

Abstract
Gas-phase H/D exchange is widely used for characterizing the structure of ions. However, many structural parameters that affect the rate of H/D exchange are poorly understood, which complicates the interpretation of experimental data. Here, the effects of sodium ion adduction on the rate of H/D exchange with D2O for a series of peptides and peptide dimers with varying numbers of acidic residues are described. The maximum number of sodium ion adducts that can be accommodated by the peptides and peptide dimers in this study is N + 1, where N is the number of free carboxylic acid groups. The formation of methyl-esters at all carboxylic acid groups, or the replacement of all the acidic hydrogens with sodium ions, effectively shuts down H/D exchange with D2O. In contrast, both the rate and the extent of H/D exchange with D2O are increased for most of the peptides and peptide dimers by the adduction of an intermediate number of sodium ions. These results are consistent with the H/D exchange occurring via a salt-bridge mechanism and show that the presence of two carboxylic acid groups is much better than one. The results with peptide dimers also indicate that surface accessibility may not be a dominant factor in the extent of H/D exchange for these ions.
In solution, H/D exchange can be used to obtain information about the solvent accessibility of amino acid residues in a protein [1]. Sites of H/D exchange can be localized to the individual amino acid with NMR or to larger portions of the protein by using proteolitic enzymes (typically pepsin) and mass spectrometric analysis of the products to determine the extent of H/D exchange [2]. From these experiments, information about the tertiary structure of proteins in solution can be deduced.
In the gas phase, the rate and maximal extents of H/D exchange have been used to differentiate protein conformers. In a series of pioneering experiments of McLafferty and coworkers, as many as seven different conformers of gas-phase cytochrome c were separated on the basis of their H/D exchange rates and extents of exchange with D2O [3–5]. Conformers that exchanged rapidly with D2O were designated as “open” conformations due to the relatively high accessibility of exchange reagent to active hydrogen/protonation sites. Smith and coworkers showed that disulfide intact bovine proinsulin and α-lactalbumin undergo more extensive H/D exchange than their disulfide reduced analogues even though the oxidized proteins are expected to have a more compact gas-phase conformation [6]. The higher H/D exchange reactivity for proteins with intact disulfide bonds was attributed to effects of higher coulombic repulsion in the constrained proteins. Cassady and Wang studied H/D exchange of three multiply protonated peptides, each having one disulfide bond, and found that the extent of H/D exchange increased, decreased, or stayed the same upon reduction of the disulfide bond, depending on the peptide [7]. Results from molecular modeling suggested that increased H/D exchange is observed for the peptide that has a more extended gas-phase conformation upon the reduction of the disulfide bond whereas decreased exchange is observed for a peptide that has a more compact conformation after disulfide bond reduction. Many other reports of gas-phase H/D exchange of peptides and proteins have been published [8–25]. The ultimate goal of these H/D exchange experiments is to determine information about the structure and conformation of these ions in the gas phase. Other methods to determine information about gas-phase protein conformation include proton-transfer reactivity [26–29] and ion-mobility experiments [30–36].
In order to infer structural information from H/D exchange, it is important to understand the mechanism by which exchange occurs. Fundamental studies by Hunt and coworkers [37] as well as Ausloos and Lias [38] showed that for small protonated compounds, H/D exchange does not occur when the proton affinity of the neutral base is greater than the deuterated reagent by more than 20 kcal/mol. However, Lebrilla and coworkers clearly showed that H/D exchange can occur even when the basicity difference is greater than 20 kcal/mol for compounds that have multiple sites of similar basicity [8, 39]. They proposed a mechanism for exchange in which deuterated methanol forms two simultaneous hydrogen bonds with basic groups of the protonated compound, e.g., the carbonyl oxygen of a carboxylic acid group and the protonated amino terminus, forming a bridge between two basic sites.
Beauchamp and coworkers have investigated the H/D exchange reactivity of protonated glycine oligomers with a variety of exchange reagents [11, 12]. They found that the rate and extent of H/D exchange with D2O is a strong function of polymer length. They proposed several mechanisms by which exchange might occur. For D2O, the preferred mechanism is a “relay” mechanism similar to the mechanism proposed by Lebrilla for exchange with CH3OD [8], in which D2O is coordinated by both the protonated amino terminus and a carbonyl oxygen. The exchange occurs by shuttling a proton from the protonated amino terminus to the oxygen of D2O while simultaneously transferring a deuteron to a carbonyl oxygen of the amide backbone. Although Gly3 exchanges rapidly with D2O, Gly4 exchanges very slowly. For Gly4, the authors suggested that the energy released by the formation of two hydrogen bonds is insufficient to alter the gas-phase conformation of the peptide to allow binding of D2O and subsequent H/D exchange to occur.
Noncovalent complexes have also been investigated using H/D exchange. Lebrilla and coworkers found that methyl-esters of several amino acids complexed with glycerol and glucose undergo no measurable gas-phase H/D exchange, indicating that a free carboxylic acid is important for H/D exchange with CH3OD for these complexes [9]. They also found that complexes of amino acids and monosaccharides undergo less H/D exchange with CH3OD than the individual amino acids themselves [9], indicating that the protonated amino acid is extensively coordinated with the monosaccharide and is not able to interact sufficiently with methanol to promote H/D exchange. Beauchamp and coworkers observed a similar result when they measured a decrease in H/D exchange with ND3 for a series of protonated glycines upon complexation with crown ethers [13]. Liftshitz and coworkers found that doubly protonated dimers of leucine enkephalin exchanged more slowly with ND3 than singly protonated monomers and attributed this phenomenon to the doubly protonated leucine enkephalin dimer forming a more compact structure [23]. In contrast, Green et al. [10] and Reid et al. [40] working with the exchange of homodimers of butylamine with CH3OD and the exchange of dimers of dipeptides with D2O respectively, found that dimer formation increased exchange. The authors suggested that dimer formation increases the rate of H/D exchange as the reagent molecule is able to insert itself into the hydrogen bond linking the dimer and undergo facile exchange with a bridging or relay exchange mechanism [8, 11]. Heck and coworkers also measured an increase in the extent of H/D exchange with ND3 for a series of vancomycin family antibiotics upon complex formation with their bacterial cell wall mimicking peptides [20]. For example, uncomplexed ristocetin exchanges 40% of its exchangeable hydrogens, but 70% when it is complexed with ac-GA. The authors suggested that the change in H/D exchange is due to changes in the higher order structure of the antibiotic upon complexation of the peptide.
Effects of metal ion adduction on H/D exchange have also been investigated. Cotter and coworkers found that reactions of singly protonated gramicidin S, a cyclic decapeptide, with deuterated ammonia resulted in the exchange of 5–6 out of 13 labile hydrogens [17]. This could be increased to 13 by collisionally activating the ions with off-resonance excitation. However, the singly sodiated ion only exchanged two hydrogens under the same conditions, and heating the sodiated peptide reduced the exchange rate. Protonated Glyn, n = 2–5 undergoes more exchange with ND3 than do their sodiated counterparts [41]. Similar results were reported by Reyzer and Brodbelt who found that H/D exchange reactions between the alkali metal cationized complexes of noncovalent polyamine complexes and ND3 are nearly quenched compared to the singly and doubly protonated complexes [42]. In contrast to the results for polyamines, singly sodiated bradykinin ions exchange with D2O more than 1000 times faster than singly protonated bradykinin, rapidly exchanging all 17 labile hydrogens [18, 25]. Also, protonated bradykinin undergoes slower H/D exchange than protonated bradykinin methyl-ester [18]. These results indicate that protonated and sodiated peptides containing basic residues, and their methyl-esters, are able to undergo H/D exchange with D2O, although the rates of exchange are lower than those reported for short glycine polymers and their analogs [8, 9, 11, 12, 14, 19, 39].
