

Abstract
The role of cardiac myosin binding protein-C (cMyBP-C) phosphorylation in cardiac physiology or pathophysiology is unclear. To investigate the status of cMyBP-C phosphorylation in vivo, we determined its phosphorylation state in stressed and unstressed mouse hearts. cMyBP-C phosphorylation is significantly decreased during the development of heart failure or pathologic hypertrophy. We then generated transgenic (TG) mice in which cMyBP-C’s phosphorylation sites were changed to nonphosphorylatable alanines (MyBP-CAllP−). A TG line showing ~40% replacement with MyBP-CAllP− showed no changes in morbidity or mortality but displayed depressed cardiac contractility, altered sarcomeric structure and upregulation of transcripts associated with a hypertrophic response. To explore the effect of complete replacement of endogenous cMyBP-C with MyBP-CAllP−, the mice were bred into the MyBP-C(t/t) background, in which less than 10% of normal levels of a truncated MyBP-C are present. Although MyBP-CAllP− was incorporated into the sarcomere and expressed at normal levels, the mutant protein could not rescue the MyBP-C(t/t) phenotype. The mice developed significant cardiac hypertrophy with myofibrillar disarray and fibrosis, similar to what was observed in the MyBP-C(t/t) animals. In contrast, when the MyBP-C(t/t) mice were bred to a TG line expressing normal MyBP-C (MyBP-CWT), the MyBP-C(t/t) phenotype was rescued. These data suggest that cMyBP-C phosphorylation is essential for normal cardiac function.
Introduction
Understanding the structure-function relations for cardiac myosin binding protein-C (cMyBP-C) is clinically relevant as cMyBP-C mutations are a widely recognized cause of familial hypertrophic cardiomyopathy.1 Various cMyBP-C transgenic (TG) and gene-targeted mouse models have demonstrated the importance of the protein for long-term integrity of sarcomeric structure and for maintaining normal cardiac contractility.2,3 Functional insight can be gained from appreciating the crucial structural differences between cMyBP-C and the skeletal isoform. Only the cardiac isoform contains an extra immunoglobulin domain at the N-terminus (C0), an insertion of 28 residues within the C5 domain, and three phosphorylation sites (Ser-273, -282, -302) that are substrates for cAMP-dependent protein kinase A (PKA), Ca2+-calmodulin-activated kinase (CaMKII) and protein kinase C (PKC).
In vivo, PKA-mediated phosphorylation of cMyBP-C is linked to modulation of cardiac contraction.4 Upon adrenergic stimulation, PKA phosphorylates Ser-273, -282 and -302, whereas PKC phosphorylates only Ser-273 and -302.5 These residues, located near the N-terminus of the protein, are of particular interest as this region binds to the S2 segment of the myosin heavy chain (MHC),6,7 which is close to myosin’s lever arm domain. It has been hypothesized that cMyBP-C-MHC interactions are dynamically regulated by the phosphorylation/dephosphorylation of cMyBP-C.8 In vitro experiments showed that after phosphorylation of cMyBP-C by PKA, the thick filaments exhibited a relative loose structure,9 preventing binding to myosin, and thus changing the maximum Ca2+-activated force (Fmax). Electron microscopy of isolated thick filaments confirmed that phosphorylation of cMyBP-C initiates crossbridge movement away from the thick-filament backbone, although this interaction may also be determined by the MHC isoform that is present.10 In theory, cMyBP-C phosphorylation could change both filament orientation and contractile mechanics,7,11 although cMyBP-C phosphorylation did not alter the Ca2+ sensitivity of Mg2+-ATPase activity in reconstituted contractile protein systems.12,13 Additionally, phosphorylation of cMyBP-C did not affect the Ca2+ sensitivity of Mg2+-ATPase activity, force-Ca2+ relationship and sarcomere length dependency of contraction in intact skinned fiber experiments,14,15 suggesting that MyBP-C phosphorylation plays a relatively minor role in regulating contraction. Given the above body of data, the role that cMyBP-C phosphorylation plays in either normal or pathological cardiac function remains unclear.
We wished to determine if cMyBP-C phosphorylation played an essential role in the maintenance of cardiac function. We first defined the phosphorylation status of cMyBP-C in the normal and diseased heart and showed that significant changes occurred during development of cardiac pathology. We then generated a TG mouse model in which the known phosphorylation sites in cMyBP-C were converted to nonphosphorylatable alanines (MyBP-CAllP−) and compared this model to a model with comparable levels of TG expression of normal cMyBP-C (MyBP-CWT). Both TG models were subsequently bred into the homozygous cMyBP-C(t/t) background.3 The data establish that cMyBP-C phosphorylation is essential for normal cardiac function.
Materials and Methods
An expanded Materials and Methods section can be found in the Online Supplement.
Transgenic and Targeted Lines
The cDNA for mouse cMyBP-C (MyBP-CWT) was obtained by RT-PCR using RNA isolated from the mouse cardiac ventricle. The cMyBP-C phosphorylation sites (Ser-273, Ser-282, -302), along with two adjacent sites that could be potentially phosphorylated (Thr-272, Thr-281) were converted to alanine, and a sequence encoding the myc epitope incorporated as described previously.2 The cDNA was subcloned into the mouse α-MHC promoter and used to generate multiple lines of TG mice for each construct. To ensure the absence of endogenous phosphorylatable cMyBP-C, mice showing germline transmission of the transgene, termed MyBP-CAllP−, were bred with mice that expressed less than 10% of normal cMyBP-C levels (MyBP-C(t/t)). The small amount of protein that is present in these animals is truncated and unstable.3 An animal that expressed the normal cardiac isoform at equivalent levels to MyBP-CAllP−, MyBP-CWT,2 was also bred to the nulls to serve as a control.
Molecular and Protein Analyses
For assessment of cMyBP-C transcript size, expression levels, and expression of hypertrophic marker genes, RNA transcript and blot analyses were performed as described.2,16 Enriched myofibrillar proteins were isolated using F60 buffer and solubilized in urea buffer.17 MyBP-CAllP− was identified via SDS-PAGE followed by western blots using anti-myc monoclonal antibodies and anti-cMyBP-C rabbit polyclonal antibodies raised against the C0-C1 domains.18
Phosphorylation and Isoelectric Focusing
Myofibrillar proteins were treated with phosphatase and PKA as described.17 One-dimensional isoelectric focusing (IEF) was performed to identify the phosphorylated forms.18
Cardiac Function
In vivo cardiac function was assessed both by noninvasive echocardiography in unsedated or sedated animals and by invasive catheterization.19
Results
Phosphorylation States of cMyBP-C in Normal and Heart Failure Mouse Models
As is the case for a number of other contractile proteins, cMyBP-C can be reversibly phosphorylated at multiple sites in response to altered physiological conditions.20 However, the in vivo phosphorylation levels of the protein under basal and stressed conditions are not well defined. In order to understand the level of phosphorylation in the nontransgenic (NTG) animals before we perturbed the cMyBP-C protein complement, cMyBP-C phosphorylation under normal and stressed conditions was determined using IEF followed by westerns using a cMyBP-C C0-C1 motif specific antibody.18 Total enriched myofibrillar proteins were extracted from unstressed, NTG adults, animals that had undergone transverse aortic constriction (TAC) to produce pressure overload hypertrophy, animals in failure as a result of cardiac specific expression of calcineurin,21 MLP-deficient mice that develop dilated heart failure,22 and animals that had been subjected to ischemia-reperfusion. Because the degree to which cMyBP-C and its phosphorylated forms regulate cardiac function may depend, in part, upon the MHC isoform that is present,23 we also determined cMyBP-C phosphorylation levels in a mouse model in which we had replaced approximately 70% of the normal α-MHC with β-MHC.24
Under basal conditions, the mono-, bi- and tri-phosphorylated species of cMyBP-C made up >90% of the cMyBP-C population (Figure 1A, lane 5). A β-MHC shift, in of itself, had no effect on the normal phosphorylation pattern failure (Figure 1B, lane 8). In all of the models in which cardiac function was significantly compromised by either surgical or genetic manipulation, total cMyBP-C phosphorylation, particularly the triphosphorylated species, was decreased (Figure 1A and 1C, Table 1S, Online Supplement). Invariably, in the animals that displayed overt cardiac failure as determined by labored breathing, anasarca and failure to groom, the tri-phosphorylated state was reduced or absent (Figure 1, lanes 4, 6, 7 and 12). These data show that increased dephosphorylation of cMyBP-C is associated with contractile dysfunction and heart failure.
