bp (1–4 spliced product). Radiolabelled splicing products were visualised on a Molecular Dynamics Storm 840 phosphorimager. The relative ratios of the splice products were determined using Molecular Dynamics ImageQuant software (v.1.11) applying volume integration for quantitation with histogram peak background correction. Similar results were obtained using line graph quantitation with manual checking of background corrections. The results were usually displayed graphically using Microsoft Excel (Office 98 edition) software. The percentage of exon 3 skipping was expressed as [1–4/(1–3–4 + 1–4)] × 100. All experiments were repeated at least three times independently to ascertain the reproducibility of results. In all figures except Figure 5, the per cent exon skipping is shown as the mean ± standard deviation of data from a single set of triplicate experiments conducted simultaneously. Figure 5 shows data from a single experiment; the behaviour of all constructs in this experiment has been reproduced in independent experiments.
Figure 5.
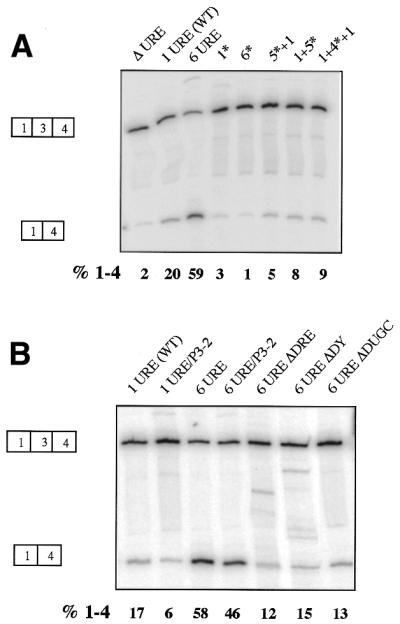
Enhanced exon skipping by URE multimers depends on the wild-type URE sequence and other regulatory elements. PAC-1 SM cells were transiently transfected with constructs containing (A) 0, 1 or 6 UREs and various mutants containing different combinations of the wild-type and mutant URE sequences or (B) constructs combining the 6URE mutation with deleterious mutations in the other regulatory elements (DRE and P3). Data were analysed as in Figure 2. The values for per cent exon 3 skipping (%1–4) correspond to this particular experiment. Enhanced exon skipping of URE multimers required multimers of the wild-type URE (A) and the other defined regulatory elements (B).
RESULTS
URE mutations disrupt α-TM splicing
Negative regulation of α-TM exon 3 occurs independently of exon 2 and the cis elements can therefore be analysed in constructs lacking exon 2 (19,21,29,30). URE mutations were introduced into the construct pTWT (Fig. 1B and C), which contains α-TM exons 1, 3 and 4 in the context of the flanking regulatory intron sequences and is driven by the SV40 early promoter and enhancer (19,21). pTWT contains two point mutations upstream of exon 3 that introduce convenient restriction sites for cloning (Fig. 1B), but which do not affect splicing regulation (C.Gooding and C.W.J.Smith, unpublished observations). Splicing of pTWT and derived constructs was analysed by RT–PCR after transient transfection into PAC-1 cells, a partially differentiated SM cell line (26), and HeLa cells. In PAC-1 SM cells exon 3 was skipped in ∼20% of pTWT transcripts (Fig. 2B, lane WT), compared with ∼2% in HeLa cells (Fig. 2C).
Figure 2.

URE mutations disrupt α-TM splicing. (A) URE sequences of mutant constructs. (B) PAC-1 SM cells and (C) HeLa cells were transiently transfected with wild-type reporter construct pTWT and various URE mutants. In lane MOCK no DNA was transfected into the cells. Harvested RNA was analysed by RT–PCR. Positions of the amplified bands corresponding to the 1–3–4 and 1–4 splicing patterns are indicated. The percentage of exon 3 skipping is shown immediately below each lane and represents the mean ± standard deviation of triplicate experiments.