Solouki et al. investigated the effects of one and two metal ion adducts on the H/D exchange rate of Arg-Gly-Asp (RGD) using ND3 [24]. They found that the rate of exchange decreases in the order H+ > Cs+ > K+ > Na+. Ab initio calculations indicate that the sodium ion forms a trivalently coordinated structure with RGD due to its relatively small size and this presumably influences the H/D exchange. However, although the doubly adducted RGD peptides continue to exchange more slowly than the singly protonated peptide, the relative rates of exchange of the doubly adducted metal ions are reversed and follow the order 2Na+ > 2K+ > 2Cs+ [24].
Cassady and coworkers investigated the effect of the position of lysines in the exchange with CH3OD and D2O of three quadruply protonated dodecapeptides, (KGG)4, (K2G4)2, and (K4G8) [16]. They found that even though molecular modeling calculations indicate that K4G8 has a more compact structure than the other peptides, it exchanges faster and to a greater extent. Molecular modeling indicates that the protonated sites of the several lysine residues of K4G8 are in close enough proximity for a molecule of the exchange reagent to form hydrogen bonds with both a protonated lysine residue and a peptide backbone carbonyl oxygen providing a suitable orientation for exchange by a bridge or relay mechanism. This suggests that factors other than the compactness of a gas-phase peptide may play a significant role in determining the rate and extent of H/D exchange.
Here, we investigate the effects of both metal ion adduction and the number of carboxylic acid groups on H/D exchange with D2O for a series of peptides having 5–6 amino acids and no basic groups other than the amino terminus. Their methyl-ester derivatives and peptide dimers are also studied. We demonstrate that the extent of sodium adduction is directly related to the number of acidic residues, and that a free carboxylic acid group is critical for H/D exchange with D2O in these peptides. We show that multiple metal ion adducts can greatly increase H/D exchange, and we demonstrate that dimers of these peptide ions, which are presumably more globular or compact than the corresponding monomer, do not necessarily exchange more slowly than their corresponding monomers.
Experimental
Mass Spectrometry
Experiments were conducted with a 110 mm bore 9.4 T Fourier-transform mass spectrometer that was constructed in collaboration with Bruker Daltonics (Billerica, MA) (Figure 1). This instrument has been described elsewhere [43]. The ion optics and introduction system are a standard Bruker design. Ions are generated by electrospray or nanoelectrospray. This is done using either a Bruker Apollo source electrospray emitter with a syringe pump flow rate of 1.0 μL/min and nitrogen assisted nebulization, or nanoelectrospray. Nanoelectrospray is performed using borosilicate capillaries that are pulled to a ~4 μm tip with a model P-87 capillary puller (Sutter Instruments, Novato, CA). A small volume of solution (4 –10 μL) is injected into the borosilicate capillary and a platinum wire is inserted into the solution at the end of the capillary. The capillary is positioned ~2 mm from the source inlet and a potential of ~900 V is applied to the platinum wire.
Figure 1.
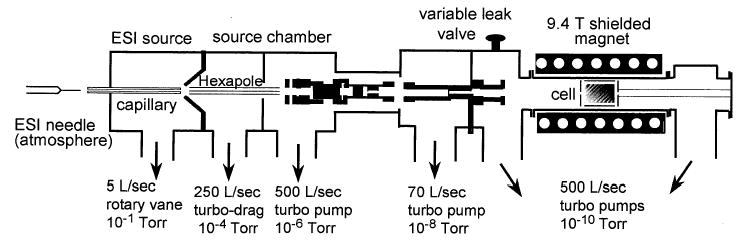
Schematic diagram of the Berkeley-Bruker FTMS instrument with a 110 mm bore magnet. Peptide ions are stored in the UHV cell and undergo exchange reactions with D2O introduced by the variable leak valve.
The electrospray generated ions are accumulated in a storage hexapole prior to injection through multiple ion lenses and three stages of differential pumping into the ultra-high vacuum chamber of the instrument where the ions are trapped in a cylindrical Bruker Infinity cell. Each ion injection is accompanied by a pulse of nitrogen gas introduced into the vacuum chamber at a pressure of ~2 × 10−6 torr to enhance trapping and to damp the motion of the ions in the cell. All spectra shown are from single detection events. No signal averaging was done.
For H/D exchange studies of peptide dimer ions, the ions were selectively accumulated in the external accumulation hexapole by increasing ion load times from 1 to 3 s, and increasing the hexapole dc offset voltage from ~2.7 V to 3.5–4.5 V. Multiple hexapole accumulations were injected into the ion cell prior to H/D exchange.
Chemicals and Chemical Modifications
D2O, leucine enkephalin (YGGFL), Ala5, Val-Glu-Pro-Ile-Pro-Tyr (VEPIPY) and Phe-Leu-Glu-Glu-Leu (FLEEL) were purchased from Sigma-Aldrich Co. (St. Louis, MO) and were used without further purification. Methanol and hydrochloric acid (37%) were purchased from EM Science (Gibbstown, NJ). Sodium chloride was purchased from Fisher Scientific (Fairlawn, NJ), and potassium chloride was purchased from Mallinckrodt (Paris, KT).
Solutions used for ESI were 1:1 water:MeOH + 2% acetic acid, and the concentration of the peptides was typically 0.5–1 × 10−3 M. High peptide concentrations were used to enhance the formation of noncovalent peptide dimers. To form abundant sodium adducts, sodium chloride was added to a concentration of 1–5 × 10−3 M. For some solutions of YGGFL, 1 × 10−3 M potassium chloride was added to compare the effects of sodium and potassium adduction on H/D exchange with D2O.
Peptide methyl-esters were synthesized by dissolving the peptide in methanol until the solution was saturated or a concentration of 2 × 10−3 M was reached. Hydrochloric acid (37% vol/vol) was then added at 1% by volume. Complete esterification of the peptides of interest occurred overnight at room temperature. To prepare partially esterified FLEEL, the reaction was allowed to proceed at room temperature for 30 minutes. After reacting with hydrochloric acid for the desired amount of time, the esterification reaction was quenched by the addition of 1/1 vol/vol water.
H/D Exchange
Gaseous D2O is introduced into the vacuum chamber of the instrument through a variable leak valve (Varian, Lexington, MA) for 50 s during which time H/D exchange occurs. The pressure of the D2O was varied between 1 × 10−8 and 1 × 10−6 torr, as measured by an uncalibrated ion gauge located ~1.1 m from the cell. After 50 s, the leak valve was closed for 50 s to allow the pressure in the vacuum chamber to decrease prior to excitation and broad-band detection so that reasonable signal could be obtained.
Results
The gas-phase H/D exchange characteristics of a class of peptides, their dimers, their methyl-esters, and all of the sodium adducts that they form are reported. Peptides with a varying number of acidic residues were selected to determine the effects of acidic sites on the rate and extent of H/D exchange with D2O. All peptides in this study have only one basic site, (the amino terminus), and from zero to three carboxylic acid groups (listed in Table 1 in the order of increasing carboxylic acid groups). In Table 1, the amount of H/D exchange that each protonated or sodium adducted species undergoes is expressed as the weighted average fraction of labile hydrogens exchanged for deuteriums after a 50 s reaction time with D2O at 1 × 10−6 torr. For example, (YGGFL)2 with one and two adducted sodium ions has 16 and 15 labile hydrogens, respectively. These ions exchange on average, 2% and 49% of their labile hydrogens, respectively, under these conditions. Peptide ions listed with -N ME represent ions with N carboxylic acids converted to methyl-esters. Blank entries in Table 1 are due to overlapping isotopic distributions of various ions in the mass spectra after the 50 s reaction time, which makes it difficult to determine the weighted average fraction of hydrogens exchanged. For these cases, the general exchange characteristics, i.e., slow or rapid exchange, with the same 50 s reaction time but at lower pressures of D2O, are described in specific instances below. Gas-phase H/D exchange studies were also done on protonated and sodium adducted peptides and peptide dimers that were fully methyl-esterified (Table 2). The exchange conditions and the method of reporting the extent of H/D exchange are identical in Tables 1 and 2.