Figure 1.
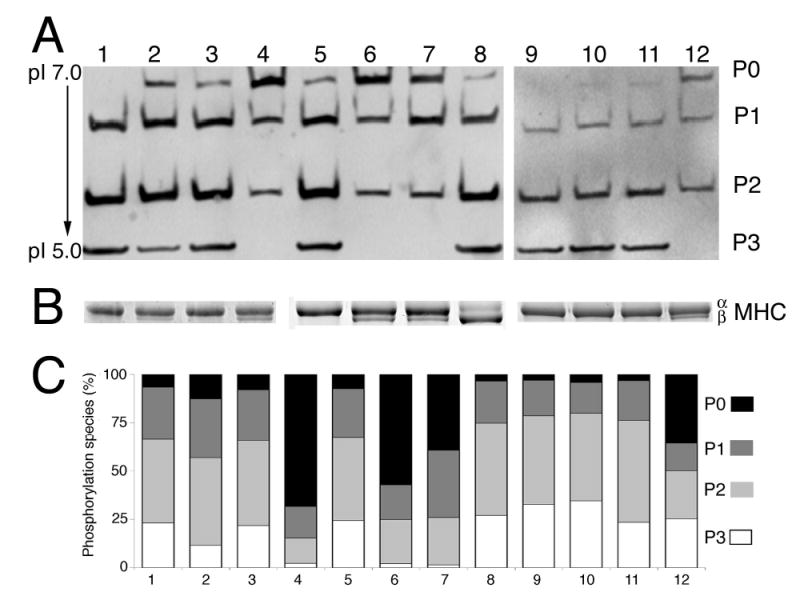
Cardiac MyBP-C phosphorylation levels in normal and pathological states. A, One dimensional IEF (pH 5–7) of total myofilament proteins followed by western blot analysis using cMyBP-C antibody.18 Proteins were obtained from the following samples: (lane 1) hearts that had undergone sham operation for TAC, 24 hours. (lane 2) 24 hours post-TAC. (lane 3) 18 day sham. (lane 4) 18 day post-TAC. (lane 5) a NTG control. (lane 6) 8 weeks old calcineurin TG mouse. (lane 7) 7–8 months old MLP deficient mouse. (lane 8) a β-MHC TG mouse.24 (lane 9) Ischemic reperfusion (IR), 24 hours sham. (lane 10) 24 hours post-IR. (lane 11) 5 day post-IR sham. (lane 12) 5 day post-IR. The four forms of cMyBP-C based on its phosphorylation status are shown as P0, P1, P2 and P3, which correspond to the de-, mono-, bi- and tri-phosphorylated forms, respectively with isoelectric points of 6.1, 5.9, 5.7 and 5.5. B, MHC isoform shifts. Using the samples from panel A, the α- and β-MHC isoforms were separated in a 5% glycerol gel. The data show the expected isoform shifts characteristic of activation of the fetal gene program in the disease models (lanes 4, 6, 7 and 12). C, cMyBP-C phosphorylation in cardiac disease. Samples are as in panel A and average values are shown as a percentage of total cMyBPC (n = 3). The quantitative data are shown in tabular form in Table 1S, Online Supplement.
Genetic Modulation of cMyBP-C
The phosphorylation motif of cMyBP-C is conserved among the human, mouse, chicken and frog (Figure 2A). To investigate the role of cMyBP-C phosphorylation in relation to cardiac function, we generated a construct (MyBP-CAllP−) in which the three phosphorylation sites (Ser-273, -282 and -302) and two neighboring potential alternative phosphorylation sites (Thr-272 and -281) were altered to alanine (Figure 2A). Previously, we prepared a TG mouse that expressed myc-tagged, normal cMyBP-C (line 21, MyBP-CWT),2 and mice that expressed MyBP-CAllP− at approximately the same level (line 262) were used along with NTG littermates for comparison of transcript and protein levels for the MyBP-CAllP− mice (Figure 2B, 2C and 2E). Despite the increase in transcript levels in both the MyBP-CAllP− and MyBP-CWT hearts (Figure 2B and 2C), no absolute increase in the level of cMyBP-C was observed, nor were the levels of the other contractile proteins overtly affected (Figure 2D and 2E).
Figure 2.
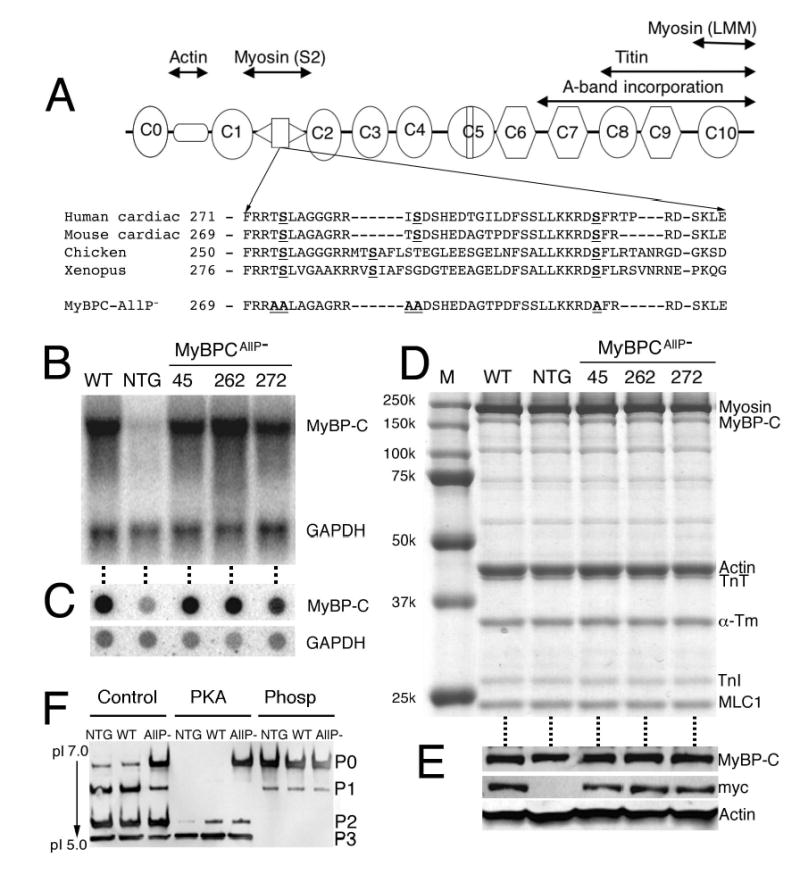
MyBP-CAllP− transgene expression. A, Schematic diagram of cMyBP-C. The eight IgI-like domains are shown as ovals and the three F3 domains as octagons. Interaction sites are shown above the diagram. The cardiac-specific phosphorylation motif, which is highly conserved between species, is located between domains C1 and C2 and the sequence shown. The three known phosphorylation sites (Ser-273, -282 and -302) and two potential alternative phosphorylation sites (Thr-272 and -281) were each altered to a non-phosphorylatable alanine. B, Northern blot analyses of RNAs from left ventricles derived from 8–12 week old MyBP-CWT, NTG and three lines of MyBP-CAllP− mice. The RNAs were hybridized with cMyBP-C and GAPDH probes to confirm expression of the intact and correctly sized transcript. C, Dot-blot analyses using the same RNAs and probes as in panel B were used to quantitate the levels of TG expression. D, Western blot analyses of myofibrillar proteins from NTG, MyBP-CWT (WT); expresses TG normal cMyBP-C such that approximately 40% of the endogenous protein is replaced with the transgenically encoded protein2 and three MyBP-CAllP− (lines 45, 262 and 272) hearts. Note the conservation of normal levels of total cMyBP-C expression. E, Total cMyBP-C levels as well as the presence of myc-tagged cMyBP-C protein in the same mice was confirmed by western blot analysis. α-sarcomeric actin was used as a loading control. F, Phosphorylation state of NTG, MyBP-CWT (WT) and MyBP-CAllP− (AllP−) cMyBP-C in myofibrillar extracts assessed by one-dimensional IEF. Normal cMyBP-C in these extracts could be further phosphorylated by PKA but MyBP-CAllP− was not a PKA substrate. Phosphatase treatment (Phosp) of myofibrillar extracts dephosphorylated normal cMyBP-C.
