

Abstract
It is increasingly accepted that steroidal receptor coregulators may also function in the cytoplasmic compartment. Proline-, glutamic acid–, and leucine-rich protein-1 (PELP1) is a novel coregulator that plays a role in both the genomic and extranuclear actions of estrogen receptors (ER) in hormonally responsive tissues. In this study using breast tumor arrays, we found that PELP1 was localized only in the cytoplasm in 58% of the PELP1-positive breast tumors. To help explain the significance of the cytoplasmic localization of PELP1 in human breast tumors, we created a mutant protein that was expressed only in the cytoplasm (PELP1-cyto) and then generated a model system wherein MCF-7 breast cancer cells were engineered to specifically express this mutant. We found that PELP1-cyto cells were hypersensitive to estrogen but resistant to tamoxifen. PELP1-cyto cells, but not parental MCF-7 cells, formed xenograft tumors in nude mice. In addition, PELP1-cyto cells exhibited increased association of PELP1 with Src, enhanced mitogen-activated protein kinase (MAPK) activation, and constitutive activation of AKT. The altered localization of PELP1 was sufficient to trigger the interaction of PELP1 with the p85 subunit of phosphatidylinositol-3-kinase (PI3K), leading to PI3K activation. In addition, PELP1 interacted with epidermal growth factor receptors and participated in growth factor–mediated ER transactivation functions. Our results suggest that the altered localization of PELP1 modulates sensitivity to antiestrogens, potentiates tumorigenicity, presumably via the stimulation of extranuclear estrogen responses, such as the activation of MAPK and AKT, and also enhance cross-regulation of ER transactivation activity by growth factors.
Introduction
The estrogen receptor (ER), a ligand-dependent transcription factor that modulates the transcription of a number of genes and contributes to genomic responses, has been implicated in the progression of breast cancer, and this is borne out by the finding that ~60% to 70% of breast tumors are ER-positive at presentation (1, 2). Although antiestrogens and selective ER modulators (SERM) are effective in curbing the progression of ER-positive breast tumors to more invasive phenotypes (3), many patients with metastatic breast tumors eventually become resistant to this treatment (4). Several mechanisms have been proposed to explain this resistance to hormonal therapy, including the expression of variant or mutant ER, the ligand-independent activation of ER, the adaptation of tumors to lower concentrations of estrogen, and pharmacologic alterations (4, 5). Emerging data also suggest that ER coregulators play a role in hormonal responsiveness and tumor progression (6, 7). However, the causes of ER coregulator–linked resistance to hormonal therapy and ways to interfere with this phenomenon remain elusive.
The answer may be in the fact that, in addition to its well-studied nuclear functions, ER also participates in cytoplasmic and membrane-mediated signaling events (nongenomic signaling; refs. 8, 9). Such nongenomic signaling has been linked to rapid responses to estrogen and generally involves the stimulation of the Src kinase, mitogen-activated protein kinase (MAPK), phosphatidylinositol-3-kinase (PI3K), and protein kinase C pathways in the cytosol (10, 11). Further, ER-activated nongenomic pathways have been shown to modify ER or its coactivators by phosphorylation, resulting in the altered topology of ER and its coregulator proteins and eventually leading to ligand-independent activation or differential responses to SERMs (9, 12).
Other factors implicated in the development and progression of breast cancer are deregulated epidermal growth factor receptor (EGFR) signaling and the constitutive activation of cytosolic pathways (MAPK, PI3K, and AKT; ref. 13). Now, there is also emerging evidence that resistance to endocrine therapies may stem from complex interactions between ER and EGFR signaling components (14). The phosphorylation of ER and its associated coregulatory proteins has been suggested as one such mechanism by which growth factor signaling contributes to hormonal resistance (15). However, very little is known about the molecular mechanisms that lead to EGFR-ER crosstalk and the molecular adaptors that facilitate EGFR-ER signaling crosstalk.
Proline-, glutamic acid–, and leucine-rich protein-1 (PELP1)/modulator of nongenomic activity of estrogen receptor (MNAR) is a novel ER coactivator (16) that plays an important role in the genomic (17) and nongenomic actions of ER (18), and promotes cell proliferation by sensitizing cells to G1-to-S progression (19). PELP1 expression is up-regulated by estradiol (E2)-ER signaling and differentially modulated by SERMs (20). PELP1 is widely deregulated in hormone-responsive cancers, including breast and endometrial cancers (16, 21). Although PELP1 is predominantly localized in the nucleus in hormonally responsive tissues (16, 17), recent studies have suggested that under certain conditions, PELP1/MNAR could be exclusively localized in the cytoplasm of cancer cells (21). However, the functional implications of the localization of PELP1 are unknown. Therefore, in the present study, we investigated the localization and functional consequences of PELP1 in human breast tumor specimens. We found that PELP1 was localized in the cytoplasm only in 58% of PELP1-positive tumors. To mimic this situation to gain further insight into the functional consequences of this event, we generated a novel MCF-7 model cell line to specifically express PELP1 in the cytoplasm. We found that the cytoplasmic localization of PELP1 made these cells hypersensitive to estrogen but resistant to SERMs and promoted tumorigenesis in nude mice. Our findings suggest that the altered localization of the ER coactivator PELP1 might lead to the excessive stimulation of cytosolic signaling pathways, thereby causing hormonal independence and increasing tumorigenic potential of breast cancer cells.
Materials and Methods
Cell cultures and reagents
MCF-7, HeLa, and COS-1 cells were maintained in DMEM-F12 (1:1) supplemented with 10% FCS. Antibodies against vinculin, actin, and the steroid hormone 17β-estradiol were purchased from Sigma Chemical Co. (St. Louis, MO). Antibodies against ERα and the phosphotyrosine (pY20) were purchased from Lab Vision (Fremont, CA). Anti–T7-epitope antibody was purchased from Novagen (Milwaukee, WI). Antibodies against AKT, phospho-AKT, MAPK, phospho-MAPK, phospho-Src Tyr416, and phospho-ER-Ser167, ER-Ser118 were purchased from Cell Signaling (Beverly, MA). Antibody for Src and specific Src kinase inhibitor PP2 was purchased from Calbiochem (La Jolla, CA). Antibodies for EGFR were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Charcoal-stripped serum (DCC serum), ICI 182,780, and LY294002 were purchased from Sigma.
Human samples and tumor array staining
Tissue sample from patients who had undergone routine surgery for breast cancer were snap frozen in liquid nitrogen and stored at −80°C in the M. D. Anderson Breast Core Pathology Laboratory. Paraffin-embedded archival breast tissue specimen array was obtained from the pathology core at University of Texas M. D. Anderson Cancer Center. The utilization of human tissue samples and array used in this study was approved by M. D. Anderson Cancer Center institutional human research committee. All tissue classifications were verified by light microscopic examination of H&E-stained slides by a breast pathologist (A.A. Sahin). For immunohistochemical detection of PELP1, sections were deparaffinized with xylene and rehydrated using graded ethanol. The tumor array section was boiled for 10 minutes in 0.01 mol/L citrate buffer and cooled for 30 minutes at room temperature to expose antigenic epitopes. The tumor array was blocked with 2% normal goat serum in 1% bovine serum albumin and PBS for 30 minutes. The tumor array section was incubated with polyclonal rabbit antihuman-PELP1 antiserum at a dilution of 1:500 and incubated overnight at room temperature. PELP1 antibody was developed in our laboratory, well characterized, and used in immunohistochemistry in earlier published studies (16, 21). The sections were washed thrice with 0.05% Tween in PBS for 10 minutes, incubated with secondary antibody for 1 hour, washed thrice with 0.05% Tween in PBS for 10 minutes, developed with 3,3′-diaminobenzidine-H2O2, and then counterstained with Mayer’s hematoxylin. Negative controls were done by replacing the primary antibody with control rabbit IgG or peptide-absorbed PELP1 antibody. The finding that no cells or <10% of cells were immunoreactive was considered as a negative, and the finding that >10% of the cells were immunoreactive was considered positive.