Table 1.
Average fractions of labile hydrogens exchanged for a series of peptides, peptide homodimers and their sodium adducts after 50 s reaction time at 1 × 10−6 torr D2O
| Weighted average fraction of hydrogens exchanged
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. of carboxylic acid groups | Peptide | Labile hydrogens (neutral) | Protonated | + 1Na | + 2Na | + 3Na | + 4Na | + 5Na | + 6Na | + 7Na |
| 1 | YGGFL | 8 | 0.09 | 0.01 | 0.00 | |||||
| 1 | Ala5 | 7 | 0.14 | 0.15 | 0.01 | |||||
| 1 | (YGGFL + YGGFL-2ME) | 15 | 0.05 | 0.01 | 0.00b | |||||
| 1 | FLEEL-2ME | 7 | —a | —a | 0.00 | |||||
| 2 | (YGGFL)2 | 16 | 0.01 | 0.02 | 0.49 | 0.04 | ||||
| 2 | (Ala5)2 | 14 | 0.35 | 0.10 | 0.28 | 0.00 | ||||
| 2 | VEPIPY | 8 | 0.01 | 0.55 | 0.38 | 0.00 | ||||
| 2 | FLEEL-ME | 8 | 0.10 | 0.18 | 0.43 | 0.01 | ||||
| 3 | FLEEL | 9 | 0.10 | 0.51 | 0.55 | 0.37 | 0.00 | |||
| 4 | (VEPIPY)2 | 16 | 0.02 | 0.11 | 0.07 | 0.58 | 0.34 | 0.01 | ||
| 6 | (FLEEL)2 | 18 | 0.35 | 0.43 | 0.55 | 0.44 | 0.45 | 0.55 | 0.05 | 0.00 |
Not included due to overlapping isotopic distributions.
One adducted sodium and one adducted potassium ion.
Table 2.
The average fraction of labile hydrogens exchanged for the peptides and peptide homodimers listed in Table 1 after methyl-esterification at all of their carboxylic acid groups
| Weighted average fraction of hydrogens exchanged
|
|||
|---|---|---|---|
| Peptide | Labile hydrogens | Protonated | 1Na |
| YGGFL-ME | 7 | 0.03 | 0.01 |
| (YGGFL-ME)2 | 14 | 0.02 | 0.00 |
| Ala5-ME | 6 | 0.04 | 0.00 |
| (Ala5-ME)2 | 12 | 0.02 | 0.02 |
| VEPIPY-2ME | 6 | 0.02 | 0.01 |
| (VEPIPY-2ME)2 | 12 | 0.01 | 0.00 |
| FLEEL-3ME | 6 | 0.04 | 0.02 |
| (FLEEL-3ME)2 | 12 | 0.03 | 0.00 |
The Maximum Number of Adducted Sodium Ions
For all of the peptides and peptide dimers in the study, the maximum number of sodium adducts observed in the ESI mass spectra is equal to N + 1, where N is the number of carboxylic acid groups in the peptide or peptide dimer. For example, the peptide FLEEL has three carboxylic acid groups and adducts up to four sodium ions. The partially esterified FLEEL peptides adduct a maximum number of sodium ions that decreases by one sodium for each methyl-ester formed at a carboxylic acid. For the peptide YGGFL, potassium adducts were formed as well as sodium adducts by adding potassium chloride to the solutions. In the case of YGGFL and (YGGFL)2, sodium and potassium are interchangeable for the purpose of determining maximal adduction.
As has been observed previously, the first sodium adduct displaces the proton typically added in the ESI process to form either a charge solvated or salt-bridge form of the adducted peptide [41, 44 –48]. Subsequent adducted sodium ions appear to replace carboxylic acid hydrogens and likely form ion pairs with the resulting carboxylate anions. It should be noted that the maximum number of adducted metal ions can be greater than N + 1 for peptides with more basic sites than the amino terminus. For example, adducts with up to four sodium ions have been observed for doubly charged gramicidin S (cyclo[-Pro-Val-Orn-Leu-D-Phe-]2), a peptide that has no acidic groups [49].
Pressure Dependence of H/D Exchange
The extent of H/D exchange after 50 s of reaction time was measured at four pressures of D2O ranging from 1 × 10−8 to 1 × 10−6 torr, for all peptides listed in Tables 1 and 2, to determine trends in the extent of H/D exchange and to study the general exchange characteristics of species that could not be distinguished at 1 × 10−6 torr due to overlapping isotopic distributions. Seven of the peptides and peptide dimers had multiple adducted states (protonated, singly sodiated, doubly sodiated, etc.) that exchanged more than one hydrogen during 50 s exchange at ~1 × 10−6 torr. For all seven of these peptides, the average rate of H/D exchange for the duration of the 50 s changed with the pressure. For example, (VEPIPY + 2Na + H)+ has two fast exchanging hydrogens that exchange during 50 s at ~1 × 10−8 torr, the lowest pressure investigated (Figure 2a). Little additional exchange occurs for (VEPIPY + 2Na – H)+ during 50 s of exchange even when the pressure of D2O is two orders of magnitude higher. In contrast, the exchange of (VEPIPY + Na)+ is virtually zero at low pressure, but increases with increasing pressure, exchanging an average of 4.4 hydrogens at 1 × 10−6 torr of D2O. The situation becomes increasingly complex with peptides and peptide dimers that have more carboxylic acid groups and thus a greater number of possible sodium adducts. For example, FLEEL dimers form between zero and seven sodium ion adducts (Figure 2b). The relative amount of exchange of each species changes greatly with reaction pressure. For example, (FLEEL2 + 6Na − 5H)+ and (FLEEL2 + 4Na − 3H)+ both exchange an average of ~1 hydrogen at 1 × 10−7 torr. However, (FLEEL2 + 4Na − 3H)+ exchanges ~8 hydrogens at 1 × 10−6 torr while (FLEEL2 + 6Na − 5H)+ exchanges an average of less than two.
Figure 2.
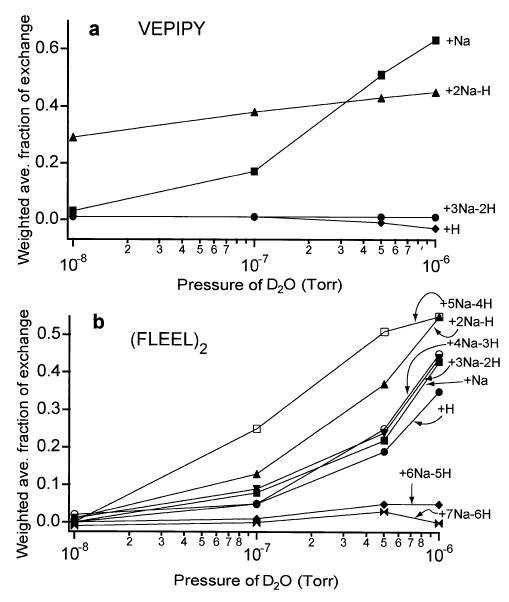
Average fraction of labile hydrogens exchanged with D2O during a 50 s reaction time at varying pressures for protonated and multiply sodiated adducts of the peptide (a) VEPIPY and (b) dimers of the peptide FLEEL.