To evaluate the level of MyBP-CAllP− replacement and define the phosphorylation status of cMyBP-C in the TG mice, cMyBP-C derived from NTG, MyBP-CWT and MyBP-CAllP− hearts were treated with either protein phosphatase or PKA and resolved in IEF gels for western analysis. Cardiac MyBP-C treated with protein phosphatase migrated at a pI of 6.06, as expected for the completely dephosphorylated species (Figure 2F). The introduction of the myc tag did not result in a detectable difference in migration from the endogenous protein and the dephosphorylated, endogenous form comigrated with MyBP-CAllP−. PKA treatment resulted in essentially complete conversion of endogenous cMyBP-C to the di- and tri-phosphorylated species and expression of MyBP-CWT had no effect on the overall levels of phosphorylation (Figure 2F, lane 2). As expected, cMyBP-C isolated from the MyBP-CAllP− hearts showed two species when treated with PKA, the nonphosphorylated MyBP-CAllP− (P0) and the phosphorylated endogenous cMyBP-C (P2, P3), allowing us to estimate the degree of replacement of the endogenous cMyBP-C with the transgenically-encoded species. Approximately 40% replacement with the MyBP-CAllP− occurred in line 262, which is equivalent to the level of replacement previously measured in the MyBP-CWT-expressing hearts derived from line 21.2
Histological and Ultrastructural Consequences of MyBP-CAllP− Expression
The gross histology of 3 month MyBP-CAllP− hearts was unremarkable with no obvious abnormalities, fibrosis, calcification or disarray compared to NTG controls (Figure 3A). However, transmission electron microscopy revealed subtle ultrastructural changes. While NTG hearts showed typical, well-organized and aligned sarcomeres with regularly distributed mitochondria, sarcoplasmic reticulum, and T-tubules (Figure 3B, C), MyBP-CAllP− hearts occasionally displayed regions that lacked the regular sarcomere-mitochondria distribution and some sarcomeres showed altered H-zones and M-lines that were relatively ill-defined (Figure 3B, lower panel, Figure 3C, lower panels).
Figure 3.
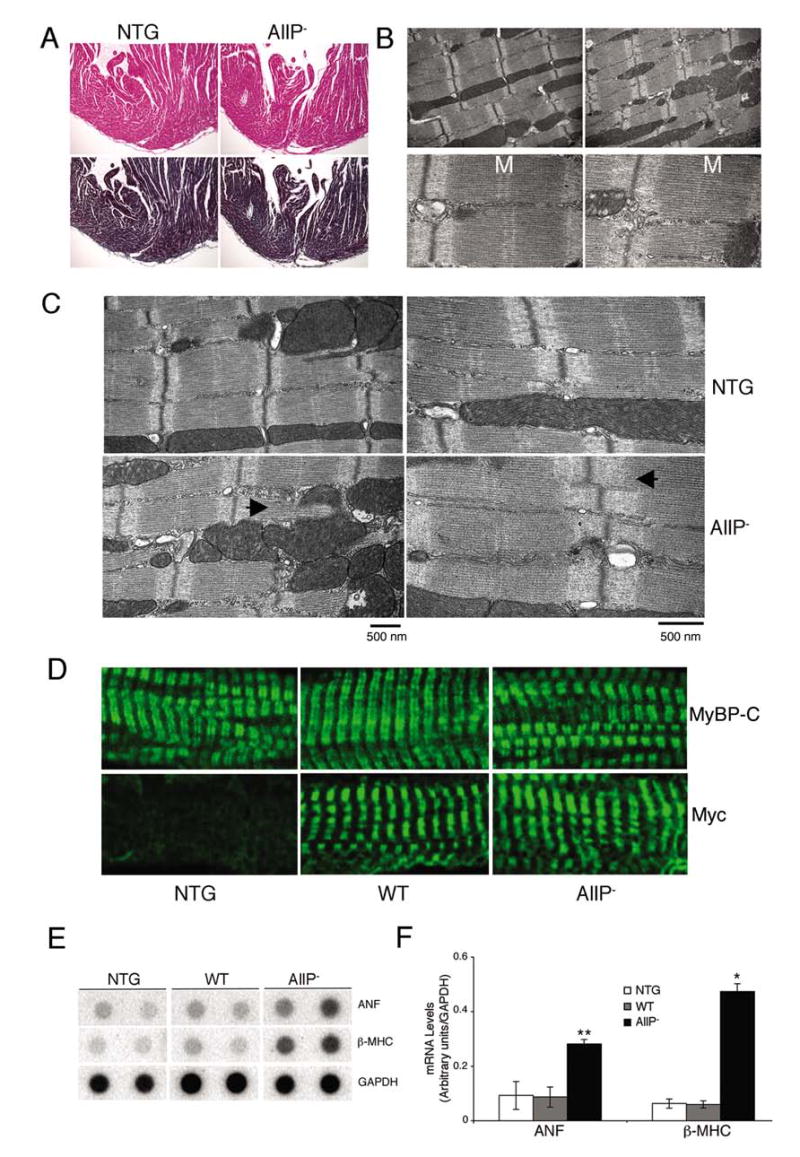
Phenotypic analyses of MyBP-CAllP− (AllP−) mouse hearts. A, Longitudinal sections derived from 3 months old left ventricle stained with hematoxylin-eosin (top) or Masson trichrome (bottom) demonstrate the absence of obvious pathology (×10). B, Transmission electron micrographs of the sarcomeres. Areas of the MyBP-CAllP− ventricles occasionally show altered sarcomeric organization, with high magnification (lower panels) showing altered M-line definition (M). Magnification: upper panels, ×10000; lower panels, ×30000. C, Transmission electron micrographs of sarcomeres from 12-week-old NTG and MyBP-CAllP− TG hearts. The abnormal H-lines and I-zones (right bottom panel) and perturbation of the normal sarcomere-mitochondria architecture (left bottom panel) in MyBP-CAllP− are indicated (◂). D, Immunofluorescent staining of cMyBP-C with either anti-cMyBP-C (top) or anti-myc antibodies (bottom) show normal incorporation. E, RNA dot blot analyses of hypertrophic markers. Four μg of total RNA was loaded in each dot. The quantitative data are summarized in F. *P<0.001, **P<0.01 MyBP-CAllP− (AllP−) vs NTG or MyBP-CWT (WT).