A series of URE mutants was created containing various point mutations within the 15 nt URE core, which contains three UGC repeats (Fig. 1C). Complete deletion of the URE (ΔURE) led to a 3- to 5-fold decrease in exon skipping in both cell types (Fig. 2). Point mutations of the U, G or C residues of the URE caused reductions in exon skipping to variable extents, with one exception. Transition of the three U residues to C residues (3U→C) or transversion of the three C residues to G residues (3C→G) led to a modest reduction in exon skipping, while transversion of the four G residues to C residues (4G→C) had an effect equal to the complete URE deletion. In contrast, transversion of the three U residues to G residues (3U→G) led to a 2- to 4-fold increase in exon skipping in both cell types. These data allow a number of conclusions to be drawn. First, the point mutations that impair exon skipping confirm the importance of the URE as a negative regulatory element. Second, none of the mutations has a cell-specific effect. Mutations that decreased exon skipping in SM cells also caused a decrease in HeLa and L cells (data not shown). This suggests that the URE is not an intrinsically SM-specific element and that it interacts with factors that are not restricted to SM cells. Furthermore, the behaviour of the 3U→G mutant, which increased exon skipping, suggests that the URE is not optimised for induction of exon skipping. It is possible that the effect of the 3U→G mutation may be to create a binding site for a repressor protein that does not usually bind to the URE, rather than strengthening binding of the authentic repressors. This possibility is supported by the observation that enhanced exon skipping was only observed with mutation of all three G residues, but not with individual or pairwise changes (data not shown).
Substitution of the URE and DUGC elements
We have previously shown that the complete DRE (UGC region and pyrimidine tract) was not functionally interchangeable with the URE and its adjacent pyrimidine tract (21). We next tested whether the UGC regions alone (URE and DUGC) were functionally interchangeable (Fig. 3). In mutant URE/URE, the DUGC sequence, which contains four UGC motifs, was replaced by the URE. In DUGC/DUGC, the URE was substituted by the DUGC sequence, while the two elements were swapped in mutant DUGC/URE (Fig. 3A). All these constructs showed decreased exon 3 skipping in both cell types (Fig. 3B and C), comparable with the levels in the URE deletion mutant (ΔURE). Similar results were obtained when short UGC-rich regions from the α-actinin gene were substituted for the URE (data not shown). These data demonstrate that the two UGC-rich α-TM regulatory elements are functionally distinct, despite the superficial similarity of clusters of UGC/CUG motifs.
Figure 3.
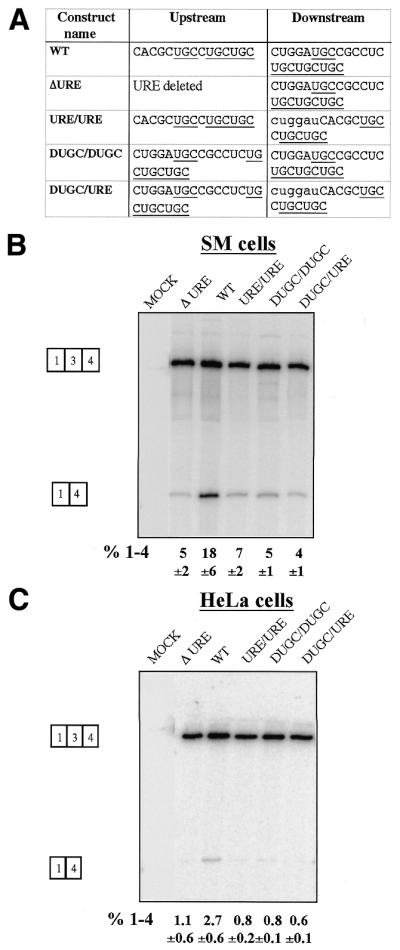
Substitution of the URE and DUGC elements. (A) Sequences at the URE and DUGC positions of each construct. UGC motifs are underlined. Bases in lower case in the downstream sequences represent additional BstXI compatible ends that were used to clone the URE sequence into the DUGC position. PAC-1 SM (B) and HeLa (C) cells were transiently transfected with URE substitution constructs and analysed as in Figure 2. The URE and DUGC elements of α-TM are not functionally interchangeable.