The fraction of labile hydrogens exchanged for the peptides listed in Table 1 are for a reaction time of 50 s at a pressure of 1 × 10−6 torr. The individual exchange rate constants for each hydrogen exchanged could be obtained by measuring the exchange as a function of reaction time. However, this is beyond the scope of the current study.
Average Exchange Rates
In order to quantitatively compare H/D exchange results of the peptides listed in Table 1 with previous reports of other peptides [12, 14, 18], the average rate constants of several of the peptides were approximated. This was done by modeling a series of linear, coupled differential equations with identical rate constants, graphing the solutions, and comparing the solutions with the experimental data. Some of the peptide-metal complexes had fast exchanging sites including [VEPIPY + 2Na − H]+ (Figure 2b) and [FLEEL + 3Na − 2H]+ which each exchanged three hydrogens during 50 s exchange at ~1 × 10−8 torr corresponding to an average rate constant of ~2 × 10−10 cm3/mol. These are similar rate constants to those measured for the first three exchanges of protonated Gly2 and Gly3 polymers [12]. Most of the average rate constants approximated for the peptides in Table 1 are two or three orders of magnitude slower. These range from protonated YGGFL, (~1 × 10−13 cm3/mol), to [Ala5 2Na + H]+, (~2 × 10−12 cm3/mol), to [FLEEL2 + 2Na − H]+, (~5 × 10−12 cm3/mol). Thus, the average rate constants for the peptide-metal complexes that exchanged the greatest number of hydrogens in Table 1 correspond to the slowest rate constants listed by Beauchamp and coworkers for the exchange of protonated glycine polymers with D2O.
H/D Exchange of Protonated Peptides and Peptide Dimmers
The protonated monomers of the peptides in this study undergo very little H/D exchange with D2O, exchanging on average less than one hydrogen. Two proton-bound peptide dimers, [(Ala5)2 + H]+ (Figure 3a, b) and [(FLEEL)2 + H]+, do undergo more rapid H/D exchange, whereas [(YGGFL)2 + H]+ and [(VEPIPY)2 + H]+ do not.
Figure 3.
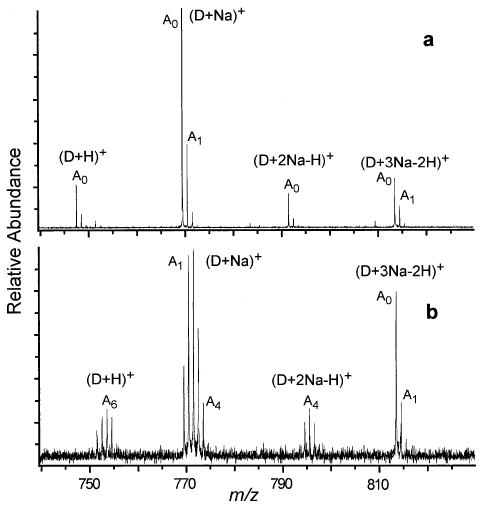
ESI mass spectra of (Ala5)2 formed from in 0.5 mM NaCl and 1:1 water: methanol solution (a) without exchange and (b) after 50 s exchange with D2O at a pressure of ~1 × 10−6 torr. The notation A0 represents the monoisotopic peak, A1 the monoisotopic peak + 1 Da, and so forth.
In contrast, none of the protonated peptides or peptide dimers with full or partial methyl-ester formation exchanges more than an average of one hydrogen after 50 s exposure to 1 × 10−6 torr D2O. A similar observation was reported by Lebrilla and coworkers [9], who compared H/D exchange of protonated Gly and Gly-ME in complexes with glycerol. Under exchange conditions where all seven hydrogens in the Gly complexes exchanged with deuterated methanol, no exchange was observed for the Gly-ME complexes [9]. Beauchamp and coworkers observed slow H/D exchange of one hydrogen for (Gly4 + H)+ and (Gly4-ME + H)+ for reactions up to 20 s at 1 × 10−7 torr D2O [12]. Interestingly, Beauchamp and coworkers found that (Gly3-ME + H)+ exchanges all five of its labile hydrogens with D2O within a reaction time of 20 s at ~1 x 10−7 torr [12]. This suggests that smaller peptides than the ones in this study may exchange relatively rapidly with or without a carboxylic acid group. Methyl-esters of larger peptides with multiple basic sites can also undergo relatively rapid exchange with D2O. Freitas and Marshall formed singly protonated bradykinin methyl-esters (BK-ME + H)+ by collisionally charge stripping (BK-ME + 2H)2+ (a necessary procedure because only the doubly protonated methyl-ester was initially present) [18]. Because ions formed by collisional charge stripping are initially ‘hot’, they were allowed to cool by collisions with a pulse of He buffer gas introduced into the vacuum chamber of the instrument prior to H/D exchange. After collisional cooling, (BK-ME + H)+ exchanged 12–14 out of 17 exchangeable hydrogens with D2O in 6 s at 1 × 10−5 torr (a comparable number of collisions as the data presented in Table 1). In contrast, (BK-ME + 2H)2+ does not exchange any hydrogens with D2O after 1 hr of reaction at 1 × 10−5 torr [18].
H/D Exchange of Singly Sodiated Peptides and Peptide Dimers
Peptide monomers with a single sodium adduct can exchange slowly, as was observed for (YGGFL + Na)+ and (Ala5 + Na)+, or rapidly, as was the case for (VEPIPY + Na)+ and (FLEEL + Na)+ (Figure 4a, b, c, d). The methyl-esterified peptides (Table 2) undergo very little H/D exchange when adducted with a single sodium ion. There are only a few studies on the H/D exchange of noncovalent complexes of metal ions and peptides [17, 18, 24, 25], and D2O was used as the exchange reagent in only two of these [18, 25]. Freitas and Marshall found that (BK + Na)+ exchanges rapidly, incorporating all 17 exchangeable hydrogens within 10 min [18]. In contrast, (BK + H)+ exchanges less than one hydrogen during an hour of reaction at 1 × 10−5 torr [18]. Thus, the addition of a single metal ion can significantly increase the H/D exchange of peptides with D2O.
Figure 4.
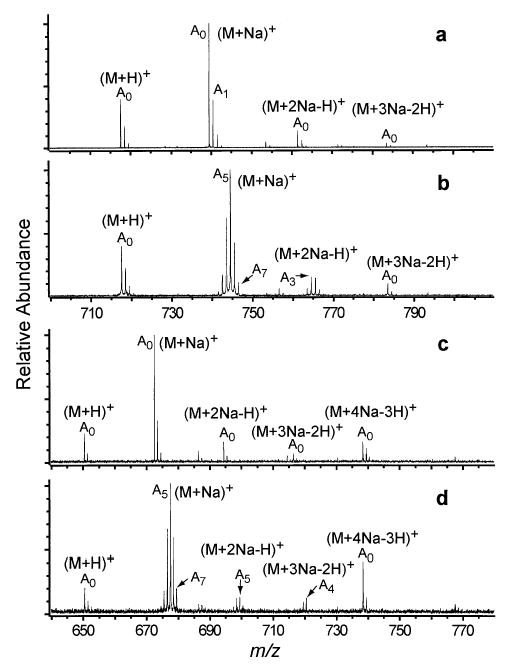
ESI mass spectra of VEPIPY formed from ~1 mM sodium chloride and 1:1 water: methanol solution (a) without exchange and (b) after 50 s exchange with D2O at a pressure of 1 × 10−6 torr and FLEEL (c) without exchange and (d) after 50 s exchange with D2O at a pressure of 1 × 10−6 torr.