The lack of definition at the central part of the sarcomere is reminiscent of the altered sarcomeres that were observed in mice in which cMyBP-C expression was ablated.3 To confirm that MyBP-CAllP− was being incorporated normally into the sarcomere, we carried out immunohistochemical analyses with both anti-myc and polyclonal anti-cMyBP-C-antibodies. Both MyBP-CAllP− and MyBP-CWT proteins were incorporated normally into the sarcomere (Figure 3C).
Despite the subtle ultrastructural changes, no cardiac hypertrophy and/or dilation in the MyBP-CAllP− could be detected. The heart/body weight ratios did not significantly differ between NTG (0.53 ± 0.05, n = 8) and MyBP-CAllP− (0.60 ± 0.05, n = 8; P <0.08) littermates at 3 months. However, we reasoned that the ultrastructural changes reflected a structural deficit that might result in subtle alterations in the transcriptional patterns and indeed, found that β-MHC and atrial natriuretic factor (ANF) transcript levels, which can serve as sensitive molecular markers for cardiac stress, were significantly increased in MyBP-CAllP− hearts when compared to either NTG or MyBP-CWT samples (Figure 3D and 3E).
Depressed Cardiac Function in MyBP-CAllP− Mice
On the basis of the altered ultrastructure, we considered that partial replacement of cMyBP-C with a nonphosphorylatable form might affect whole organ function. Cardiac function was evaluated both noninvasively and by catheterization in the intact animals at 3 months. M-mode echocardiography in unsedated animals showed that the MyBP-CAllP− animals had normal cardiac dimensions and function under baseline conditions (Table 2S, Online Supplement). As cMyBP-C phosphorylation occurs in response to alterations in intracellular calcium levels (activating calcium/calmodulin dependent kinase) and β- adrenergic stress (activating PKA), we reasoned that adrenergic stimulation might reveal a deficit in the MyBP-CAllP− animals’ ability to respond to dobutamine. Hemodynamic load can be significantly altered via β-adrenergic stimulation, activation of PKA and the subsequent phosphorylation of phospholamban, cTnI and MyBP-C. Phosphorylation of cTnI and phospholamban leads to increases in crossbridge cycling and enhanced relaxation but the role that cMyBP-C plays in these processes, if any, is unclear. Cardiac function was assessed by in vivo catheterization and dobutamine stimulation. The complete data are shown in Table 3S, Online Supplement. At baseline in this model, we were able to detect significant differences in the basal LV end systolic pressure (83.6 ± 1.0), dP/dtmax (6051.1 ± 241.5) and dP/dtmin (−5541.1 ± 151.2) in the MyBP-CAllP− hearts, compared to the NTG and MyBP-CWT groups. The MyBP-CAllP− hearts had decreased contractile performance (peak dP/dtmax) at maximum dobutamine stimulation, suggesting that a lack of cMyBP-C phosphorylation inhibits maximum contractility. To confirm that these changes were not due to compensatory phosphorylation of the other contractile proteins, such as the myosin light chains, the phosphorylation status of these proteins was examined using 2-dimensional electrophoresis and found to be unchanged in the MyBP-CAllP− mouse hearts (Figure 1S, Online Supplement).
MyBP-CAllP− Fails to Rescue the MyBP-C(t/t) Phenotype
The data indicated that partial replacement with a nonphosphorylatable cMyBP-C led to both structural and functional deficits. If phosphorylatable cMyBP-C serves an essential function in the heart, complete replacement of endogenous cMyBP-C with MyBP-CAllP− should lead to significant functional deficits or even death. To test this hypothesis, line 262 was bred with MyBP-C(t/t),3 which should result in a homogenous complement of MyBP-CAllP− as MyBP-C(t/t) produces only a small amount of protein that is truncated and nonfunctional. The MyBP-CWT line was also bred to MyBP-C(t/t) in order to confirm that a transgenic strategy could, in fact rescue the MyBP-C(t/t) phenotype. Northern and dot blot analysis confirmed robust expression of both MyBP-CWT and MyBP-CAllP− in the MyBP-C(t/t) background (Figure 4A). SDS-polyacrylamide gel electrophoresis (Figure 4B) and western blot analysis (Figure 4C) using both anti-myc and anti-MyBP-C antibodies demonstrated the absence of cMyBP-C in MyBP-C(t/t) hearts and the presence of MyBP-CWT and MyBP-CAllP− in the MyBP-C(t/t) background, at approximately the levels observed for the endogenous protein in NTG hearts. IEF was subsequently performed on untreated, PKA-treated and phosphatase-treated samples in order to define the phosphorylation states of the total cMyBP-C complement in the mice and confirmed that MyBP-CAllP− “replacement” in the MyBP-C(t/t) background was complete (Figure 4D).
Figure 4.
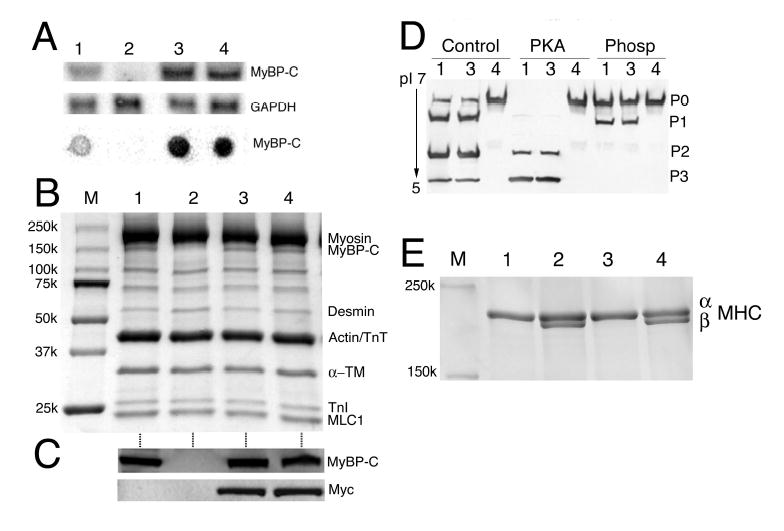
MyBP-CAllP− fails to rescue the MyBP-C(t/t) cardiac phenotype. A, Representative Northern (upper two panels) and dot-blot (lower panel) analyses of cMyBP-C mRNA from the left ventricles of 12-week old NTG (lane 1), homozygous MyBP-C(t/t) (lane 2), MyBP-CWT:(t/t) (lane 3) and MyBP-CAllP−:(t/t) (lane 4) mice hybridized with a cMyBP-C probe. GAPDH was used as a loading control. B, SDS-PAGE analysis shows the absence of cMyBP-C in the homozygous MyBP-C(t/t) hearts, replacement of endogenous cMyBP-C at normal levels in the MyBP-CWT:(t/t) and MyBP-CAllP−:(t/t) crosses and conservation of the other sarcomeric protein levels in these animals. Lanes are numbered as in panel A. C, Western blot analysis shows the absence of endogenous cMyBP-C in the homozygous MyBP-C(t/t) hearts and the presence of myc-tagged TG cMyBP-C in MyBP-CWT:(t/t) and MyBP-CAllP−:(t/t) mice. D, One dimensional IEF showing the complete replacement of MyBP-CAllP− in the cMyBP-C null background. Samples from NTG (lane 1), MyBP-CWT:(t/t) (lane 3) and MyBP-CAllP−:(t/t) (lane 4) were either untreated (Control), or treated with PKA or phosphatase (Phosp). PKA treated myofilaments show levels of MyBP-CAllP− in the MyBP-C(t/t) background that are essentially identical to NTG levels. Lane 2, which corresponds to the protein derived from MyBP-C(t/t) hearts, showed no detectable signal and was omitted. E, Myosin isoform shifts in the samples shown in panel D. MyBP-CWT expression completely rescued the cardiac phenotype observed in MyBP-C(t/t) hearts in terms of the shift to the fetal hypertrophic pattern as evidenced by β-MHC expression. In contrast, MyBP-CAllP− failed to prevent reversion to the fetal MHC isoform program. M, size markers.