Generation of PELP1 cytoplasmic mutant cells
PELP1 cytoplasmic mutant (PELP1-cyto) lacking nuclear localization signal was generated using a Quick Change kit (Stratagene, La Jolla, CA) by mutating PELP1 amino acid 495KKLK498 to EELE using T7-tagged wild-type PELP1 as a template. The sequence of the primer used for mutagenesis was 5′-CCTAGCGCCCCCGAGGAGCTAGAGCTGGATGTG-3′. MCF-7 cells stably expressing PELP1-cyto were generated by transfecting PELP1-cyto using FuGENE-6 transfection reagent (Roche Molecular Biochemicals, Indian-apolis, IN). Stable clones were selected using G418 selection (1 mg/mL).
Tissue and cell extracts, immunoblotting, immunoprecipitation, and kinase assays
Tissue and cell lysates were prepared using Triton X-100 buffer [50 mmol/L Tris-HCl (pH 7.5), 100 mmol/L NaCl, 0.5% Triton X-100, 1× protease inhibitor mixture, 1 mmol/L sodium vanadate] for 15 minutes on ice. The lysates were centrifuged in an Eppendorf centrifuge at 4°C for 15 minutes. Cell lysates containing equal amounts of protein (~200 μg) were resolved on SDS-polyacrylamide gels (8% acrylamide), transferred to nitrocellulose membranes, probed with the appropriate antibodies, and developed using either the enhanced chemiluminescence method or the alkaline phosphatase–based color reaction method. Immunoprecipitation was done for 2 hours at 4°C using 1 μg of antibody per milligram of protein. For PI3K assay, cell lysates (1 mg) were immunoprecipitated with antiphosphotyrosine monoclonal antibody (mAb) PY20 (Labvision, Fremont, CA) and subjected to in vitro kinase reaction in 50 μL of kinase buffer containing 0.2 mg/mL phosphatidylinositol (Sigma) and 20 μCi of [γ-32P]ATP and 20 mmol/L MgCl2. The reaction products were separated on TLC plates using chloroform/methanol/ammonium hydroxide/water (87:76:10:14) buffer and visualized by autoradiography.
Cell proliferation, soft agar, and tumorigenesis assays
For the cell proliferation assays, cells were grown in phenol red–free medium supplemented with 5% DCC serum for 48 hours, and then estrogen or tamoxifen was added. The proliferation rate of the cells was measured by counting them in a Beckman Coulter Counter as previously described (19). Soft-agar colony-growth assays were done as previously described (22). Briefly, 1 mL of 0.6% Difco agar in DMEM supplemented with 5% DCC serum and insulin was layered onto tissue culture plates. Test cells (1 × 104) mixed with 1 mL of 0.36% bactoagar solution in DMEM were layered on top of the 0.6% bactoagar layer. The plates were incubated at 37°C in 5% CO2 for 21 days. For tumorigenesis studies, 5 × 107 cells were implanted s.c. into the mammary fat pads of eight nude mice as previously described (23) and allowed to grow for 8 weeks. Tumor size was then measured.
Reporter gene assay
For reporter gene transient transfections, COS-1 cells were cultured for 48 hours in minimal essential medium without phenol red–containing 5% DCC serum. Estrogen response element (ERE)-luciferase reporter constructs were cotransfected with ER in the presence or absence of pcDNA vector, wild-type PELP1 (PELP1-WT), or PELP1-cyto expression plasmids using FuGENE-6 according to the instructions of the manufacturer. Twenty-four hours later, cells were treated with EGF for 12 hours. Cells were then lysed with passive lysis buffer, and the luciferase assay was done using a luciferase reporter assay kit (Promega, Madison, WI). The total amount of DNA used in the transfections was kept constant by adding a parental vector. Each transfection was carried out in six-well plates in triplicate wells.
Immunofluorescence and confocal microscopy
The cellular localization of PELP1 was determined using indirect immunofluorescence as previously described (17).
Statistical analysis
Statistical analysis was done using Student’s t test and values with P < 0.05 were considered statistically significant.
Results
PELP1 localization is altered in a subset of breast tumors
To examine the localization of PELP1 in human breast tumors, we stained 62 breast tumor specimens using a well-characterized PELP1 antibody (Fig. 1A; refs. 16, 21). The staining intensity was scored in both nuclear and cytoplasmic compartments. Normal breast epithelium (n = 8) was used as a control. All normal breast epithelium stained positively; PELP1 staining was strong in the nucleus and modest in the cytoplasm. In tumors, however, PELP1 staining intensity and localization was variable. PELP1 staining was observed in 38 (61%) of the 62 analyzed tumors. In 22 (58%) of the 38 PELP1-positive tumors, PELP1 was localized in the cytoplasm only; in one tumor (3%), intense nuclear staining was observed and in 15 tumors (39%), both nuclear and cytoplasmic staining were observed (Table 1). We then correlated the expression and localization of PELP1 with the clinical ER status of these tumors. ER staining was present in 19 (50%) of 38 PELP1-positive tumors. ER expression was observed in the one tumor that showed nuclear PELP1 expression, in 8 of 22 (36%) cytoplasmic PELP1-expressing tumors and in 10 of 15 (66.6%) nuclear and cytoplasmic PELP1-expressing tumors. These results suggest that in a subset of breast tumors, PELP1 is localized in the cytoplasm only and that a substantial number of breast tumors coexpress PELP1 and ER.
Figure 1.