H/D Exchange of Multiply Sodiated Ppeptides and Peptide Dimers
A general trend in the extent of exchange with increasing sodium adduction is observed. The peptides and peptide dimers in Table 1 tend to exchange slowly when protonated. With increasing numbers of sodium adducts, exchange increases until maximal exchange is reached, after which, a decrease in exchange occurs. For ions with the maximum number of sodium ions adducted, the exchange is very slow. This trend is shown graphically for VEPIPY monomers and FLEEL dimers (Figure 5a, b). The exception is [(Ala5)2 + H]+ which exchanges slightly faster and to a greater extent than [(Ala5)2 + 2Na − H]+ (Figure 3a, b and Table 1).
Figure 5.
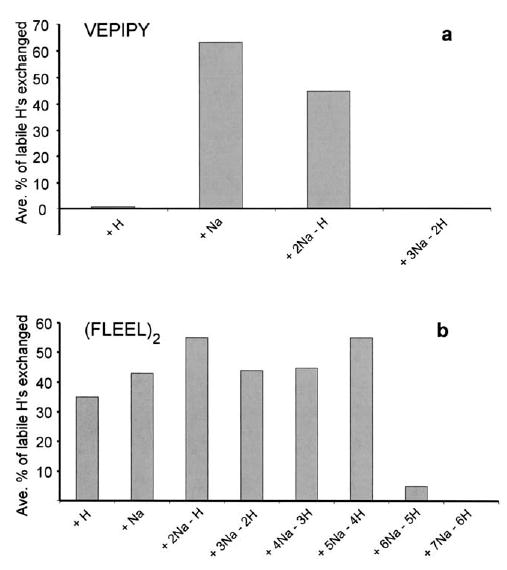
Bar graph of the average percent exchange of labile hydrogens after 50 s H/D exchange with D2O at a pressure of 1 × 10−6 torr for protonated and all of the sodiated ions that form for (a) VEPIPY peptides and (b) FLEEL peptide dimers.
The peptide and peptide dimers that have the maximum number of adducted sodium ions (N + 1) are presumably lacking acidic hydrogens. In order for H/D exchange to occur with these N + 1 adducted ions, D2O would need to exchange directly with labile but non-acidic hydrogens on the peptide. Note that no H/D exchange occurs for fully methyl-esterified peptides and peptide dimers (Table 2) indicating that a carboxylic acid hydrogen plays a crucial role in the H/D exchange that occurs in these ions.
Sodium and Potassium Adducted (YGGFL)2 Ions
To compare the effects of the adduction of a different alkali metal ion on the rate of H/D exchange, (YGGFL)2 ions were formed from an electrospray solution containing 1 mM of both sodium chloride and potassium chloride. Various peptide-metal ion complexes are readily formed, and H/D exchange was done for 50 s at several different D2O pressures (Figure 6a, b, c). Due to overlapping isotopic distributions, it is not possible to determine the fraction of labile hydrogens exchanged for all metal adducted (YGGFL)2 ions at the highest D2O pressure. Nonetheless, for (YGGFL)2 it is clear that dimers having one adducted metal exchange slowly, any two adducted metal ions exchange rapidly, and any three adducted metal ions exchange slowly. The dimer ion species with three adducted potassium ions was formed in low abundance using our experimental conditions.
Figure 6.
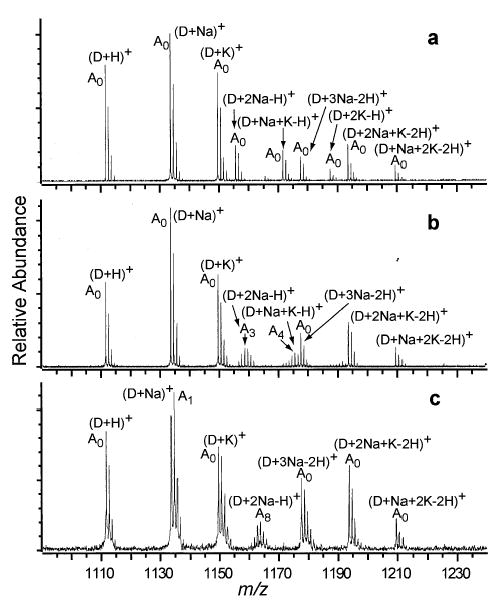
ESI mass spectra of (YGGFL)2 in 1 mM NaCl and KCl and 1:1 water: methanol (a) without exchange, and after 50 s H/D exchange with D2O at a pressure of (b) ~1 × 10−7 torr, and (c) ~1 × 10−6 torr.
Partially Esterified FLEEL Exchange
In addition to fully methyl-esterified FLEEL, which has methyl-esters formed at all three carboxylic acid groups, FLEEL was also partially methyl-esterified by allowing the peptide esterification reaction to proceed for 30 min. prior to quenching. Under these conditions, methyl-esters form at zero to two carboxylic acid groups. The partially esterified FLEEL monomers were exchanged with D2O in the gas phase to determine the effects of removing some of the carboxylic acid groups on the H/D exchange rates of the peptide-sodium ion complexes. FLEEL, with one of the three carboxylic acid groups methyl-esterified, exchanges similarly to VE-PIPY (two carboxylic acid groups). Both exchange slowly in the protonated form, rapidly when one or two sodium ions are adducted (Figure 7a, b, c), and do not exchange when three sodium ions are adducted. FLEEL, with methyl-esters at two of the three carboxylic acid positions exchanges similarly to YGGFL (one carboxylic acid group). Monomers exchange slowly in both the protonated form and with one or two adducted sodium ions.
Figure 7.
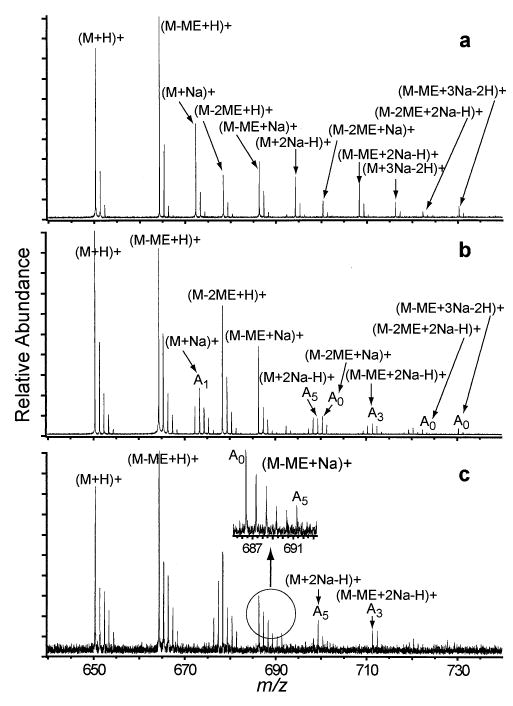
ESI mass spectra of partially esterified FLEEL in 60 mM NaCl and 1:1 water: methanol (a) without exchange and (b) after 50 s exchange with D2O at a pressure of (b) ~1 × 10−7 torr and (c) ~1 × 10−6 torr. The inset shows isomeric separation for FLEEL with one methyl-ester and one noncovalently adducted sodium ion.
A noticeable feature in the exchange spectrum of partially esterified FLEEL is the appearance of a bimodal isotopic distribution for (FLEEL-ME + Na)+ (Figure 7c insert). These results clearly indicate the presence of two or more isomers or conformers. The methyl-esterification reaction is relatively non-specific, so that methyl-esterification can occur at any one of the three carboxylic acids, resulting in potentially three isomers. The positional isomers for methyl-esterification of FLEEL-ME do not appear to be resolvable by H/D exchange. Only a single isotopic pattern is measured for (FLEEL-ME + H)+ and for (FLEEL-ME + 2Na − H)+. Each of these ions also has the potential for three isomers. For the latter ion, each isomer has only two free carboxylic acids available to form ion pairs with the two sodium ions. In contrast, each positional isomer for (FLEEL-ME + Na)+ has two carboxylic acids available to form ion pairs with the adducted sodium ion and thus six different isomers of this ion can be produced. Thus, it seems reasonable that the bimodal isotopic distribution observed for (FLEEL-ME + Na)+ results from different positions of sodium adduction.