The MyBP-C(t/t) mice have been characterized previously.3 The homozygous animals are viable but soon after birth display a progressive dilated cardiomyopathy. Myocyte hypertrophy, disarray, fibrosis and calcification are observed and these progress as the animals mature. The effectiveness of MyBP-CWT expression in preventing re-activation of a fetal transcription program was underscored by the lack of β-MHC expression, which serves as a sensitive marker for a nascent hypertrophic response (Figure 4E).
TG expression of MyBP-CWT also effectively rescued the overt hypertrophy displayed by the MyBP-C(t/t) hearts. In contrast, equal levels of MyBP-CAllP− expression did not rescue the MyBP-C(t/t) induced hypertrophy (Figure 5A). The MyBP-CAllP−:(t/t) and MyBP-C(t/t) mouse hearts showed markedly enlarged chambers with significant cardiac hypertrophy and myocyte disarray, as compared to the NTG and MyBP-CWT:(t/t) mice (Figure 5B). Light microscopic analyses showed pathology typical of cardiac hypertrophy in both the MyBP-C(t/t) and MyBP-CAllP−:(t/t) ventricles, while the MyBP-CWT:(t/t) derived sections appeared normal (Figure 5C and 5D). Heart/body weights in the MyBP-C(t/t) and MyBP-CAllP−:(t/t) mice were significantly elevated while, in contrast, the values derived from the MyBP-CWT:(t/t) mice were essentially normal, confirming the absence of physiological hypertrophy (Figure 5E).
Figure 5.
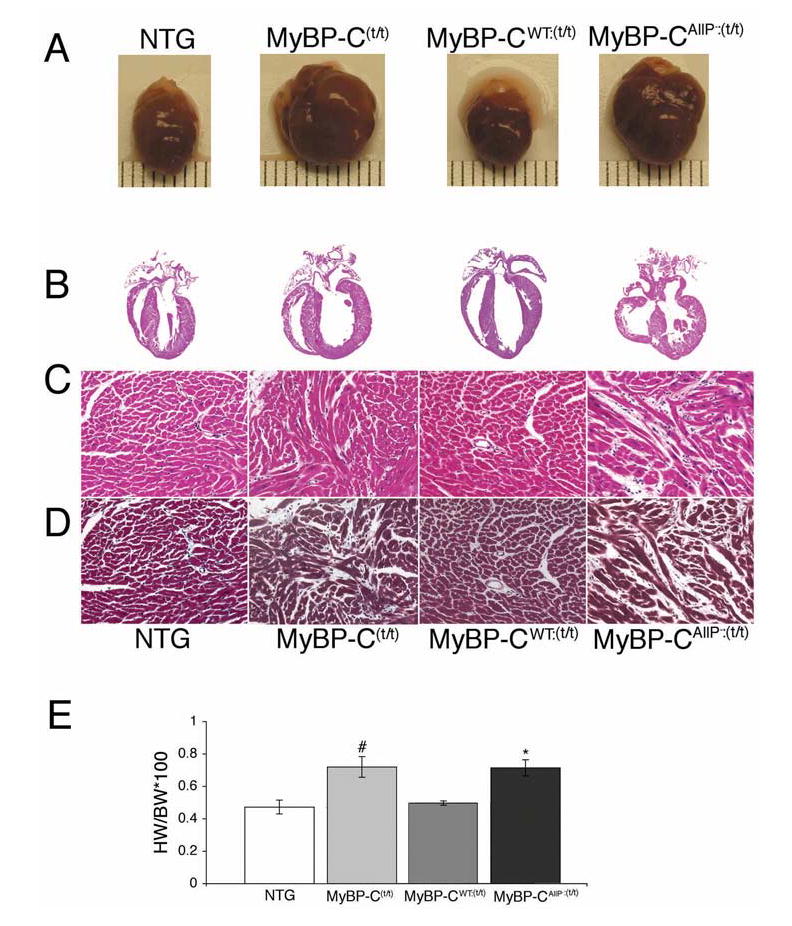
Gross cardiac morphology and histological comparisons. A, Rescue and non-rescue of overt hypertrophy by MyBP-CWT and MyBP-CAllP−, respectively. Gross morphology of 12–14 week old hearts. B, Longitudinal sections of the entire heart stained with hematoxylin-eosin (×4). C, Hematoxylin-eosin; D, Masson trichrome stained myocardial sections to assess fibrosis (×20). Shown are representative paraffin-embedded sections prepared from 10% buffered formalin perfusion-fixed hearts from 12-week-old mice. Interstitial fibrosis, calcification and myocardial disarray are seen in the MyBP-C(t/t) and MyBP-CAllP−:(t/t) sections. E, Heart weight/body weight ratios. All data are presented as means±SE (n=3). #Significant difference vs NTG (P<0.05), *Significant difference vs MyBP-CWT:(t/t) (P<0.05).
Ultrastructural analysis showed the expected lack of M-band definition in the MyBP-C(t/t) sarcomeres, as described previously in this model.3 In contrast, regular A- and I-bands and M-lines in both the MyBP-CAllP−:(t/t) and MyBP-CWT:(t/t) compared to MyBP-C(t/t) sarcomeres were apparent (Figure 6A). To confirm correct incorporation of MyBP-CAllP− and MyBP-CWT in the MyBP-C(t/t) background, we performed immunohistochemistry with both cMyBP-C and myc antibody. As expected, cMyBP-C was absent in the MyBP-C(t/t) hearts but each transgenically-encoded species showed the expected pattern of incorporation at approximately equal levels (Figure 6B).
Figure 6.