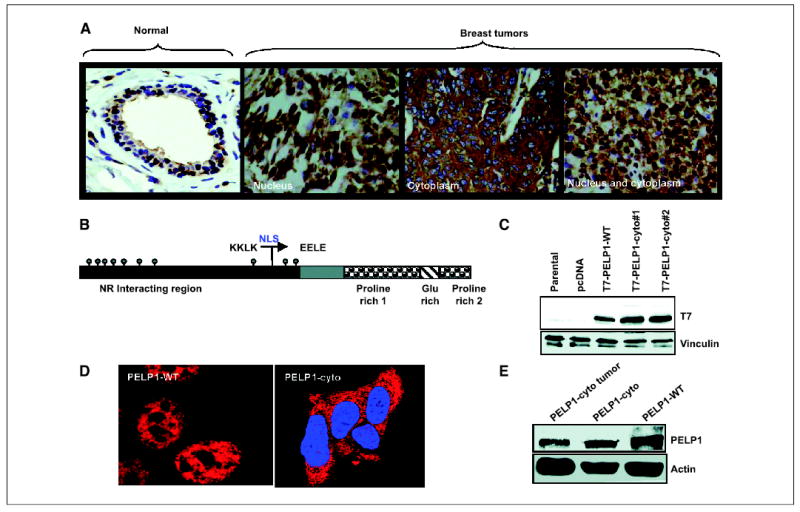
Localization of PELP1 in human breast tissues and breast cancer model cells. A, a tissue array containing samples of 62 breast tumors was stained with PELP1 antibody. Three representative PELP1 localizations (nuclear, cytoplasmic, and nuclear and cytoplasmic) observed in breast tumors are shown. Magnification, ×200. Normal breast tissue was used as a control. B, schematic representation of PELP1-cyto construct. C, total cellular lysates were obtained from MCF-7 cells or MCF-7 clones stably expressing pcDNA, PELP1-WT, and PELP1-cyto. Expression of T7-tagged PELP1-cyto was analyzed by Western blotting using anti–T7-epitope monoclonal antibody (T7 mAb). Vinculin was used as a loading control. D, MCF-7 cells expressing PELP1-WT or PELP1-cyto were cultured in DCC medium and fixed in methanol. The localization of T7-tagged PELP1 in these clones was analyzed by confocal microscopy using T7 mAb. E, total lysates obtained from breast tumors or MCF-7 clones that stably express PELP1-WT and PELP1-cyto were analyzed by Western blotting using PELP1 antibody (top) and subsequently reprobed with an actin antibody, which served as a loading control (bottom).
Table 1.
Summary of the immunoreactive staining of PELP1 in breast tumors
| No staining | Positive staining | Nuclear staining | Cytoplasmic staining | Nuclear and cytoplasmic staining |
|---|---|---|---|---|
| 38.7% (24 of 62) | 61.3% (38 of 62) | 2.63% (1 of 38) | 57.8% (22 of 38) | 39.5% (15 of 38) |
Generation of PELP1-cyto model cells
To examine the functional consequences of the cytoplasmic localization of PELP1, we next generated a mutant of PELP1 (PELP1-cyto) in which the nuclear localization signal was mutated from KKLK to EELE (Fig. 1B). Preliminary analysis using confocal microscopy showed that PELP1-cyto was predominantly localized in the cytosol. Using this construct, we generated MCF-7 cells that stably expressed PELP1-cyto. To avoid potential clonal variations, we developed two pooled clones. Western blotting showed expression of T7-tagged PELP1-cyto in these clones (Fig. 1C). We used a previously characterized PELP1-WT pooled clone as the control (19). Confocal scanning analysis showed that PELP1-cyto was localized to the cytoplasm, whereas PELP1-WT was localized to the nucleus, as expected (Fig. 1D). To examine whether the cytoplasmic PELP1 observed in the tumors corresponded to the full-length PELP1, we did Western blot analysis of the lysates of the tumors that expressed PELP1 in the cytoplasm only. Lysates from MCF-7 cells expressing both PELP1-WT and PELP1-cyto were used as controls. Results showed that PELP1 isolated from lysates of the tumors that exclusively expressed PELP1 in the cytoplasm migrated in a similar fashion as PELP1-WT and PELP1-cyto that were expressed in MCF-7 cells (Fig. 1E). These findings suggest that PELP1 expressed in the cytoplasm of tumors represents the full-length PELP1.
Cytoplasmic retention enhances PELP1 interactions with Src kinase
Recent studies have shown that in addition to its suggested nuclear functions (17), PELP1 participates in nongenomic signaling activities, such as activation of Src (18, 24). Therefore, we examined whether the localization of PELP1 to the cytoplasmic compartment, a physiologic situation existing in a subset of breast tumors, promotes PELP1 interaction with Src. Results from confocal microscopy showed that a substantial amount of PELP1 was localized in the nuclear compartment of PELP1-WT–expressing cells and that E2 treatment stimulated colocalization of PELP1 with Src at the membrane in some cells (Fig. 2A, top). In contrast, in PELP1-cyto–expressing cells, E2 treatment promoted substantial colocalization of PELP1-cyto with Src both at the membrane and in the cytoplasm (Fig. 2A, bottom). Another study found that PELP1 interaction with Src promotes Src activation (24); this raised the possibility that the cytoplasmic localization of PELP1 might provide an opportunity for PELP1 to interact with Src, thereby leading to enhanced Src kinase activation. To test whether PELP1-cyto interacts with Src kinase, immunoprecipitation was done by using total cellular lysates from pcDNA- and PELP1-cyto–expressing cells. Results showed that PELP1-cyto interacts with Src kinase (Fig. 2B, left). To confirm that overexpression of PELP1-cyto activates Src kinase, we did Western blotting with a phosphospecific antibody (phospho-Src Tyr416) that recognizes activated Src kinase. This showed that PELP1-cyto–expressing cells increased the activation of Src kinase compared with pcDNA-expressing clones (Fig. 2B, right). These findings suggest that PELP1 cytolocalization promotes PELP1 interaction with Src kinase. Because PELP1-WT functions as a scaffolding protein for the coupling of ER with Src kinase, we then investigated whether PELP1-cyto also interacts with ER. The immunoprecipitation of PELP1-cyto showed that PELP1-cyto could interact with ER (Fig. 2C).
Figure 2.
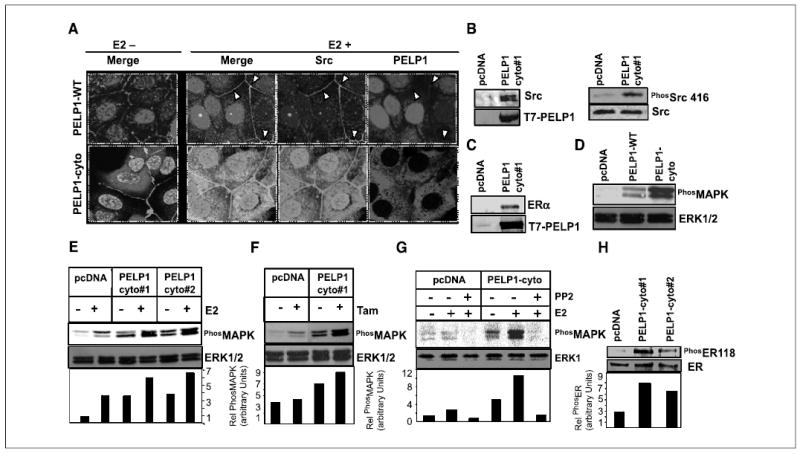
PELP1 cytoplasmic retention promotes PELP1-Src kinase interaction and MAPK activation. A, MCF-7 cells stably expressing T7-tagged PELP1-WT and PELP1-cyto were treated with or without E2 (10−9 mol/L) for 10 minutes. Cells were fixed, and the colocalization of PELP1 (red) and Src (green) was analyzed by confocal microscopy. Colocalization of PELP1 and Src is shown in yellow. B, pcDNA-and PELP1-cyto clones were cultured in 5% DCC serum. Total cellular lysates (2 mg) were subjected to immunoprecipitation using T7 antibody followed by Western blot analysis with Src antibody (left). Total lysate (100 μg) was analyzed by Western blotting using phospho-Src Tyr416 antibody (right). C, total cellular lysate (2 mg) from pcDNA or PELP1-cyto clones was subjected to immunoprecipitation by using T7 antibody followed by Western blot analysis with ERα antibody. D, MCF-7 clones expressing pcDNA, PELP1-WT, and PELP1-cyto were cultured in 5% serum, and total lysates were subjected to Western blotting by using phospho-MAPK antibody. Blots were stripped and reprobed with total MAPK as the loading control. E and F, MCF-7 clones expressing pcDNA or PELP1-cyto were treated with E2 (10−9 mol/L) or tamoxifen (10−8 mol/L) and total lysates were subjected to Western blotting by using a phospho-MAPK antibody. Blots were stripped and reprobed with total MAPK as the loading control. G, pcDNA and PELP1-cyto clones were pretreated with Src kinase inhibitor PP2 30 minutes before the addition of E2 (10−9 mol/L). Activation status of MAPK pathway was analyzed by Western blotting. H, MCF-7 clones expressing pcDNA and PELP1-cyto were cultured in 5% serum, and total lysates were subjected to Western blotting using phospho-ER Ser118 and subsequently reprobed with total ER.