URE multimerisation leads to increased exon skipping
The ability of the 3U→G mutation to increase exon skipping (Fig. 2) suggested that the wild-type URE is not optimised for exon skipping. To investigate this further we tested the effects of systematically altering the copy number of the URE (Fig. 4). Multimerisation of the URE led to a progressive increase in exon skipping in all cell types tested. In SM cells, exon skipping reached a maximum of ∼70% with four and six UREs (Fig. 4A and E). In L, HeLa and C2C12 myoblast cells a lower maximum was reached at four or six copies, but its level was variable between the cell types and appeared to decrease in the order SM > C2C12 > L ≈ HeLa (Fig. 4A–E). Cell-specific regulation, expressed as the ratio of exon skipping in SM cells compared with the various non-SM cell lines decreased with increasing copy number (Fig. 4F). This was primarily due to the fact that the increase in exon skipping was proportionately less in the SM cells compared with the other cell lines. The greatest degree of regulation was observed between HeLa and PAC-1 cells with a single copy of the URE.
The progressive increase in exon 3 skipping caused by URE multimerisation could be due to increased binding of negative regulatory factors. However, the branch point (BP3) of α-TM exon 3 is located 175 nt upstream from its 3′ splice site and 301 nt upstream of the exon 5′ splice site. This corresponds to the ‘exon definition’ threshold of 300 nt, above which exon skipping is induced (31,32). Insertion of multiple URE copies upstream of exon 3 increases this separation to 376 nt in the case of six UREs. Therefore, an alternative explanation for the high degree of exon skipping in the presence of URE multimers is disruption of exon definition (32). To control for this possibility constructs with multimers of the 4G→C URE mutation were tested (Fig. 5A). These constructs all contained a 90 nt insertion corresponding to either six copies of the mutated URE (6*) or combinations of mutant and wild-type UREs (1+5*, 5*+1 and 1+4*+1). All these mutants demonstrated lower exon skipping than both the 6URE and 1URE constructs. This suggests that disruption of exon definition is not the cause of enhanced exon skipping by URE multimers, and supports the concept that the multimerised UREs constitute a high affinity binding site for regulatory factors.
We also tested the dependence of the enhanced exon skipping conferred by URE multimerisation upon the other defined negative regulatory elements by combining the 6URE mutation with deleterious mutations in the other elements. Three previously characterised deletion mutations of the DRE were combined with the 6URE mutation (Fig. 5B). These either contained a complete deletion of the DRE (6UREΔDRE) or of just the DY (6UREΔDY) or DUGC (6UREΔDUGC) regions. All the DRE mutations (ΔDRE, ΔDUGC and ΔDY) reduced exon skipping caused by the 6UREs. This demonstrates that exon skipping induced by URE multimerisation requires the presence of the complete wild-type DRE element. The polypyrimidine tract associated with the 3′ splice site (P3) contains three UCUU motifs (P3-1, P3-2, P3-3) (Fig. 1B). Previously, mutation of either of the PTB-binding sites P3-2 or P3-3 in the exon 3 polypyrimidine tract from UCUU→CCCC resulted in a switch to exon 2 inclusion, with P3-2 having the greatest quantitative effect (22). Likewise, introduction of the P3-2 mutation into pTWT also led to a 3-fold decrease in skipping of exon 3 (Fig. 5B, compare 1URE with 1URE/P3-2). Exon skipping was also reduced when the P3-2 mutation was combined with the 6URE mutation. The enhanced exon skipping induced by URE multimerisation therefore does not override a requirement for the other defined negative regulatory sequences. Taken together, the data in Figures 4 and 5 suggest that the enhanced exon 3 skipping demonstrated by URE multimers is due to enhanced negative regulation of exon 3 rather than to simple disruption of exon definition. Furthermore, the ability of the URE multimers to enhance exon skipping in non-SM cells suggests that the factors that mediate skipping of α-TM exon 3 via the URE are not restricted to smooth muscle.
Downstream UGC multimerisation leads to increased exon skipping
We next tested the effects of multimerising the downstream UGC element (DUGC, Fig. 6). Insertion of two DUGC copies had no effect in the four cell lines tested, but three or four DUGC copies increased exon skipping by approximately 2-fold in all cell types. In contrast, insertion of two, three or six copies of the URE in place of the DUGC element impaired exon skipping (lanes 5–7 from left side in Fig. 6A–D). The URE multimer insertions demonstrate that the effect of DUGC multimerisation was not due to non-specific spacing effects. These data emphasise again the non-equivalence of the URE and DUGC elements and indicate that, like the URE, the DUGC is not optimised for general exon skipping. The effects of DUGC multimerisation also apparently differed from URE multimerisation in that the degree of regulation was not reduced with higher copy numbers (compare Figs 6F and 4F). The reason for this contrast is that DUGC multimerisation had the same proportionate effect in all cell types.