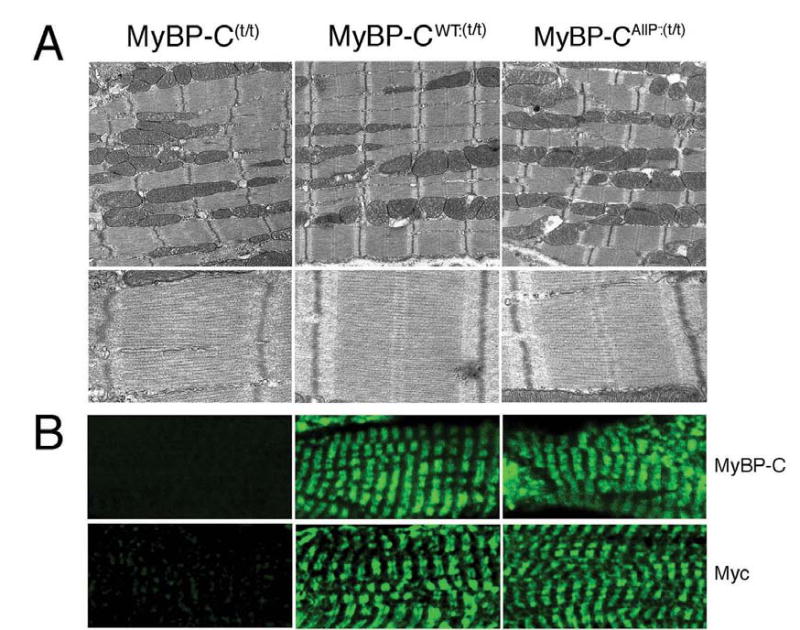
Ultrastructural and immunohistochemical analyses. A, Transmission electron micrographs showing ultrastructure of sarcomeres in MyBP-C(t/t), MyBP-CWT:(t/t) and MyBP-CAllP−:(t/t) at 12-weeks. Note the loss of M-lines in the MyBP-C(t/t). Although the MyBP-CAllP−:(t/t) hearts displayed regular A- and I-bands, and M-lines, the overall architecture was disrupted, with sarcomeres slightly misaligned. Magnification: upper panels, ×10000; lower panels, ×30000. B, Incorporation of MyBP-CAllP− into the MyBP-C(t/t) background. Ventricular myocardial sections were immunolabeled for cMyBP-C using either anti-cMyBP-C (top) or anti-myc antibodies (bottom). There is no staining of cMyBP-C in the MyBP-C(t/t) background (×60).
The inability of MyBP-CAllP− to rescue the MyBP-C(t/t) phenotype was confirmed at the functional level. M-mode echocardiography showed the MyBP-CAllP−:(t/t) and MyBP-C(t/t) mice had increased LV end diastolic and systolic end dimensions as well as reduced fractional shortening while normal shortening fractions were observed in the MyBP-CWT:(t/t) hearts (Figure 7A, Table 1). Although both MyBP-CAllP− and MyBP-CWT incorporate normally into the sarcomere, MyBP-CWT expression appears to be able to rescue the MyBP-C(t/t) phenotype while MyBP-CAllP− cannot. To define this more completely, we looked for activation of the fetal gene program in these mice as up-regulation of ANF, BNP, β-MHC and skeletal α-actin, and down-regulation of α-MHC, phospholamban (PLN) and the sarcoplasmic reticulum Ca2+ pump (SERCA) often serve as sensitive markers for hypertrophy or cardiac stress.25 The data illustrate the completeness of the rescue as no differences, even at the molecular level, were detected between the NTG and MyBP-CWT:(t/t) groups (Figure 7B and 7C). In contrast, ANF, β-MHC, BNP, skeletal α-actin were upregulated while α-MHC, PLN, and SERCA were significantly down-regulated in both the MyBP-C(t/t) and MyBP-CAllP−:(t/t) groups (Figure 7B and 7C), a pattern consistent with compromised cardiac function.
Figure 7.
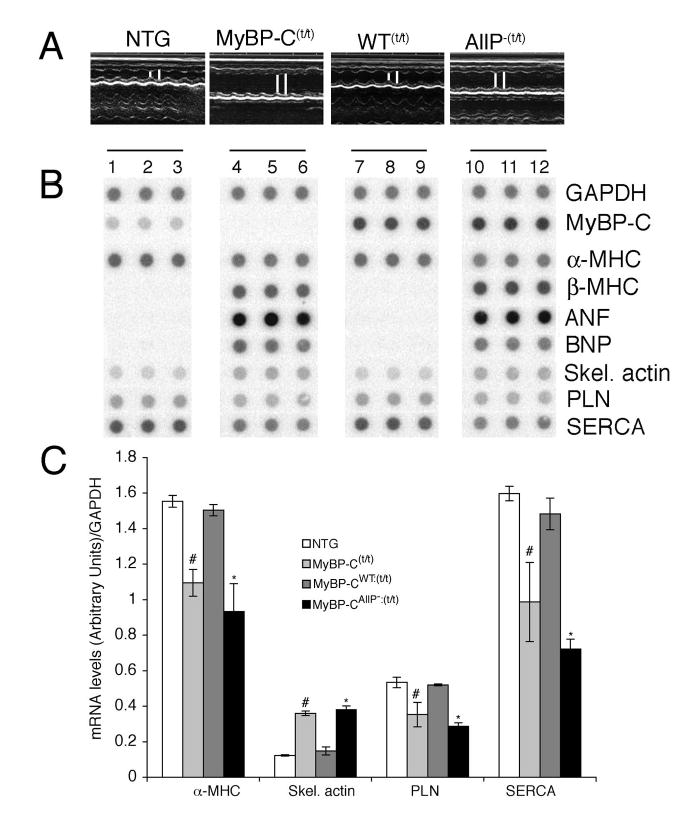
In vivo cardiac function and analysis of molecular responses. A, M-mode echocardiographic tracings show left ventricular dilation in the MyBP-Ct/t and MyBP-CAllP−:(t/t) hearts. The MyBP-CWT:(t/t) hearts show relatively preserved cardiac function. B, Dot-blot analyses of hypertrophic markers. Two μg of total mRNA from NTG (lanes 1–3), homozygous MyBP-C(t/t) (lanes 4–6), MyBP-CWT:(t/t) (lanes 7–9) and MyBP-CAllP−:(t/t) (lanes 10–12) were dotted and hybridized with GAPDH, cMyBP-C, α- and β-MHC, atrial natriuretic factor (ANF), brain natriuretic peptide (BNP), α-skeletal-actin, phospholamban (PLN) and sarcoplasmic reticulum Ca2+-ATPase 2a (SERCA) γ-32P labeled probes. Hypertrophic molecular markers such as ANF, BNP, skeletal actin and β-MHC were significantly up regulated in the MyBP-Ct/t and MyBP-CAllP−:(t/t) groups, while the NTG pattern of expression is conserved in the MyBP-CWT:(t/t) hearts. The quantitative data are summarized in C.
Table 1.
Cardiac Function Assessed by M-mode Echocardiography
| Age (weeks) | sd (mm) | LVED (mm) | hd (mm) | LVES (mm) | hs (mm) | HR (mm) | FS (%) | |
|---|---|---|---|---|---|---|---|---|
| NTG | 12 (n=6) | 0.89 ± 0.16 | 3.88 ± 0.49 | 0.78 ± 0.15 | 2.24 ± 0.29 | 1.2 ± 0.17 | 445 ± 49 | 42 ± 3 |
| MyBP-C(t/t) | 12 (n=7) | 1.02 ± 0.16## | 4.98 ± 0.7# | 0.9 ± 0.24 | 4.23 ± 0.62# | 1.16 ± 0.2 | 405 ± 53 | 40 ± 3 |
| MyBP-CWT(t/t) | 12 (n=7) | 0.95 ± 0.1 | 3.7 ± 0.39 | 0.91 ± 0.23 | 2.21 ± 0.24 | 1.4 ± 0.28 | 455 ± 54 | 40 ± 3 |
| MyBP-CAllP−:(tt) | 12 (n=8) | 1.02 ± 0.13** | 4.5 ± 0.51*¶¶ | 0.96 ± 0.16 | 3.75 ± 0.51*¶ | 1.29 ± 0.18¶¶ | 469 ± 34¶¶ | 21 ± 5*¶ |
Echocardiographic measurements were averaged from at least 3 separate cardiac cycles: septal thickness in diastole (sd), LV end diastolic thickness (LVED), free wall thickness during diastole (hd), LV end systolic thickness (LVES), septal thickness in systole (hs), heart rates (HR) and fractional shortening (FS). The MyBP-C(t/t) and MyBP-CAllP−:(t/t) hearts demonstrated increased LV diastolic and systolic diameter as well as reduced fractional shortening. The statistical significance of differences was determined by unpaired Student’s t-tests. Data are expressed as means±SE.