Cytoplasmic retention of PELP1 promotes activation of the mitogen-activated protein kinase pathway
Next, we examined whether the interaction we observed between PELP1 and Src in the PELP1-cyto–expressing clones leads to the functional activation of MAPK, a well-established downstream end point of ER nongenomic signaling (10, 11). Western blot analysis of cells grown in 5% serum revealed that PELP1-WT and PELP1-cyto–expressing cells showed increased activation of the MAPK pathway compared with pcDNA-expressing clones. However, the PELP1-cyto–expressing clones exhibited significantly more MAPK activation than did the PELP1-WT clones (Fig. 2D). When these clones were cultured in DCC serum, the PELP1-cyto–expressing clones exhibited increased basal MAPK activity compared with pcDNA-vector–overexpressing clones, and E2 treatment substantially enhanced MAPK activation in PELP1-cyto clones (Fig. 2E). PELP1-cyto–expressing cells also showed greater MAPK activity in response to tamoxifen treatment than did pcDNA-expressing clones (Fig. 2F). To confirm that the increased MAPK activation seen in PELP1-cyto clones was due to PELP1-cyto interactions with Src kinase, we pretreated cells with PP2, a specific Src kinase inhibitor. PP2 reduced the basal as well as E2-induced MAPK activation seen in the PELP1-cyto clones (Fig. 2G). These results suggest that PELP1 cytoplasmic localization enhances MAPK activation via its interactions with Src kinase. Because MAPK activation has been shown to phosphorylate ER on Ser118, we then examined whether ER was phosphorylated on ER-Ser118 in the PELP1-cyto clones. Western blot analysis of cells grown in 5% serum revealed that PELP1-cyto–expressing clones showed increased ER-Ser118 phosphorylation compared with pcDNA-expressing clones (Fig. 2H).
Cytoplasmic retention of PELP1 promotes constitutive activation of the AKT pathway
Because the activation of PI3K is another recognized nongenomic signaling activator of ER (13), we examined the functionality of PI3K signaling in PELP1-cyto clones by using phospho-AKT as a downstream marker of this pathway. Interestingly, PELP1-cyto clones showed excessive constitutive activation of the AKT pathway that could not be further induced by E2 treatment (Fig. 3A). Because the AKT pathway has also been shown to promote tamoxifen resistance (25), we examined the status of the AKT pathway in PELP1-cyto clones after treating the clones with tamoxifen. PELP1-cyto–expressing cells showed increased AKT activity compared with pcDNA-expressing clones (Fig. 3B), and the status of already elevated levels of AKT in PELP1-cyto clones was not affected by tamoxifen. Our finding that PELP1-cyto clones exhibit significant constitutive stimulation of the AKT pathway even in the absence of E2 treatment raised the possibility that PELP1 activation of the AKT pathway might be independent of the ability of PELP1 to interact with ER. To examine this possibility, we treated pcDNA and PELP1-cyto clones with the antiestrogen ICI 182,780 for 3 days to down regulate ER expression and activity. The ICI 182,780 did not affect the levels of constitutive AKT stimulation observed in the PELP1-cyto clones (Fig. 3C), suggesting that the increased activation seen in the PELP1-cyto clones is independent of ER. We then examined the possibility that increased AKT was due to the activation of PI3K, an upstream activator of AKT. Treatment of PELP1-cyto clones with LY294002, a widely used inhibitor of PI3K, blocked the constitutive activation of AKT (Fig. 3D). These findings suggested that the cytoplasmic localization of PELP1 might activate PI3K, thereby contributing to enhanced AKT signaling.
Figure 3.
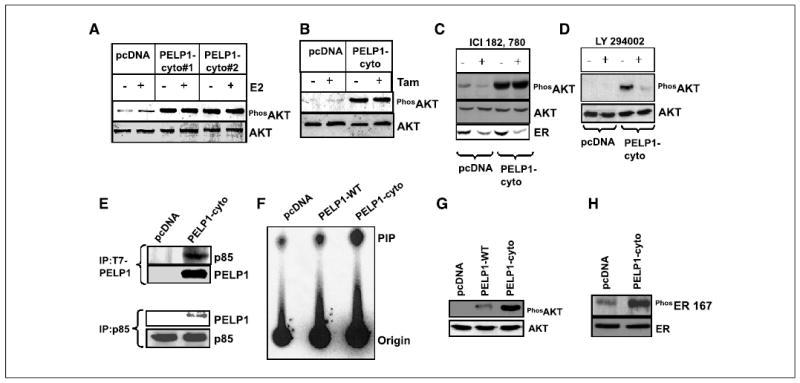
PELP1 cytoplasmic retention promotes PELP1-p85 interaction and constitutive AKT activation. A and B, MCF-7 clones expressing pcDNA or PELP1-cyto were treated with E2 (10−9 mol/L) or tamoxifen (10−8 mol/L), and total lysates were subjected to Western blotting by using a phospho-AKT antibody. Blots were stripped and reprobed with total AKT as a loading control. C, pcDNA and PELP1-cyto clones were treated with or without ICI 182,780 for 3 days, and total lysates were Western blotted with a phospho-AKT antibody. The blot was reprobed with total AKT and ER antibody. D, pcDNA and PELP1-cyto clones were treated with or without LY294002 for 60 minutes, and the total cell lysates were immunoblotted with phospho-AKT antibody. E, pcDNA and PELP1-cyto cells were cultured in 5% DCC serum, and total cell lysates (2 mg) were subjected to immunoprecipitation by using an antibody that recognizes the p85 subunit of PI3K (top) or T7 mAb (bottom), which recognizes T7-tagged PELP1. F, pcDNA, PELP1-WT, and PELP1-cyto clones were cultured in 5% DCC serum, and total lysates were subjected to immunoprecipitation by using antityrosine mAb PY-20 followed by in vitro PI3K assay. G, MCF-7 clones expressing pcDNA, PELP1-WT, and PELP1-cyto were cultured in 5% DCC serum, and total lysates were subjected to Western blotting using phospho-AKT followed by total AKT as the loading control. H, pcDNA or PELP1-cyto clones were cultured in 5% DCC serum and total lysates were subjected to Western blotting using phospho-ER Ser167 antibody and reprobed with total ER as the loading control.