Discussion
H/D Exchange Mechanism
On the basis of experimental results with glycine homopolymers and semi-empirical calculations, Beauchamp and coworkers proposed several mechanisms for gas-phase H/D exchange with D2O [12]. The “relay” mechanism [11, 12], which is very similar to a bridging mechanism proposed independently [8], has become a popular way to mechanistically describe and explain H/D exchange data [25, 50]. In the “relay” mechanism, D2O coordinates between a protonated site and a remote basic site, forming two hydrogen bonds. H/D exchange occurs by the transfer of a deuteron from the water to the basic site coincident with the transfer of a proton from the protonated site to the water. Intramolecular deuterium-hydrogen rearrangement subsequently distributes deuterium to other sites on the molecule. Alternatively, a “salt-bridge” mechanism was proposed involving proton transfer from a carboxylic acid to D2O and the stabilization of the resulting carboxylate anion by a nearby charge site [12]. While not conclusive, our results are most consistent with H/D exchange occurring primarily by a “salt-bridge” mechanism for the small peptide-metal complexes listed in Table 1.
Requisite for exchange by the “salt-bridge” mechanism is the presence of charge sites. For these peptides, we observe the complexation of a maximum of N + 1 metal ions, where N is the number of carboxylic acid groups in the peptide or peptide dimer. The maximal value of N is consistent with the first metal ion displacing the proton normally added by the ESI process and then subsequent metal ions displacing carboxylic acid hydrogens and forming ion pairs. Complexation of metal ions provides multiple charge sites that are available to stabilize carboxylate anions formed by proton transfer to D2O. For peptides and peptide dimers in Table 1 that have multiple carboxylic acid groups, the stepwise addition of metal ions typically increases the rate of exchange until a maximum is reached and additional metal ions begin to decrease the rate of exchange, presumably due to a scarcity or absence of acidic protons.
While the complexation of metal ions is expected to facilitate salt-bridge formation and tends to increase the rate of H/D exchange, methyl-esterification of the carboxylic acid positions blocks salt-bridge formation and virtually eliminates H/D exchange for the protonated and sodiated peptides studied (Table 2). This result is consistent with H/D exchange by the “salt-bridge” mechanism as there are no carboxylic acids available for proton transfer in the methyl-esterified peptides. The absence of H/D exchange of the peptide methyl-esters with one adducted sodium is also consistent with the “relay” mechanism and other mechanisms that have been proposed [12] as no acidic protons remain for proton transfer to the exchange reagent.
Several distinct groups of H/D exchange rates are measured for these peptide-metal ion complexes, consistent with exchange by multiple mechanisms. Three of the peptides exchange an average of 2–3 hydrogens after 50 s exchange at ~1 × 10−8 torr which corresponds to average exchange rates of 1–2 × 10−10 cm3/mol. These exchange rates are very similar to three fast-exchanging sites measured for protonated Gly2, Gly3, and Gly3-ME that were attributed to exchange by the “relay” mechanism [12]. The rest of the protonated peptide and peptide-metal ion complexes in Table 1 that exchange more than one hydrogen during 50 s at ~1 × 10−6 torr have average rate constants of 2–5 × 10−12 cm3/molecule. These are approximately two orders of magnitude smaller than the rate constants for exchange processes attributed to the “relay” mechanism. However, rate constants on the order of 2–5 × 10−12 cm3/molecule correspond quite well to rate constants measured for single exchanges of betaine, and protonated Gly1, Gly4, and Gly5 that were proposed to exchange by the “salt-bridge” or “flip-flop” mechanisms [12]. Thus, the measured rate constants, the general increase in exchange rate with the adduction of metal ions and the dramatic decrease in exchange rate upon complete methyl-esterification are all consistent with exchange by the “salt-bridge” mechanism. It should be noted that these results do not rule out exchange by other mechanisms or a combination of mechanisms.
Cooperative Effects of Multiple Carboxylic Acids
The observed results in Tables 1 and 2 are well described by placing all of the protonated peptides and peptide dimers and their metal ion complexes into two categories, those having two or more carboxylic acid groups, and those having less than two carboxylic acid groups. All of the peptides and peptide dimers having two or more carboxylic acid groups undergo rapid H/D exchange when they are either protonated or complexed with one or more metal ions. All of the peptides and peptide dimers that have less than two carboxylic acids exchange no more than one hydrogen with D2O. This classification holds true for all of the species studied including FLEEL that has been partially methyl-esterified. Protonated or metal ion complexed FLEEL with only one remaining carboxylic acid does not exchange with or without adducted sodium ions, while metal ion complexes of FLEEL, with two remaining carboxylic acids, exchanges multiple hydrogens. More data are necessary to determine if this “two are much better than one” mechanism holds true for H/D exchange with D2O for this class of peptides.
Conclusions
The rate and extent of gas-phase H/D exchange with D2O have been measured for a series of peptides with 5–6 amino acids residues, their noncovalent homodimers and all of the peptide-sodium ion complexes that they form. All of the peptides studied have a single basic site at the amino terminus and 1–3 acidic sites. A maximum of N + 1 sodium ions form complexes with these peptides and peptide homodimers, where N is the total number of carboxylic acids. This maximal adduction is consistent with sodium ion displacement of the proton normally added in the electrospray process and all of the acidic carboxylic acid hydrogens. Presumably, the adducted sodium ions form ion pairs with the carboxyl anions. Protonated and sodium adducted peptides having only one carboxylic acid exchange less than one hydrogen during 50 s of exchange at 1 × 10−6 torr. Peptides and peptide dimers with multiple carboxylic acid groups tend to exchange more rapidly when complexed with one or more sodium ions than when protonated. Peptides and peptide dimers with N + 1 adducted sodium ions or with all carboxylic acid groups converted to methyl-esters exchanged on average less than one hydrogen in 50 s at 1 × 10−6 torr. With the same exposure time and pressure, multiple hydrogens are exchanged for sodium complexes of FLEEL peptides with only one esterified carboxylic acid.
Results of these experiments are consistent with H/D exchange of these peptides occurring, at least in part, by a “salt-bridge” mechanism [12] as sodium ion adduction is seen to generally increase the rate of H/D exchange. The presence of metal ions could be expected to assist in the stabilization of carboxylate anions formed by proton transfer from a carboxylic acid to D2O. The average rate constants for the exchange of peptide and peptide-metal ion complexes that undergo multiple exchanges are 1–5 × 10−12 cm3/molecule which are very similar to the rate constants for slow exchange of betaine and Gly4 and Gly5 that were suggested to exchange by a “salt-bridge” mechanism [12]. Three of the peptides exchanged 2–3 hydrogens with average rate constants two orders of magnitude larger, suggesting that H/D exchange may be occurring by multiple mechanisms.
A general goal of gas-phase H/D exchange is to reveal aspects of gas-phase conformation. The experiments presented here suggest that properties not necessarily related to conformation, such as the inclusion of acidic groups in the primary structure, are very significant in determining the rates of H/D exchange with D2O. The adduction of one or more sodium ions also significantly affects the rate of H/D exchange, but the impact of sodium adduction on the conformation of these peptides is not known. Peptide dimers showed exchange that could be either faster or slower than the corresponding monomer despite the fact that the dimers would be expected to have lower surface-accessibility normalized to their mass. Information about the gas-phase conformations of these peptides and their various dimers and adducts could be obtained from ion mobility cross-sectional measurements and molecular modeling [30 –32, 41]. Such studies may help to determine the role of conformation on the rate of H/D exchange measured for these peptides and peptide dimers.