Figure 6.

Enhanced exon skipping by DUGC multimers. PAC-1, L, HeLa and C2C12 cells (A–D) were transiently transfected with constructs containing 1–4 copies of the DUGC element or 2, 3 or 6 copies of the URE sequence in the DUGC position. The nomenclature of the mutant constructs shows that each construct contains a wild-type URE, indicated before the forward slash. The number and type of sequence element in the DUGC position is indicated after the forward slash. For example, URE/4DUGC has four copies of the DUGC element. Data were analysed as in Figure 2. (E) Histogram of per cent exon skipping for each construct in each cell type. (F) Histogram of regulation of each construct, expressed as per cent exon skipping in SM cells/per cent exon skipping in HeLa, L or C2C12 cells.
CELF proteins antagonise exon skipping
The CELF proteins (CUG-BP and Etr3-like factors) (25,33) are good candidate factors that might recognise the URE and DUGC elements. They are known to regulate some alternative splicing decisions and they can bind to sequences containing CUG repeats (34–36). We therefore tested the effects of three widely expressed CELF proteins, CUG-BP, Etr3 and CELF4 (25), upon splicing of co-transfected wild-type and 6URE tropomyosin constructs in PAC-1 and L cells (Fig. 7). The overexpressed proteins had no clear effect upon splicing of the wild-type construct in either cell type. However, with the 6URE construct all three CELF proteins caused a significant decrease in exon skipping, especially in L cells. This result is not consistent with these proteins acting as negative regulators of TM exon 3. The result could be explained by direct activation of TM exon 3 by CELF protein binding to the UGC elements. A simpler explanation is that they are able to bind to these elements and thereby prevent binding of the endogenous negative regulators. The more pronounced effect of the CELFs in L cells would be consistent with the expectation that the levels of endogenous regulators would be lower than in PAC-1 cells.
Figure 7.
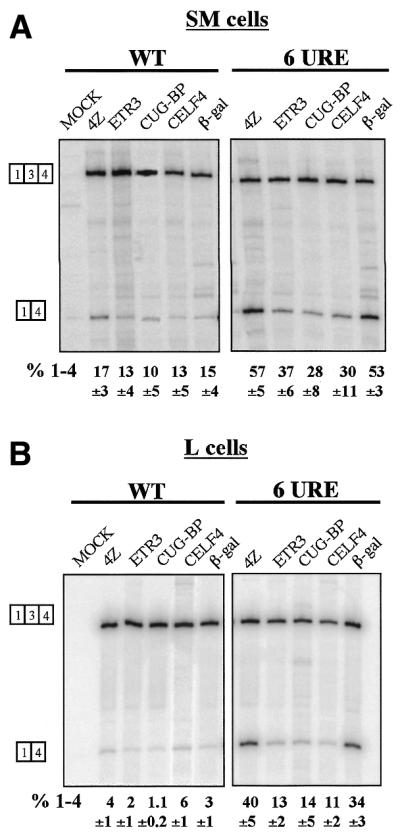
Co-transfected CELF proteins decrease skipping of α-TM exon 3. The wild-type and 6URE constructs were transfected into PAC-1 SM cells (A) and L cells (B) along with expression vectors for the CELF proteins ETR3, CUG-BP and CELF4. Negative control co-transfections contained pGEM4Z (lanes marked 4Z) or a construct expressing LacZ (lanes β-gal). Data were analysed as in Figure 2. The CELFs had no significant effect upon the wild-type construct, but caused a significant decrease in exon skipping from the 6URE constructs in both cell types. The effect was most emphatic in L cells (right hand panel in B).