PELP1 interacts with the p85 subunit of phosphatidylinositol-3-kinase
Analysis of the motif scan (http://scansite.mit.edu) of the PELP1 amino acid sequence revealed that PELP1 contains two potential p85-SH3 model 1 sites (HPPNRSAPHLPGLMC; PSPFRAPPFHPPGPM) and one potential p85-SH3 model 2 site (QTGKPSAPKKLKLDV). Therefore, we examined whether PELP1 directly interacts with the p85 subunit of PI3K and whether the observed altered PELP1 localization facilitates constitutive interaction of PI3K. Immunoprecipitation of pcDNA and PELP1-cyto lysates with antibodies p85 and T7 showed that PELP1-cyto interacts with the p85 subunit of PI3K (Fig. 3E). Immunoprecipitation and in vitro PI3K assays also confirmed the constitutive activation of the PI3K pathway in the PELP1-cyto clones (Fig. 3F). Western blot analysis of PELP1-WT clones also revealed mild activation of AKT; however, the activation was significantly lower than the magnitude of AKT activation seen in PELP1-cyto clones (Fig. 3G). Because AKT activation has been shown to phosphorylate ER on Ser167, we examined whether ER was phosphorylated in the PELP1-cyto clones. Western blotting using a phosphospecific antibody that recognizes ER Ser167 confirmed that ER was constitutively phosphorylated on Ser167 in PELP1-cyto clones (Fig. 3H). Collectively, these findings suggested that cytoplasmic localization of PELP1 promotes constitutive activation of the PI3K-AKT pathway, which can phosphorylate ER.
PELP1 associates with epidermal growth factor receptor
Previous studies have suggested bidirectional signaling between ER and EGFR pathways, and growth factors promote ER phosphorylation through the PI3K-AKT pathway (13). Because we observed that PELP1 interacts with PI3K, whose activity is modulated by growth factor signaling, we examined whether PELP1 interacts with growth factor receptors as well. To determine whether there is physiologic evidence of endogenous PELP1-EGFR interactions, we initially examined this in HeLa cells, which abundantly express EGFR and PELP1. HeLa cells were treated with EGF, and the colocalization of PELP1 and EGFR was analyzed by confocal microscopy. Our results showed that EGF stimulation caused a small portion of PELP1 to associate with membranous regions, and, in some areas, the colocalization of PELP1 with EGFR was observed (Fig. 4A, left). We next examined whether PELP1-cyto also interacts with EGFR. Confocal microscopy examination of EGF-treated PELP1-cyto clones revealed colocalization of PELP1-cyto with EGFR (Fig. 4A, right). To confirm these results, we did immunoprecipitation experiments in COS-1 cells, which were treated with or without EGF. Immunoprecipitation of EGFR followed by Western blot analysis with PELP1 confirmed that PELP1 indeed associates with EGFR upon EGF stimulation (Fig. 4B).
Figure 4.
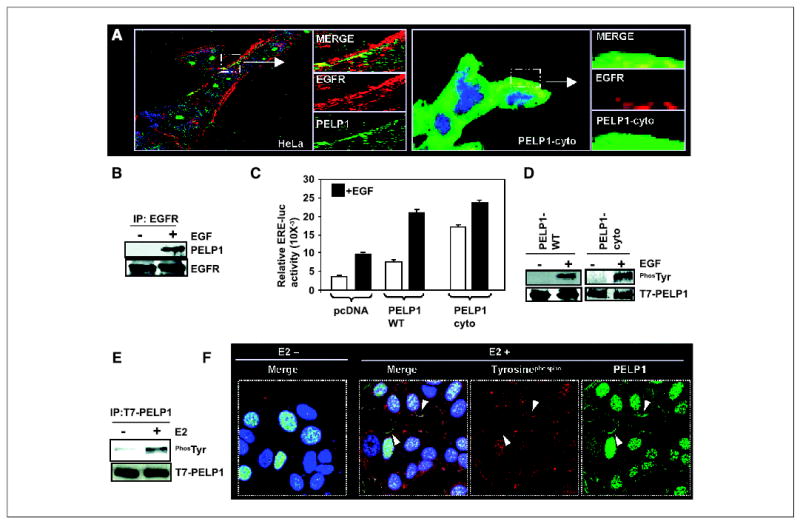
PELP1 interacts with EGFR and promotes EGF-mediated, ligand-independent ER transactivation functions. A, HeLa cells were treated with EGF (50 ng/mL). The cells were then fixed and costained with antibodies against PELP1 (green) and EGFR (red). The images were analyzed by confocal microscopy. Arrows, area of colocalization (yellow) of PELP1 and EGFR (left). PELP1-cyto cells were treated with EGF (50 ng/mL). Cells were stained with antibodies against T7 tag (to detect T7-tagged PELP1-cyto, green) and EGFR (red; right). A small portion of the image showing the colocalization (yellow) was enlarged and shown in the side panels. B, COS-1 cells were serum starved for 24 hours and treated with or without EGF. EGFR was immunoprecipitated by using an EGFR antibody; the presence of PELP1 in the immunoprecipitates was analyzed by Western blotting by using an anti-PELP1 antibody. C, HeLa cells were cotransfected with ER and ERE-luc reporter gene along with pcDNA or PELP1-WT or PELP1-cyto. After 48 hours, cells were stimulated with EGF for 12 hours and ERE-luc reporter activity was measured. D, COS-1 cells were transfected with T7-tagged PELP1-WT and after 72 hours, treated with or without EGF. Total lysates were immunoprecipitated with T7-epitope specific antibody and blotted with phosphotyrosine and T7 antibodies (left). MCF-7 cells that stably express T7-tagged PELP1-cyto were treated with or without EGF, and T7-PELP1-cyto was immunoprecipitated, followed by Western blotting with the phosphotyrosine antibody (right). E, MCF-7 cells that stably express T7-tagged PELP1-WT were treated with or without E2 for indicated periods of time and T7-PELP1 was immunoprecipitated; this was followed by Western blotting with the phosphotyrosine antibody. F, MCF-7 cells that stably express T7-tagged PELP1-WT were treated with or without E2, and colocalization of PELP1 (green) and phosphotyrosine (red) was analyzed by confocal microscopy. Arrows, areas of colocalization of PELP1 and phosphotyrosine at the membrane.
Because EGF has been shown to promote ligand-independent activation of ER transactivation functions (26, 27), we examined whether PELP1 promotes EGF-mediated ER transactivation functions by performing ERE reporter gene assays using HeLa cells cotransfected with ER in the presence or absence of pcDNA vector, PELP1-WT, or PELP1-cyto constructs. Results showed that pcDNA-transfected clones exhibited 2.3-fold activation of ERE reporter upon addition of EGF. PELP1-WT transfection modestly enhanced the basal activity of the ERE reporter; however, EGF treatment induced a 3.2-fold induction of ERE reporter gene activity over untreated control. Interestingly, PELP1-cyto transfection substantially increased the ERE reporter gene activity in the absence of ligand to a level similar to EGF-treated, PELP1-WT–transfected cells. EGF treatment further increased ERE reporter activity in PELP1-cyto–transfected cells (Fig. 4C). Increased basal activity of the ERE reporter in PELP1-cyto–transfected cells could be due to the constitutive activation of AKT pathway seen in the PELP1-cyto cells. These results suggest that PELP1-WT has a potential to enhance EGF-mediated ER transactivation functions and mislocalization of PELP1 in the cytoplasm can potentially activate ER basal activation by excessively coupling the EGFR-PI3K signaling pathways.