P<0.001,
P<0.05, Significant difference between MyBP-C(t/t) and NTG
P<0.001,
P<0.05 Significant difference between MyBP-CAllP−:(t/t) and MyBP-CWT:(t/t)
P<0.001,
P<0.05 Significant difference between MyBP-CAllP−:(t/t) and NTG
Discussion
Heart failure is associated with diminished β-adrenergic responsiveness, loss of cardiac contractility, abnormalities in Ca2+-handling26,27 and altered contractile protein phosphorylation.18,28 Although cMyBP-C is extensively phosphorylated under basal conditions, it becomes dephosphorylated during the development of heart failure or pathological hypertrophy with the tri-phosphorylated form largely or completely absent in the advanced stages of heart failure (Figure 1). This phenomenon appears to be relatively independent of the type of cardiac stress as pressure overload, ischemic-reperfusion injury and various genetic alterations in the cardiac machinery all resulted in significantly decreased phosphorylation. Basal levels of cMyBP-C phosphorylation may be necessary for maintaining thick filament orientation, dynamic regulation and contractile mechanics,4,7,11 and compromised phosphorylation patterns almost certainly reflect alterations in these cardiac parameters. Protein phosphatase activities appear to be increased in heart failure,29 and this increased activity, along with decreased β-adrenergic responsiveness during heart failure, may be responsible for the decrease in cMyBP-C phosphorylation.
We next addressed whether altered cMyBP-C phosphorylation patterns could actually cause cardiac disease. Mice in which approximately 40% of the endogenous cMyBP-C was replaced with MyBP-CAllP− appeared overtly normal. However, ultrastructural analysis did show subtle alterations at multiple foci, consistent with the hypothesis that normal cMyBP-C phosphorylation levels are needed to maintain normal myofibril structure.18 These mice also showed up-regulation of transcripts usually associated with a nascent hypertrophic response and invasive catheterization showed that contraction and relaxation were significantly decreased. Rapid and reversible changes in thick filament structure and ordering of myosin heads can be produced in cardiac muscle by changes in the degree of cMyBP-C phosphorylation,10,30 and these changes in structure are accompanied by changes in force production.31 Cardiac MyBP-C and its phosphorylation state may play an important role in determining thick-thin filament interaction, with the force of contraction and time to half-relaxation dependent upon the C1-C2 domains’ phosphorylated state.4,10,32 While it is not clear that dephosphorylated cMyBP-C directly causes cardiac disease, our data suggest that cMyBP-C phosphorylation patterns can have a significant effect on whole heart function and compromise cardiac hemodynamics.
The inability of MyBP-CAllP− to rescue the MyBP-C(t/t) phenotype is consistent with the hypothesis that cMyBP-C phosphorylation is essential for normal cardiac function. Strikingly, equivalent TG expression of MyBP-CWT effectively rescued the MyBP-C(t/t) mice, resulting in restoration of normal morphology, preventing activation of the fetal gene program and resulting in normal cardiac hemodynamics (Table 1). Both MyBP-CAllP− and MyBP-CWT appear to incorporate normally into the sarcomere yet only the phosphorylatable form is effective in suppressing the MyBP-C(t/t) phenotype. In addition to cMyBP-C’s structural roles,32 and ability to bind to titin,33 phosphorylation of cMyBP-C extends the crossbridges from the backbone of the thick filament, changes their orientation, increases the degree of order of the crossbridges, and decreases crossbridge flexibility.23,31,34,35 While our data do not address crossbridge mechanics directly, it is clear that the mutant cMyBP-C is present in the sarcomere in a pattern that is indistinguishable from normal protein, but appears to lack some critical function necessary for maintaining the overall sarcomere architecture as manifested by the alterations observed in the sarcomere-mitochondrial spatial relationships. Further studies utilizing these mouse models should provide valuable insight into the mechanical consequences of cMyBP-C phosphorylation.
Acknowledgments
This research was supported by National Institutes of Health grants HL69799, HL60546, HL52318, HL60546, HL56370 (J.R.) and by the American Heart Association, Ohio Valley Affiliate (S.S.). The authors would like to thank Dr Robert S. Decker for sharing his technical expertise in isolating and analyzing the phosphorylated forms of cMyBP-C.
References
- 1.Watkins H, Conner D, Thierfelder L, Jarcho JA, MacRae C, McKenna WJ, Maron BJ, Seidman JG, Seidman CE. Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat Genet. 1995;11:434–437. doi: 10.1038/ng1295-434. [DOI] [PubMed] [Google Scholar]
- 2.Yang Q, Sanbe A, Osinska H, Hewett TE, Klevitsky R, Robbins J. A mouse model of myosin binding protein C human familial hypertrophic cardiomyopathy. J Clin Invest. 1998;102:1292–1300. doi: 10.1172/JCI3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McConnell BK, Jones KA, Fatkin D, Arroyo LH, Lee RT, Aristizabal O, Turnbull DH, Georgakopoulos D, Kass D, Bond M, Niimura H, Schoen FJ, Conner D, Fischman DA, Seidman CE, Seidman JG. Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J Clin Invest. 1999;104:1771. doi: 10.1172/JCI7377C1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gautel M, Zuffardi O, Freiburg A, Labeit S. Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? Embo J. 1995;14:1952–1960. doi: 10.1002/j.1460-2075.1995.tb07187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mohamed AS, Dignam JD, Schlender KK. Cardiac myosin-binding protein C (MyBP-C): identification of protein kinase A and protein kinase C phosphorylation sites. Arch Biochem Biophys. 1998;358:313–319. doi: 10.1006/abbi.1998.0857. [DOI] [PubMed] [Google Scholar]
- 6.Gruen M, Gautel M. Mutations in beta-myosin S2 that cause familial hypertrophic cardiomyopathy (FHC) abolish the interaction with the regulatory domain of myosin-binding protein-C. J Mol Biol. 1999;286:933–949. doi: 10.1006/jmbi.1998.2522. [DOI] [PubMed] [Google Scholar]
- 7.Winegrad S. Myosin binding protein C, a potential regulator of cardiac contractility. Circ Res. 2000;86:6–7. doi: 10.1161/01.res.86.1.6. [DOI] [PubMed] [Google Scholar]
- 8.Gruen M, Prinz H, Gautel M. cAPK-phosphorylation controls the interaction of the regulatory domain of cardiac myosin binding protein C with myosin-S2 in an on-off fashion. FEBS Lett. 1999;453:254–259. doi: 10.1016/s0014-5793(99)00727-9. [DOI] [PubMed] [Google Scholar]