DISCUSSION
The data reported here confirm the URE as an important element necessary for SM cell-specific repression of α-TM exon 3, but indicate that it is not itself a cell-specific element and neither is it optimised for inducing exon skipping. This is consistent with previous analyses, none of which has identified any elements that mediate an effect solely in SM cells (19,21). Other than the UGC/CUG regulatory elements, the other well defined elements are binding sites for PTB (21,23,37), a widespread protein that is not restricted to SM cells (38). Thus the analysis of regulatory elements points towards a mechanism that does not involve dedicated SM cell-specific regulators. Instead, it appears more likely that the particular arrangement and functional strength of cis-acting elements is fine tuned to respond to variations in the activities of various widespread factors such that exon exclusion is most pronounced in SM cells. This would explain why some mutations were able to increase exon skipping but at the same time decrease regulation (Figs 2 and 4). This type of combinatorial control appears to be a common strategy for tissue-specific splicing, as best exemplified by the regulation of the neuron-specific splicing of the c-src N1 exon by the members of hnRNP/hnRNP-like family of proteins (39–42).
Mutations that increased exon skipping generally led to a decrease in regulation (Figs 2 and 4). In particular, comparing the effects of the URE multimers between PAC-1 SM and HeLa cells, optimal regulation occurred with the wild-type single copy URE (Fig. 4F). The comparison of PAC-1 with C2C12 or L cells was not as clear cut. Although regulation decreased with URE number, it was no better in the wild-type than in the URE deletion construct. The underlying reason why some of these trends are not more clear cut is the partially differentiated state of the PAC-1 SM cells. In intact SM tissues, inclusion of TM exon 2 and skipping of exon 3 approaches 100%. In contrast, the PAC-1 cells, which are the best available cell line for regulated TM splicing, behave variably and rarely achieve better than 25% of the regulated splicing pattern. If a fully differentiated SM cell line were available, it is likely that the wild-type single URE would clearly demonstrate regulation better than any of the other mutants. We are currently testing the effects of a limited number of selected mutations using reporter constructs in transgenic mice (P.Ellis, A.Speed, P.Kemp and C.W.J.Smith, unpublished observations). This has the major advantage that the mutants will be tested in multiple cell types, including fully differentiated SM tissues. Despite the limitations of the PAC-1 cells the general trend for increased exon skipping with URE multimerisation is clear. Thus it appears that the functional strength of the URE is fine tuned to transduce quantitative differences in the activities of regulatory factors between SM and other cells to give an essentially qualitative switch in exon selection. The URE can be converted into a stronger repressor element, but only at the cost of proportionally greater repression in all cells and, hence, decreased cell-specific regulation. The experiments with multimerised wild-type and mutant UREs also indicated that the relative spacing of the URE from the other regulatory elements, both upstream (P3) and downstream (DRE), is important. If the distance separating the URE only from P3 or the DRE were crucial, then one of the constructs 1+5*, 5*+1 or 1+4*+1 would be expected to behave similarly to the wild-type. The fact that all three mutants produced less exon skipping than a single wild-type URE suggests that the URE facilitates assembly of a repressive complex that extends in both directions.
The effects of multimerisation of the URE show some similarity with analysis of elements that regulate selection of the c-src N1 exon (16). In that case, duplication of either an upstream repressor or a downstream enhancer led to decreased neuron-specific regulation due to overall low or high levels of exon selection, respectively. In contrast, a balanced combination of repressor and enhancer elements gave a 40-fold difference in the levels of exon inclusion in neuronal and non-neuronal cells. In contrast to these cases, multimers of a muscle-specific enhancer (MSE2) that positively regulates cTnT splicing were sufficient to confer a muscle-specific splicing pattern, whereas a single MSE2 copy was inactive (17).
Despite the superficial sequence similarity suggested by clusters of UGC motifs, the URE and DUGC elements are not functionally interchangeable (Figs 3 and 6). In addition, mutagenesis of individual guanosines within the URE indicated that the CGC motif at the 5′ end is as important as any of the UGC motifs (data not shown). Thus the precise arrangements of UGC and other motifs may dictate different preferences for binding of regulatory factors. The non-equivalence of the URE and DUGC elements contrasts with results from the FGFR-1 gene, where the UGC-rich repressor sequences surrounding the α-exon (ISS-1 and ISS-2) are functionally redundant and act in a position-independent manner (43).