Growth factor signaling has been shown to modulate coactivator functions by posttranslation modifications (14). Because PELP1 interacted with EGFR and promoted EGF-mediated ERE transactivation functions, we next examined whether EGF stimulation promotes the phosphorylation of PELP1. COS-1 cells were transfected with T7-tagged PELP1, and after 48 hours cells were treated with or without EGF. Total lysates were immunoprecipitated with T7-epitope antibody (Fig. 4D, left), and immunoprecipitates were blotted with T7 and phosphotyrosine antibodies. Results showed that EGF promotes tyrosine phosphorylation of PELP1. Similarly, immunoprecipitation of PELP1-cyto from PELP1 stable clones also showed that EGF promotes tyrosine phosphorylation of PELP1-cyto (Fig. 4D, right).
Because PELP1 interacts with ER and promotes E2-mediated nongenomic signaling pathways, we examined whether E2 can promote the tyrosine phosphorylation of PELP1. MCF-7 cells stably expressing PELP1-WT were treated with or without E2. Immunoprecipitation experiments showed that E2 also promoted the tyrosine phosphorylation of PELP1 (Fig. 4E). We confirmed these results with confocal microscopic analysis. As shown by phosphotyrosine antibody staining, E2 promoted the membrane localization of PELP1 and its colocalization with phosphotyrosine (Fig. 4F). These results suggest that both EGF and E2 signaling modulate PELP1 functions and that the cytoplasmic retention of PELP1 excessively promotes the interactions of PELP1 with components of the growth factor signaling axis.
PELP1 cytoplasm retention promotes hormonal independence
To examine the effect of the cytoplasmic localization of PELP1 on biological functions, we did cell proliferation and anchorage independence assays. Results showed that PELP1-cyto–expressing clones were hypersensitive to E2 compared with parental MCF-7– or pcDNA-vector–expressing clones (Fig. 5A). Further, PELP1-cyto clones showed an increased ability to form colonies in an anchorage-independent manner compared with pcDNA-expressing cells (Fig. 5B). Because AKT activation has been shown to promote tamoxifen partial agonist action in breast cancer cells, we repeated these biological assays in the presence or absence of tamoxifen for 7 days. MCF-7 parental and pcDNA-expressing clones showed a 50% reduction in cell number. Interestingly, PELP1-cyto–expressing clones showed resistance to tamoxifen in that the cell number was not altered (Fig. 5C). In anchorage-independence assays, PELP1-cyto–expressing cells exhibited an increased ability to form colonies in the presence of tamoxifen compared with parental or pcDNA clones (Fig. 5D). Consistent with these results, PELP1-cyto clones exhibited tumorigenic potential in nude mice, and these tumors also exhibited constitutive activation of the AKT pathway (Fig. 5E). These findings suggest that a close relationship exists between PELP1 cytoplasmic localization, AKT activation, and tumorigenesis.
Figure 5.
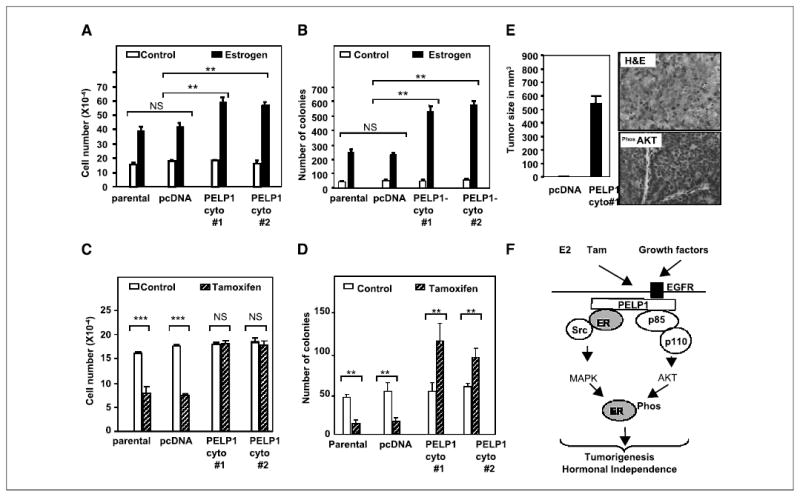
PELP1 deregulation promotes hormonal resistance and tumorigenesis. A and C, pcDNA- and PELP1-cyto–expressing clones were cultured in 5% DCC serum for 48 hours and treated with or without E2 (10−9 mol/L, A) or tamoxifen (10−8 mol/L, C) for 7 days, and the cell number was determined. B and D, the anchorage-independent growth potential of pcDNA- and PELP1-cyto–expressing clones was measured by the ability of the cells to form colonies on soft agar. Cells were treated with either E2 (10−9 mol/L, B) or tamoxifen (10−8 mol/L, D), and colonies were counted after 21 days. Similar results were obtained in three independent experiments. Columns, mean; bars, SE. **P < 0.01; ***P < 0.001; NS, not significant (Student’s t test). E, nude mice were injected in the mammary fat pad with 5 × 107 MCF-7 cells that stably express either pcDNA or PELP1-cyto, and tumor growth was measured after 8 weeks. Morphology of tumor was evaluated by H&E staining and activation of AKT pathway in the tumors was confirmed by immunohistochemistry with a phospho-AKT antibody. F, model for PELP1 regulation of ER functions. Cytoplasmic localization of PELP1 promotes its interactions with EGFR, Src kinase, and PI3K, thus leading to the activation of nongenomic pathways including MAPK and AKT. Such excessive activation of nongenomic pathways promotes hypersensitivity and tamoxifen resistance, which could in part be due to the modification of ER phosphorylation. PELP1 serves as an adaptor protein because it interacts with growth factor receptors, signaling kinases, and nuclear receptors; its deregulation may perturb ER signaling network toward tumorigenesis.
Discussion
In this study, we observed that the localization of the ER coactivator PELP1 is altered in a subset of breast tumors. Specifically in model cells that mimic PELP1 localization in tumors, we observed that the cytoplasmic localization of PELP1 promotes its interactions with EGFR, Src, and PI3K, thereby leading to the activation of the MAPK and AKT pathways. In addition, the altered localization of PELP1 promoted hypersensitivity to estrogen and anchorage independence. Our results thus indicate that the ER coactivator PELP1 may contribute to tumorigenesis in breast tumors by serving as a molecular adaptor between EGFR-ER pathways and by promoting excessive crosstalk between these pathways.