- 9.Levine R, Weisberg A, Kulikovskaya I, McClellan G, Winegrad S. Multiple structures of thick filaments in resting cardiac muscle and their influence on cross-bridge interactions. Biophys J. 2001;81:1070–1082. doi: 10.1016/S0006-3495(01)75764-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weisberg A, Winegrad S. Alteration of myosin cross bridges by phosphorylation of myosin-binding protein C in cardiac muscle. Proc Natl Acad Sci U S A. 1996;93:8999–9003. doi: 10.1073/pnas.93.17.8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kunst G, Kress KR, Gruen M, Uttenweiler D, Gautel M, Fink RH. Myosin binding protein C, a phosphorylation-dependent force regulator in muscle that controls the attachment of myosin heads by its interaction with myosin S2. Circ Res. 2000;86:51–58. doi: 10.1161/01.res.86.1.51. [DOI] [PubMed] [Google Scholar]
- 12.Hofmann PA, Lange JH., 3rd Effects of phosphorylation of troponin I and C protein on isometric tension and velocity of unloaded shortening in skinned single cardiac myocytes from rats. Circ Res. 1994;74:718–726. doi: 10.1161/01.res.74.4.718. [DOI] [PubMed] [Google Scholar]
- 13.Garvey JL, Kranias EG, Solaro RJ. Phosphorylation of C-protein, troponin I and phospholamban in isolated rabbit hearts. Biochem J. 1988;249:709–714. doi: 10.1042/bj2490709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Konhilas JP, Irving TC, Wolska BM, Jweied EE, Martin AF, Solaro RJ, de Tombe PP. Troponin I in the murine myocardium: influence on length-dependent activation and interfilament spacing. J Physiol. 2003;547:951–961. doi: 10.1113/jphysiol.2002.038117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fentzke RC, Buck SH, Patel JR, Lin H, Wolska BM, Stojanovic MO, Martin AF, Solaro RJ, Moss RL, Leiden JM. Impaired cardiomyocyte relaxation and diastolic function in transgenic mice expressing slow skeletal troponin I in the heart. J Physiol. 1999;517 (Pt 1):143–157. doi: 10.1111/j.1469-7793.1999.0143z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Q, Sanbe A, Osinska H, Hewett TE, Klevitsky R, Robbins J. In vivo modeling of myosin binding protein C familial hypertrophic cardiomyopathy. Circ Res. 1999;85:841–847. doi: 10.1161/01.res.85.9.841. [DOI] [PubMed] [Google Scholar]
- 17.Sakthivel S, Finley NL, Rosevear PR, Lorenz JN, Gulick J, Kim S, VanBuren P, Martin LA, Robbins J. In vivo and in vitro analysis of cardiac troponin I phosphorylation. J Biol Chem. 2005;280:703–714. doi: 10.1074/jbc.M409513200. [DOI] [PubMed] [Google Scholar]
- 18.Decker RS, Decker ML, Kulikovskaya I, Nakamura S, Lee DC, Harris K, Klocke FJ, Winegrad S. Myosin-binding protein C phosphorylation, myofibril structure, and contractile function during low-flow ischemia. Circulation. 2005;111:906–912. doi: 10.1161/01.CIR.0000155609.95618.75. [DOI] [PubMed] [Google Scholar]
- 19.Hahn HS, Marreez Y, Odley A, Sterbling A, Yussman MG, Hilty KC, Bodi I, Liggett SB, Schwartz A, Dorn GW., 2nd Protein kinase Calpha negatively regulates systolic and diastolic function in pathological hypertrophy. Circ Res. 2003;93:1111–1119. doi: 10.1161/01.RES.0000105087.79373.17. [DOI] [PubMed] [Google Scholar]
- 20.Hartzell HC, Glass DB. Phosphorylation of purified cardiac muscle C-protein by purified cAMP-dependent and endogenous Ca2+-calmodulin-dependent protein kinases. J Biol Chem. 1984;259:15587–15596. [PubMed] [Google Scholar]
- 21.Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arber S, Hunter JJ, Ross J, Jr, Hongo M, Sansig G, Borg J, Perriard JC, Chien KR, Caroni P. MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell. 1997;88:393–403. doi: 10.1016/s0092-8674(00)81878-4. [DOI] [PubMed] [Google Scholar]
- 23.Weisberg A, Winegrad S. Relation between crossbridge structure and actomyosin ATPase activity in rat heart. Circ Res. 1998;83:60–72. doi: 10.1161/01.res.83.1.60. [DOI] [PubMed] [Google Scholar]
- 24.Krenz M, Sanbe A, Bouyer-Dalloz F, Gulick J, Klevitsky R, Hewett TE, Osinska HE, Lorenz JN, Brosseau C, Federico A, Alpert NR, Warshaw DM, Perryman MB, Helmke SM, Robbins J. Analysis of myosin heavy chain functionality in the heart. J Biol Chem. 2003;278:17466–17474. doi: 10.1074/jbc.M210804200. [DOI] [PubMed] [Google Scholar]
- 25.Jones WK, Grupp IL, Doetschman T, Grupp G, Osinska H, Hewett TE, Boivin G, Gulick J, Ng WA, Robbins J. Ablation of the murine alpha myosin heavy chain gene leads to dosage effects and functional deficits in the heart. J Clin Invest. 1996;98:1906–1917. doi: 10.1172/JCI118992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Port JD, Bristow MR. Altered beta-adrenergic receptor gene regulation and signaling in chronic heart failure. J Mol Cell Cardiol. 2001;33:887–905. doi: 10.1006/jmcc.2001.1358. [DOI] [PubMed] [Google Scholar]
- 27.Houser SR, Piacentino V, 3rd, Weisser J. Abnormalities of calcium cycling in the hypertrophied and failing heart. J Mol Cell Cardiol. 2000;32:1595–1607. doi: 10.1006/jmcc.2000.1206. [DOI] [PubMed] [Google Scholar]
- 28.van der Velden J, Papp Z, Zaremba R, Boontje NM, de Jong JW, Owen VJ, Burton PB, Goldmann P, Jaquet K, Stienen GJ. Increased Ca2+-sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc Res. 2003;57:37–47. doi: 10.1016/s0008-6363(02)00606-5. [DOI] [PubMed] [Google Scholar]
- 29.Neumann J. Altered phosphatase activity in heart failure, influence on Ca2+ movement. Basic Res Cardiol. 2002;97 (Suppl 1):I91–95. doi: 10.1007/s003950200036. [DOI] [PubMed] [Google Scholar]
- 30.McClellan G, Weisberg A, Winegrad S. Effect of endothelin-1 on actomyosin ATPase activity. Implications for the efficiency of contraction. Circ Res. 1996;78:1044–1050. doi: 10.1161/01.res.78.6.1044. [DOI] [PubMed] [Google Scholar]
- 31.Hofmann PA, Greaser ML, Moss RL. C-protein limits shortening velocity of rabbit skeletal muscle fibres at low levels of Ca2+ activation. J Physiol. 1991;439:701–715. doi: 10.1113/jphysiol.1991.sp018689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Flashman E, Redwood C, Moolman-Smook J, Watkins H. Cardiac myosin binding protein C: its role in physiology and disease. Circ Res. 2004;94:1279–1289. doi: 10.1161/01.RES.0000127175.21818.C2. [DOI] [PubMed] [Google Scholar]
- 33.Squire JM, Luther PK, Knupp C. Structural evidence for the interaction of C-protein (MyBP-C) with actin and sequence identification of a possible actin-binding domain. J Mol Biol. 2003;331:713–724. doi: 10.1016/s0022-2836(03)00781-2. [DOI] [PubMed] [Google Scholar]
- 34.Winegrad S. Cardiac myosin binding protein C. Circ Res. 1999;84:1117–1126. doi: 10.1161/01.res.84.10.1117. [DOI] [PubMed] [Google Scholar]
- 35.Kulikovskaya I, McClellan G, Levine R, Winegrad S. Effect of extraction of myosin binding protein C on contractility of rat heart. Am J Physiol Heart Circ Physiol. 2003;285:H857–865. doi: 10.1152/ajpheart.00841.2002. [DOI] [PubMed] [Google Scholar]