The identity of the factors that interact with the URE remains an important open question. Similar elements containing UGC (or CUG) motifs have been identified in a number of other genes, such as FGFR-1 (43), cardiac troponin T (17), CLCB, NMDA (44) and α-actinin (45). In some cases the UGC/CUG elements were found to enhance exon inclusion (e.g. cTnT, CLCB and NMDA), while in others they repress the alternative exon (e.g. α-TM and FRFR-1). Recently, members of the CELF protein family (25,33) have been shown to bind UGC/CUG elements and to positively regulate cTnT exon selection (25). However, the three widely expressed CELF proteins that we tested all promoted exon 3 inclusion rather than skipping (Fig. 7), which is not consistent with their acting as URE-binding repressors. A possible explanation for these data is that the overexpressed CELF proteins are able to bind to the URE and displace the authentic repressors, thereby leading to increased exon inclusion. This is similar to the promotion of exon 3 inclusion by overexpressed PTB1, which competes with the more repressive PTB4 (23). Although they are ruled out as candidate repressors, it is still possible that these CELF proteins could modulate TM splicing in some circumstances in vivo by similar competition for URE binding. The authentic URE-binding repressor could be an alternatively spliced isoform of one of the CELF proteins, a different CELF family member or an entirely different protein. The observation that URE multimers can mediate strong exon exclusion in HeLa cells (Fig. 4) suggests an indirect approach for identification of the URE-binding regulatory factors. Biochemical identification of HeLa URE-binding factors could be followed by analysis of the isoforms or homologues that are present in differentiated SM cells. A similar approach has led us to identify a PTB homologue that is highly expressed in differentiated SM cells (C.Gooding and C.W.J.Smith, unpublished observations). The indentification and characterisation of the regulatory UGC-binding proteins remains an important goal for understanding regulation of TM splicing.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Clare Gooding for the initial tropomyosin constructs, Jim Patton for the P3-2 mutant construct and Tom Cooper for the CELF4 and ETR3 constructs. We also thank Mat Wollerton, Clare Gooding, Justine Southby and Arianne Matlin for critically reading the manuscript. This work was supported by a grant from the Wellcome Trust (059879). N.G. was supported by a Scholarship from the Darwin Trust of Edinburgh University.
REFERENCES
- 1.Black D.L. (2000) Protein diversity from alternative splicing: a challenge for bioinformatics and post-genome biology. Cell, 103, 367–370. [DOI] [PubMed] [Google Scholar]
- 2.Smith C.W.J. and Valcarcel,J. (2000) Alternative pre-mRNA splicing: the logic of combinatorial control. Trends Biochem. Sci., 25, 381–388. [DOI] [PubMed] [Google Scholar]
- 3.Graveley B.R. (2001) Alternative splicing: increasing diversity in the proteomic world. Trends Genet., 17, 100–107. [DOI] [PubMed] [Google Scholar]
- 4.Modrek B. and Lee,C. (2002) A genomic view of alternative splicing. Nature Genet., 30, 13–19. [DOI] [PubMed] [Google Scholar]
- 5.Roberts G. and Smith,C.W.J. (2002) Alternative splicing: combinatorial output from the genome. Curr. Opin. Chem. Biol., 6, 375–383. [DOI] [PubMed] [Google Scholar]
- 6.Schmucker D., Clemens,J.C., Shu,H., Worby,C.A., Xiao,J., Muda,M., Dixon,J.E. and Zipursky,S.L. (2000) Drosophila Dscam is an axon guidance receptor exhibiting extraordinary molecular diversity. Cell, 101, 671–684. [DOI] [PubMed] [Google Scholar]
- 7.Lopez A.J. (1998) Alternative splicing of pre-mRNA: developmental consequences and mechanisms of regulation. Annu. Rev. Genet., 32, 279–305. [DOI] [PubMed] [Google Scholar]
- 8.Blencowe B.J. (2000) Exonic splicing enhancers: mechanism of action, diversity and role in human genetic diseases. Trends Biochem. Sci., 25, 106–110. [DOI] [PubMed] [Google Scholar]
- 9.Manley J.L. and Tacke,R. (1996) SR proteins and splicing control. Genes Dev., 10, 1569–1579. [DOI] [PubMed] [Google Scholar]
- 10.Valcarcel J. and Green,M.R. (1996) The SR protein family: pleiotropic functions in pre-mRNA splicing. Trends Biochem. Sci., 21, 296–301. [PubMed] [Google Scholar]
- 11.Tacke R. and Manley,J.L. (1999) Determinants of SR protein specificity. Curr. Opin. Cell Biol., 11, 358–362. [DOI] [PubMed] [Google Scholar]
- 12.Graveley B.R. (2000) Sorting out the complexity of SR protein functions. RNA, 6, 1197–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dreyfuss G., Matunis,M.J., Pinol Roma,S. and Burd,C.G. (1993) hnRNP proteins and the biogenesis of mRNA. Annu. Rev. Biochem., 62, 289–321. [DOI] [PubMed] [Google Scholar]
- 14.McAfee J.G., Huang,M., Soltaninassab,S., Rech,J.E., Iyengar,S. and Lestourgeon,W.M. (1997) The packaging of pre-mRNA. In Krainer,A.R. (ed.), Eukaryotic mRNA Processing. Oxford University Press, Oxford, pp. 68–102.