Acknowledgments
The authors gratefully acknowledge the financial support of RC from the University of California Biology Fellows Program and the Howard Hughes Medical Institute’s Undergraduate Biological Science Education Initiative. Mr. Neil Shenvi (University of California, Berkeley) assisted in Matlab calculations for average rate constant determinations. Funding for this work has been provided by the National Institutes of Health (Grant R01-GM64712-01).
References
- 1.Englander SW, Mayne L, Bai Y, Sosnick TR. Hydrogen Exchange: The Modern Legacy of Linderstrom-Lang. Protein Sci. 1997;6:1101–1109. doi: 10.1002/pro.5560060517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith DL, Deng YZ, Zhang ZQ. Probing the Noncovalent Structure of Proteins by Amide Hydrogen Exchange and Mass Spectrometry. J Mass Spectrom. 1997;32:135–146. doi: 10.1002/(SICI)1096-9888(199702)32:2<135::AID-JMS486>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 3.Suckau D, Shi Y, Beu SC, Senko MW, Quinn JP, Wampler FM, McLafferty FW. Coexisting Stable Conformations of Gaseous Protein Ions. Proc Natl Acad Sci USA. 1993;90:790 –793. doi: 10.1073/pnas.90.3.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wood TD, Chorush RA, Wampler FM, Little DP, O’Connor PB, McLafferty FW. Gas-Phase Folding and Unfolding of Cytochrome-c Cations. Proc Natl Acad Sci USA. 1995;92:2451–2454. doi: 10.1073/pnas.92.7.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McLafferty FW, Guan ZQ, Haupts U, Wood TD, Kelleher NL. Gaseous Conformational Structures of Cytochrome c. J Am Chem Soc. 1998;120:4732–4740. [Google Scholar]
- 6.Winger BE, Light-Wahl KJ, Rockwood AL, Smith RD. Probing Qualitative Conformation Differences of Multiply Protonated Gas-Phase Proteins via H/D Isotopic Exchange with D2O. J Am Chem Soc. 1992;114:5897–5898. [Google Scholar]
- 7.Wang JR, Cassady CJ. Effects of Disulfide Linkages on Gas-Phase Reactions of Small Multiply Charged Peptide Ions. Int J Mass Spectrom. 1999;183:233–241. [Google Scholar]
- 8.Gard E, Green MK, Bregar J, Lebrilla CB. Gas-Phase Hydrogen/Deuterium Exchange as a Molecular Probe for the Interaction of Methanol and Protonated Peptides. J Am Soc Mass Spectrom. 1994;5:623–631. doi: 10.1016/1044-0305(94)85003-8. [DOI] [PubMed] [Google Scholar]
- 9.Green MK, Penn SG, Lebrilla CB. The Complexation of Protonated Peptides with Saccharides in the Gas Phase Decreases the Rates of Hydrogen/Deuterium Exchange Reactions. J Am Soc Mass Spectrom. 1995;6:1247–1251. doi: 10.1016/1044-0305(95)00632-X. [DOI] [PubMed] [Google Scholar]
- 10.Green MK, Lebrilla CB. The Role of Proton-Bridged Intermediates in Promoting Hydrogen/Deuterium Exchange in Gas-Phase Protonated Diamines, Peptides, and Proteins. Int J Mass Spectrom. 1998;175:15–26. [Google Scholar]
- 11.Campbell S, Rodgers MT, Marzluff EM, Beauchamp JL. Structural and Energetic Constraints on Gas-Phase Hydrogen/Deuterium Exchange Reactions of Protonated Peptides with D2O, CD3OD, CD3CO2D, and ND3. J Am Chem Soc. 1994;116:9765–9766. [Google Scholar]
- 12.Campbell S, Rodgers MT, Marzluff EM, Beauchamp JL. Deuterium Exchange Reactions as a Probe of Biomolecule Structure. Fundamental Studies of Gas Phase H/D Exchange Reactions of Protonated Glycine Oligomers with D2O, CD3OD, CD3CO2D, and ND3. J Am Chem Soc. 1995;117:12840 –12854. [Google Scholar]
- 13.Lee SW, Lee HN, Kim HS, Beauchamp JL. Selective Binding of Crown Ethers to Protonated Peptides can be Used to Probe Mechanisms of H/D Exchange and Collision-Induced Dissociation Reactions in the Gas Phase. J Am Chem Soc. 1998;120:5800 –5805. [Google Scholar]
- 14.Gur EH, Dekoning LJ, Nibbering NMM. The Bimolecular Hydrogen/Deuterium Exchange Behavior of Protonated Alkyl Dipeptides in the Gas-Phase. J Am Soc Mass Spectrom. 1995;6:466 –477. doi: 10.1016/1044-0305(95)00189-K. [DOI] [PubMed] [Google Scholar]
- 15.Cassady CJ, Carr SR. Elucidation of Isomeric Structures for Ubiquitin M + 12H (12+) Ions Produced by Electrospray Ionization Mass Spectrometry. J Mass Spectrom. 1996;31:247–254. doi: 10.1002/(SICI)1096-9888(199603)31:3<247::AID-JMS285>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 16.Zhang X, Ewing NP, Cassady CJ. Effects of Basic Site Proximity on Deprotonation and Hydrogen/Deuterium Exchange Reactions for Model Dodecapeptide Ions Containing Lysine and Glycine. Int J Mass Spectrom. 1998;175:159–171. [Google Scholar]
- 17.Kaltashov IA, Doroshenko VM, Cotter RJ. Gas Phase Hydrogen/Deuterium Exchange Reactions of Peptide Ions in a Quadrupole Ion Trap Mass Spectrometer. Proteins. 1997;28:53–58. [PubMed] [Google Scholar]
- 18.Freitas MA, Marshall AG. Rate and Extent of Gas-Phase Hydrogen/Deuterium Exchange of Bradykinins: Evidence for Peptide Zwitterions in the Gas Phase. Int J Mass Spectrom. 1999;183:221–231. [Google Scholar]
- 19.He F, Marshall AG, Freitas MA. Assignment of Gas-Phase Dipeptide Amide Hydrogen Exchange Rate Constants by Site-Specific Substitution: GlyGly. J Phys Chem B. 2001;105:2244 –2249. [Google Scholar]
- 20.Heck AJR, Jorgensen TJD, O’Sullivan M, von Raumer M, Derrick PJ. Gas-Phase Noncovalent Interactions Between Vancomycin-Group Antibiotics and Bacterial Cell-Wall Precursor Peptides Probed by Hydrogen/Deuterium Exchange. J Am Soc Mass Spectrom. 1998;9:1255–1266. [Google Scholar]
- 21.Levy-Seri E, Koster G, Kogan A, Gutman K, Reuben BG, Lifshitz C. An Electrospray Ionization-Flow Tube Study of H/D Exchange in Protonated Bradykinin. J Phys Chem A. 2001;105:5552–5559. [Google Scholar]
- 22.Ustyuzhanin P, Kogan A, Reuben BG, Lifshitz C. An Electrospray-Ionization–Flow-Tube Study of H/D Exchange in Protonated Leucine-Enkephalin. Int J Chem Kinet. 2001;33:707–714. [Google Scholar]
- 23.Kogan A, Ustyuzhanin P, Reuben BG, Lifshitz C. Hydrogen/Deuterium Exchange of Monomers and Dimers of Leucine Enkephalin. Int J Mass Spectrom. 2002;213:1–4. [Google Scholar]