The altered localization of the ER coactivator PELP1, which has the potential to stimulate nongenomic ER functions (18, 24), may alter the ratio of genomic and nongenomic signaling in breast cancer cells and thus might promote hormonal independence by modulating ER-transactivating functions and SERM actions. As our study and other studies indicate, PELP1 expression is deregulated in breast and endometrial cancers (16, 21); therefore, the altered localization of PELP1 is expected to contribute toward the excessive activation of the MAPK and PI3K-AKT pathways, leading to follow-up modifications of ER. Such modifications of the ER pathway may lead to the activation of ER target genes in a ligand-independent manner. Alternatively, the modified ER may allow the recruitment of a different set of coregulator proteins, and thus may exhibit resistance to hormonal therapy. In our study, the cytoplasmic localization of PELP1 promoted increased activation of MAPK, leading to enhanced ER-Ser118 phosphorylation. Similarly, these clones also exhibited constitutive activation of the AKT pathway, which was not further induced by E2. Therefore, the enhanced E2-mediated growth and anchorage independence seen in these clones is likely to be caused by excessive activation of the MAPK pathway, whereas tamoxifen-mediated resistance could be due to the cumulative effect of the activation of the MAPK and AKT pathways, which leads to the modification of ER and its associated proteins as well as the activation of downstream pathways by activated MAPK and AKT.
ER coregulatory proteins have been suggested to play a role in the generally observed tissue-specific effects of tamoxifen (28, 29). However, these ER coregulators are targeted by excessive ER-HER2 crosstalk leading to hormonal resistance in a subset of breast tumors (15). High levels of the ER coactivator AIB1 and HER2 in breast cancer contribute to tamoxifen resistance (30). Our findings that PELP1 interacts with EGFR and that PELP1 has the ability to enhance EGF-mediated ER transactivation functions suggest that PELP1 potentiates growth factor receptor–mediated hormonal resistance in a subset of tumors that have deregulated growth factor signaling.
Growth factor signaling promotes ER phosphorylation (both serine and tyrosine; refs. 14, 31). Recent evidence suggests that ER coactivators, in addition to ER, are targets of growth factor signaling (32). Growth factor–mediated activation of nongenomic pathways and phosphorylation of ER and ER-coregulatory proteins have been shown to have a role in tamoxifen resistance (33, 34). Our findings that PELP1 enhances tamoxifen resistance and that EGF promotes the phosphorylation of PELP1 suggest a possibility that growth factor–mediated posttranslational modification of PELP1 may play an important role in PELP1-mediated hormonal resistance functions.
Activation of the PI3K-AKT pathway has been shown to be an essential step in the estrogenic action of growth factors (35). Previous studies also showed that forced expression of constitutively active AKT in MCF-7 cells promotes E2-independent growth as well as tamoxifen response (25). A recent study found that overexpression of the ER coactivator AIB1 promoted high tumor incidence, which is associated with the activation of the PI3K-AKT pathway (36). Our findings suggest that cytoplasmic localization of PELP1 plays a role in tamoxifen resistance and that the ability of PELP1 to modulate the PI3K-AKT pathways may represent one mechanism by which PELP1-cyto cells develop resistance to hormonal treatment. Further, our analysis of human breast tissues provided the proof-of-principle that PELP1 localization is altered in a subset of human breast tumors.
Our results suggest that the cytoplasmic localization of PELP1 may play a role in the constitutive activation of AKT in tumor cells. Our findings also suggest the existence of a close relationship between cytoplasmic PELP1 localization and increased nongenomic signaling. Additionally, our findings suggest that the cytoplasmic localization of PELP1 might be sufficient to promote tumorigenic phenotypes and hormonal independence. Further, the ability of PELP1-cyto cells to form tumors in nude mice and the presence of increased AKT signaling in these tumors also supports the potential role of PELP1 localization in tumorigenesis. Because these tumors were formed in the absence of estrogen pellet implantation, PELP1-mediated constitutive activation of the AKT pathway rather than the MAPK pathway might play a role in PELP1-cyto–mediated in vivo tumorigenic functions. It will be interesting to discover the mechanisms by which PELP1 localization is altered in pathologic conditions. Because cytoplasmic PELP1 observed in the tumors migrated in a similar fashion as PELP-WT, it is likely that it represents a full-length protein and that its deregulation in localization could be because of posttranslational modifications or mutations that affect its localization to the nuclear compartment. Our future studies will be directed at identifying the mechanisms of PELP1 deregulation, including possible posttranslational modifications/mutations in tumors.
In summary, our results suggest that the localization of ER coactivators, such as PELP1, could activate nongenomic signals and may play an important role in the hormonal responses of ER-positive cancer cells (Fig. 5F). Furthermore, the ability of PELP1 to activate MAPK and AKT signaling pathways, its potential to enhance tamoxifen resistance, and its distinct localization in a subset of breast tumors suggest that PELP1 plays a role in the biology of a subset of breast cancers and that its deregulation might contribute to hormonal independence.
Acknowledgments
Grant support: Department of Defense grant DAMD17-03-1-0661 and NIH grants CA095681 (R.K. Vadlamudi) and CA65746, CA90970, and CA98823 (R. Kumar).
References
- 1.Barnes CJ, Vadlamudi RK, Kumar R. Novel estrogen receptor coregulators and signaling molecules in human diseases. Cell Mol Life Sci. 2004;61:281–91. doi: 10.1007/s00018-003-3222-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Encarnacion CA, Ciocca DR, McGuire WL, Clark GM, Fuqua SA, Osborne CK. Measurement of steroid hormone receptors in breast cancer patients on tamoxifen. Breast Cancer Res Treat. 1993;26:237–46. doi: 10.1007/BF00665801. [DOI] [PubMed] [Google Scholar]
- 3.Osborne CK, Zhao H, Fuqua SA. Selective estrogen receptor modulators: structure, function, and clinical use. J Clin Oncol. 2000;18:3172–86. doi: 10.1200/JCO.2000.18.17.3172. [DOI] [PubMed] [Google Scholar]
- 4.Clarke R, Leonessa F, Welch JN, Skaar TC. Cellular and molecular pharmacology of antiestrogen action and resistance. Pharmacol Rev. 2001;53:25–71. [PubMed] [Google Scholar]
- 5.Tonetti DA, Jordan VC. Possible mechanisms in the emergence of tamoxifen-resistant breast cancer. Anticancer Drugs. 1995;6:498–507. doi: 10.1097/00001813-199508000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Graham JD, Bain DL, Richer JK, Jackson TA, Tung L, Horwitz KB. Thoughts on tamoxifen resistant breast cancer. Are coregulators the answer or just a red herring? J Steroid Biochem Mol Biol. 2000;74:255–9. doi: 10.1016/s0960-0760(00)00101-1. [DOI] [PubMed] [Google Scholar]