- 15.Krecic A.M. and Swanson,M.S. (1999) hnRNP complexes: composition, structure and function. Curr. Opin. Cell Biol., 11, 363–371. [DOI] [PubMed] [Google Scholar]
- 16.Modafferi E.F. and Black,D.L. (1999) Combinatorial control of a neuron-specific exon. RNA, 5, 687–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cooper T.A. (1998) Muscle-specific splicing of a heterologous exon mediated by a single muscle-specific splicing enhancer from the cardiac troponin T gene. Mol. Cell. Biol., 18, 4519–4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wieczorek D.F., Smith,C.W.J. and Nadal Ginard,B. (1988) The rat alpha-tropomyosin gene generates a minimum of six different mRNAs coding for striated, smooth, and nonmuscle isoforms by alternative splicing. Mol. Cell. Biol., 8, 679–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gooding C., Roberts,G.C., Moreau,G., Nadal Ginard,B. and Smith,C.W.J. (1994) Smooth muscle-specific switching of alpha-tropomyosin mutually exclusive exon selection by specific inhibition of the strong default exon. EMBO J., 13, 3861–3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dye B.T., Buvoli,M., Mayer,S.A., Lin,C.H. and Patton,J.G. (1998) Enhancer elements activate the weak 3′ splice site of alpha-tropomyosin exon 2. RNA, 4, 1523–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gooding C.G., Roberts,G.C. and Smith,C.W.J. (1998) Role of an inhibitory pyrimidine-element and general pyrimidine-tract binding proteins in regulation of α-tropomyosin alternative splicing. RNA, 4, 85–100. [PMC free article] [PubMed] [Google Scholar]
- 22.Perez I., Lin,C.-H., McAfee,J.G. and Patton,J.G. (1997) Mutation of PTB binding sites causes misregulation of alternative 3′ splice site selection in vivo. RNA, 3, 764–778. [PMC free article] [PubMed] [Google Scholar]
- 23.Wollerton M.C., Gooding,C., Robinson,F., Brown,E.C., Jackson,R.J. and Smith,C.W.J. (2001) Differential alternative splicing activity of isoforms of polypyrimidine tract binding protein (PTB). RNA, 7, 819–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin C.H. and Patton,J.G. (1995) Regulation of alternative 3′ splice site selection by constitutive splicing factors. RNA, 1, 234–245. [PMC free article] [PubMed] [Google Scholar]
- 25.Ladd A.N., Charlet-B.,N. and Cooper,T.A. (2001) The CELF family of RNA binding proteins is implicated in cell-specific and developmentally regulated alternative splicing. Mol. Cell. Biol., 21, 1285–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rothman A., Kulik,T.J., Taubman,M.B., Berk,B.C., Smith,C.W.J. and Nadal Ginard,B. (1992) Development and characterization of a cloned rat pulmonary arterial smooth muscle cell line that maintains differentiated properties through multiple subcultures. Circulation, 86, 1977–1986. [DOI] [PubMed] [Google Scholar]
- 27.Yaffe D. and Saxel,O. (1977) Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature, 270, 725–727. [DOI] [PubMed] [Google Scholar]
- 28.Mullen M.P., Smith,C.W.J., Patton,J.G. and Nadal Ginard,B. (1991) Alpha-tropomyosin mutually exclusive exon selection: competition between branchpoint/polypyrimidine tracts determines default exon choice. Genes Dev., 5, 642–655. [DOI] [PubMed] [Google Scholar]
- 29.Roberts G.C., Gooding,C. and Smith,C.W.J. (1996) Smooth muscle alternative splicing induced in fibroblasts by heterologous expression of a regulatory gene. EMBO J., 15, 6301–6310. [PMC free article] [PubMed] [Google Scholar]