- 24.Solouki T, Fort RC, Alomary A, Fattahi A. Gas Phase Hydrogen/Deuterium Exchange Reactions of a Model Peptide: FT-ICR and Computational Analyses of Metal Induced Conformational Mutations. J Am Soc Mass Spectrom. 2001;12:1272–1285. doi: 10.1016/S1044-0305(01)00315-4. [DOI] [PubMed] [Google Scholar]
- 25.Mao D, Douglas DJ. H/D Exchange of Gas Phase Bradykinin Ions in a Linear Quadrupole Ion Trap. J Am Soc Mass Spectrom. 2003;14:85–94. doi: 10.1016/S1044-0305(02)00818-8. [DOI] [PubMed] [Google Scholar]
- 26.Loo RRO, Smith RD. Investigation of the Gas-Phase Structure of Electrosprayed Proteins Using Ion–Molecule Reactions. J Am Soc Mass Spectrom. 1994;5:207–220. doi: 10.1016/1044-0305(94)85011-9. [DOI] [PubMed] [Google Scholar]
- 27.Loo RRO, Winger BE, Smith RD. Proton-Transfer Reaction Studies of Multiply-Charged Proteins in a High Mass-to-Charge Ratio Quadrupole Mass-Spectrometer. J Am Soc Mass Spectrom. 1994;5:1064 –1071. doi: 10.1016/1044-0305(94)85067-4. [DOI] [PubMed] [Google Scholar]
- 28.Williams ER. Proton Transfer Reactivity of Large Multiply Charged Ions. J Mass Spectrom. 1996;31:831–842. doi: 10.1002/(SICI)1096-9888(199608)31:8<831::AID-JMS392>3.0.CO;2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gross DS, Schnier PD, Rodriguez-Cruz SE, Fagerquist CK, Williams ER. Conformations and Folding of Lysozyme Ions in Vacuo. Proc Natl Acad Sci USA. 1996;93:3143–3148. doi: 10.1073/pnas.93.7.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wyttenbach T, vonHelden G, Bowers MT. Gas-Phase Conformation of Biological Molecules: Bradykinin. J Am Chem Soc. 1996;118:8355–8364. [Google Scholar]
- 31.Clemmer DE, Jarrold MF. Ion Mobility Measurements and Their Applications to Clusters and Biomolecules. J Mass Spectrom. 1997;32:577–592. [Google Scholar]
- 32.Kinnear BS, Hartings MR, Jarrold MF. The Energy Landscape of Unsolvated Peptides: Helix Formation and Cold Denaturation in Ac-A(4)G(7)A4 + H+ J Am Chem Soc. 2002;124:4422–4431. doi: 10.1021/ja012150v. [DOI] [PubMed] [Google Scholar]
- 33.Valentine SJ, Anderson JG, Ellington AD, Clemmer DE. Disulfide-Intact and -Reduced Lysozyme in the Gas Phase: Conformations and Pathways of Folding and Unfolding. J Phys Chem B. 1997;101:3891–3900. [Google Scholar]
- 34.Badman ER, Hoaglund-Hyzer CS, Clemmer DE. Monitoring Structural Changes of Proteins in an Ion Trap Over Similar to 10 –200 ms: Unfolding Transitions in Cytochrome c Ions. Anal Chem. 2001;73:6000 –6007. doi: 10.1021/ac010744a. [DOI] [PubMed] [Google Scholar]
- 35.Guevremont R, Purves RW. High Field Asymmetric Waveform Ion mobility Spectrometry-Mass Spectrometry: An Investigation of Leucine Enkephalin ions Produced by Electrospray Ionization. J Am Soc Mass Spectrom. 1999;10:492–501. doi: 10.1016/S1044-0305(99)00016-1. [DOI] [PubMed] [Google Scholar]
- 36.Purves RW, Barnett DA, Guevremont R. Separation of Protein Conformers Using Electrospray-High Field Asymmetric Waveform Iion Mobility Spectrometry-Mass Spectrometry. Int J Mass Spectrom. 2000;197:163–177. [Google Scholar]
- 37.Hunt DF, Sethi SK. Gas-Phase Ion-Molecule Isotope-Exchange Reactions—Methodology for Counting Hydrogen-Atoms in Specific Organic Structural Environments by Chemical Ionization Mass-Spectrometry. J Am Chem Soc. 1980;102:6953–6963. [Google Scholar]
- 38.Ausloos P, Lias SG. Thermoneutral Isotope Exchange-Reactions of Cations in the Gas Phase. J Am Chem Soc. 1981;103:3641–3647. [Google Scholar]
- 39.Gard E, Willard D, Bregar J, Green MK, Lebrilla CB. Site-Specificity in the H/D Exchange Reactions of Gas-Phase Protonated Amino-Acids with CH3OD. Org Mass Spectrom. 1993;28:1632–1639. [Google Scholar]
- 40.Reid GE, O’Hair RAJ, Styles ML, McFadyen WD, Simpson RJ. Gas Phase Ion–Molecule Reactions in a Modified Ion Trap: H/D Exchange of Noncovalent Complexes and Coordinatively Unsaturated Platinum Complexes. Rapid Commun Mass Spectrom. 1998;12:1701–1708. [Google Scholar]
- 41.Wyttenbach T, Bushnell JE, Bowers MT. Salt-Bridge Structures in the Absence of Solvent? The Case for the Oligoglycines. J Am Chem Soc. 1998;120:5098 –5103. [Google Scholar]
- 42.Reyzer ML, Brodbelt JS. Gas-phase H/D Exchange Reactions of Polyamine Complexes: (M + H)(+), [M plus alkali metal(+)], and (M + 2H)(2+) J Am Soc Mass Spectrom. 2000;11:711–721. doi: 10.1016/S1044-0305(00)00142-2. [DOI] [PubMed] [Google Scholar]
- 43.Jurchen JC, Williams ER. Origin of Asymmetric Charge Partitioning in the Dissociation of Gas-Phase Protein Homodimers. J Am Chem Soc. 2003;125:2817–2826. doi: 10.1021/ja0211508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grese RP, Cerny RL, Gross ML. Metal–Ion Peptide Interactions in the Gas Phase—A Tandem Mass-Spectrometry Study of Alkali-Metal Cationized Peptides. J Am Chem Soc. 1989;111:2835–2842. [Google Scholar]
- 45.Hu PF, Gross ML. Gas-Phase Interactions of Transition-Metal Ions and Dipeptides and Tripeptides—A Comparison with Alkaline-Earth-Metal–Ion Interactions. J Am Chem Soc. 1993;115:8821–8828. [Google Scholar]
- 46.Schnier PD, Price WD, Jockusch RA, Williams ER. Blackbody Infrared Radiative Dissociation of Bradykinin and Its Analogues: Energetics, Dynamics, and Evidence for Salt-Bridge Structures in the Gas Phase. J Am Chem Soc. 1996;118:7178 –7189. doi: 10.1021/ja9609157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jockusch RA, Price WD, Williams ER. Structure of Cationized Arginine (Arg · M+, M = H, Li, Na, K, Rb, and Cs) in the Gas Phase: Further Evidence for Zwitterionic Arginine. J Phys Chem A. 1999;103:9266 –9274. doi: 10.1021/jp9931307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wyttenbach T, Witt M, Bowers MT. On the Stability of Amino Acid Zwitterions in the Gas Phase: The Influence of Derivatization, Proton Affinity, and Alkali Ion Addition. J Am Chem Soc. 2000;122:3458 –3464. [Google Scholar]
- 49.Gross DS, Williams ER. Structure of Gramicidin S (M + H + X)(2+) Ions (X = Li, Na, K) Probed by Proton Transfer Reactions. J Am Chem Soc. 1996;118:202–204. [Google Scholar]
- 50.Wyttenbach T, Bowers MT. Gas Phase Conformations of Biological Molecules: The Hydrogen/Deuterium Exchange Mechanism. J Am Soc Mass Spectrom. 1999;10:9–14. [Google Scholar]