- 7.Osborne CK. Mechanisms for tamoxifen resistance in breast cancer: possible role of tamoxifen metabolism. J Steroid Biochem Mol Biol. 1993;47:83–9. doi: 10.1016/0960-0760(93)90060-a. [DOI] [PubMed] [Google Scholar]
- 8.Losel R, Wehling M. Nongenomic actions of steroid hormones. Nat Rev Mol Cell Biol. 2003;4:46–56. doi: 10.1038/nrm1009. [DOI] [PubMed] [Google Scholar]
- 9.Revelli A, Massobrio M, Tesarik J. Nongenomic actions of steroid hormones in reproductive tissues. Endocr Rev. 1998;19:3–17. doi: 10.1210/edrv.19.1.0322. [DOI] [PubMed] [Google Scholar]
- 10.Levin ER. Cell localization, physiology, and nongenomic actions of estrogen receptors. J Appl Physiol. 2001;91:1860–7. doi: 10.1152/jappl.2001.91.4.1860. [DOI] [PubMed] [Google Scholar]
- 11.Mendelsohn ME. Genomic and nongenomic effects of estrogen in the vasculature. Am J Cardiol. 2002;90:3–6F. doi: 10.1016/s0002-9149(02)02418-9. [DOI] [PubMed] [Google Scholar]
- 12.Michalides R, Griekspoor A, Balkenende A, et al. Tamoxifen resistance by a conformational arrest of the estrogen receptor α after PKA activation in breast cancer. Cancer Cell. 2004;5:597–605. doi: 10.1016/j.ccr.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 13.Levin ER. Bidirectional signaling between the estrogen receptor and the epidermal growth factor receptor. Mol Endocrinol. 2003;17:309–17. doi: 10.1210/me.2002-0368. [DOI] [PubMed] [Google Scholar]
- 14.Driggers PH, Segars JH. Estrogen action and cytoplasmic signaling pathways. Part II: the role of growth factors and phosphorylation in estrogen signaling. Trends Endocrinol Metab. 2002;13:422–7. doi: 10.1016/s1043-2760(02)00634-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shou J, Massarweh S, Osborne CK, et al. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst. 2004;96:926–35. doi: 10.1093/jnci/djh166. [DOI] [PubMed] [Google Scholar]
- 16.Vadlamudi RK, Wang RA, Mazumdar A, et al. Molecular cloning and characterization of PELP1, a novel human coregulator of estrogen receptor α. J Biol Chem. 2001;276:38272–9. doi: 10.1074/jbc.M103783200. [DOI] [PubMed] [Google Scholar]
- 17.Nair SS, Mishra SK, Yang Z, Balasenthil S, Kumar R, Vadlamudi RK. Potential role of a novel transcriptional coactivator PELP1 in histone H1 displacement in cancer cells. Cancer Res. 2004;64:6416–23. doi: 10.1158/0008-5472.CAN-04-1786. [DOI] [PubMed] [Google Scholar]
- 18.Wong CW, McNally C, Nickbarg E, Komm BS, Cheskis BJ. Estrogen receptor-interacting protein that modulates its nongenomic activity—crosstalk with Src/Erk phosphorylation cascade. Proc Natl Acad Sci U S A. 2002;99:14783–8. doi: 10.1073/pnas.192569699. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Balasenthil S, Vadlamudi RK. Functional interactions between the estrogen receptor coactivator PELP1/MNAR and retinoblastoma protein. J Biol Chem. 2003;278:22119–27. doi: 10.1074/jbc.M212822200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mishra SK, Balasenthil S, Nguyen D, Vadlamudi RK. Cloning and functional characterization of PELP1/MNAR promoter. Gene. 2003;330:115–22. doi: 10.1016/j.gene.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 21.Vadlamudi RK, Balasenthil S, Broaddus RR, Gustafsson JA, Kumar R. Deregulation of estrogen receptor coactivator proline-, glutamic acid-, and leucine-rich protein-1/modulator of nongenomic activity of estrogen receptor in human endometrial tumors. J Clin Endocrinol Metab. 2004;89:6130–8. doi: 10.1210/jc.2004-0909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vadlamudi RK, Adam L, Wang RA, et al. Regulatable expression of p21-activated kinase-1 promotes anchorage-independent growth and abnormal organization of mitotic spindles in human epithelial breast cancer cells. J Biol Chem. 2000;275:36238–44. doi: 10.1074/jbc.M002138200. [DOI] [PubMed] [Google Scholar]
- 23.Vadlamudi RK, Bagheri-Yarmand R, Yang Z, et al. Dynein light chain 1, a p21-activated kinase 1-interacting substrate, promotes cancerous phenotypes. Cancer Cell. 2004;5:575–85. doi: 10.1016/j.ccr.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 24.Barletta F, Wong CW, McNally C, Komm BS, Katzenellenbogen B, Cheskis BJ. Characterization of the interactions of estrogen receptor and MNAR in the activation of cSrc. Mol Endocrinol. 2004;18:1096–108. doi: 10.1210/me.2003-0335. [DOI] [PubMed] [Google Scholar]
- 25.Faridi J, Wang L, Endemann G, Roth RA. Expression of constitutively active Akt-3 in MCF-7 breast cancer cells reverses the estrogen and tamoxifen responsivity of these cells in vivo. Clin Cancer Res. 2003;9:2933–9. [PubMed] [Google Scholar]
- 26.Kato S, Endoh H, Masuhiro Y, et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270:1491–4. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- 27.Bunone G, Briand PA, Miksicek RJ, Picard D. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J. 1996;15:2174–83. [PMC free article] [PubMed] [Google Scholar]
- 28.Schiff R, Massarweh S, Shou J, Osborne CK. Breast cancer endocrine resistance: how growth factor signaling and estrogen receptor coregulators modulate response. Clin Cancer Res. 2003;9:447–54S. [PubMed] [Google Scholar]
- 29.Graham JD, Bain DL, Richer JK, Jackson TA, Tung L, Horwitz KB. Nuclear receptor conformation, coregulators, and tamoxifen-resistant breast cancer. Steroids. 2000;65:579–84. doi: 10.1016/s0039-128x(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 30.Osborne CK, Schiff R. Growth factor receptor crosstalk with estrogen receptor as a mechanism for tamoxifen resistance in breast cancer. Breast. 2003;12:362–7. doi: 10.1016/s0960-9776(03)00137-1. [DOI] [PubMed] [Google Scholar]
- 31.Nilsson S, Makela S, Treuter E, et al. Mechanisms of estrogen action. Physiol Rev. 2001;81:1535–65. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 32.Smith CL, O’Malley BW. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr Rev. 2004;25:45–71. doi: 10.1210/er.2003-0023. [DOI] [PubMed] [Google Scholar]
- 33.Hermanson O, Glass CK, Rosenfeld MG. Nuclear receptor coregulators: multiple modes of modification. Trends Endocrinol Metab. 2002;13:55–60. doi: 10.1016/s1043-2760(01)00527-6. [DOI] [PubMed] [Google Scholar]
- 34.Hutcheson IR, Knowlden JM, Madden TA, et al. Oestrogen receptor-mediated modulation of the EGFR/MAPK pathway in tamoxifen-resistant MCF-7 cells. Breast Cancer Res Treat. 2003;81:81–93. doi: 10.1023/A:1025484908380. [DOI] [PubMed] [Google Scholar]
- 35.Martin MB, Franke TF, Stoica GE, et al. A role for Akt in mediating the estrogenic functions of epidermal growth factor and insulin-like growth factor I. Endocrinology. 2000;141:4503–11. doi: 10.1210/endo.141.12.7836. [DOI] [PubMed] [Google Scholar]
- 36.Torres-Arzayus MI, De Mora JF, Yuan J, et al. High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell. 2004;6:263–74. doi: 10.1016/j.ccr.2004.06.027. [DOI] [PubMed] [Google Scholar]