- 30.Roberts G.C., Gooding,C., Mak,H.Y., Proudfoot,N.J. and Smith,C.W.J. (1998) Co-transcriptional commitment to alternative splice site selection. Nucleic Acids Res., 26, 5568–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robberson B.L., Cote,G.J. and Berget,S.M. (1990) Exon definition may facilitate splice site selection in RNAs with multiple exons. Mol. Cell. Biol., 10, 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berget S.M. (1995) Exon recognition in vertebrate splicing. J. Biol. Chem., 270, 2411–2414. [DOI] [PubMed] [Google Scholar]
- 33.Good P.J., Chen,Q., Warner,S.J. and Herring,D.C. (2000) A family of human RNA-binding proteins related to the Drosophila Bruno translational regulator. J. Biol. Chem., 275, 28583–28592. [DOI] [PubMed] [Google Scholar]
- 34.Philips A.V., Timchenko,L.T. and Cooper,T.A. (1998) Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science, 280, 737–741. [DOI] [PubMed] [Google Scholar]
- 35.Timchenko L.T., Timchenko,N.A., Caskey,C.T. and Roberts,R. (1996) Novel proteins with binding specificity for DNA CTG repeats and RNA CUG repeats: implications for myotonic dystrophy. Hum. Mol. Genet., 5, 115–121. [DOI] [PubMed] [Google Scholar]
- 36.Savkur R.S., Philips,A.V. and Cooper,T.A. (2001) Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nature Genet., 29, 40–47. [DOI] [PubMed] [Google Scholar]
- 37.Perez I., McAfee,J.G. and Patton,J.G. (1997) Multiple RRMs contribute to RNA binding specificity and affinity for polypyrimidine tract binding protein. Biochemistry, 36, 11881–11890. [DOI] [PubMed] [Google Scholar]
- 38.Wagner E.J. and Garcia-Blanco,M.A. (2001) Polypyrimidine tract binding protein antagonizes exon definition. Mol. Cell. Biol., 21, 3281–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Markovtsov V., Nikolic,J.M., Goldman,J.A., Turck,C.W., Chou,M.Y. and Black,D.L. (2000) Cooperative assembly of an hnRNP complex induced by a tissue-specific homolog of polypyrimidine tract binding protein. Mol. Cell. Biol., 20, 7463–7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Min H., Chan,R.C. and Black,D.L. (1995) The generally expressed hnRNP F is involved in a neural-specific pre-mRNA splicing event. Genes Dev., 9, 2659–2671. [DOI] [PubMed] [Google Scholar]
- 41.Min H., Turck,C.W., Nikolic,J.M. and Black,D.L. (1997) A new regulatory protein, KSRP, mediates exon inclusion through an intronic splicing enhancer. Genes Dev., 11, 1023–1036. [DOI] [PubMed] [Google Scholar]
- 42.Chou M.Y., Rooke,N., Turck,C.W. and Black,D.L. (1999) hnRNP H is a component of a splicing enhancer complex that activates a c-src alternative exon in neuronal cells. Mol. Cell. Biol., 19, 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jin W., Huang,E.S., Bi,W. and Cote,G.J. (1999) Redundant intronic repressors function to inhibit fibroblast growth factor receptor-1 alpha-exon recognition in glioblastoma cells. J. Biol. Chem., 274, 28035–28041. [DOI] [PubMed] [Google Scholar]
- 44.Zhang L., Liu,W. and Grabowski,P.J. (1999) Coordinate repression of a trio of neuron-specific splicing events by the splicing regulator PTB. RNA, 5, 117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Southby J., Gooding,C. and Smith,C.W.J. (1999) Polypyrimidine tract binding protein functions as a repressor to regulate alternative splicing of alpha-actinin mutally exclusive exons. Mol. Cell. Biol., 19, 2699–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]