

Abstract
Aklanonic acid, an anthraquinone natural product, is a common advanced intermediate in the biosynthesis of several antitumor polyketide antibiotics, including doxorubicin and aclacinomycin A. Intensive semisynthetic and biosynthetic efforts have been directed toward developing improved analogs of these clinically important compounds. The primer unit of such polyfunctional aromatic polyketides is an attractive site for introducing novel chemical functionality, and attempts have been made to modify the primer unit by precursor directed biosynthesis or protein engineering of the polyketide synthase (PKS). We have previously demonstrated the feasibility of engineering bimodular aromatic PKSs capable of synthesizing unnatural hexaketides and octaketides. In this report we extend this ability by preparing analogs of aklanonic acid, a decaketide, and its methyl ester. For example, by recombining the R1128 initiation module with the dodecaketide-specific pradimicin PKS, the isobutyryl primed analog of aklanonic acid (YT296b, 10) and its methyl ester (YT299b, 12) were prepared. In contrast, elongation modules from dodecaketide-specific spore pigment PKSs were unable to interact with the R1128 initiation module. Thus, in addition to revealing a practical route to new anthracycline antibiotics, we also observed a fundamental incompatibility between antibiotic and spore pigment biosynthesis in the actinomycetes bacteria.
Introduction
Aklanonic acid, a member of the polyketide family of natural products1,2, is an anthraquinone compound and a biosynthetic precursor of several important antibiotics including daunorubicin, baumycin A1, nogalamycin, and the anticancer drugs doxorubicin and aclacinomycin A (Figure 1(A), Figure 2). The primary differences between these natural products lie in their substitution patterns, especially their glycosylation patterns. Over the past few decades there have been intensive attempts to prepare semi-synthetic analogs of this family of compounds.3 (Figure 1(B)) For example, anthracyclines containing a disaccharide chain with substitutions at 4′-hydroxy group or a monosaccharide with methoxymorpholino ring (PNU152243) have shown promising activity.4,5,6 Similarly, removal of the 4-methoxy group from certain anthracyclines resulted in increased drug activity.7,8 The C-9 side chain has also been modified with or without reduction of the carbonyl group.9,10,11 At least one of these analogs (MEN10755) is currently in clinical trials.3
Figure 1.
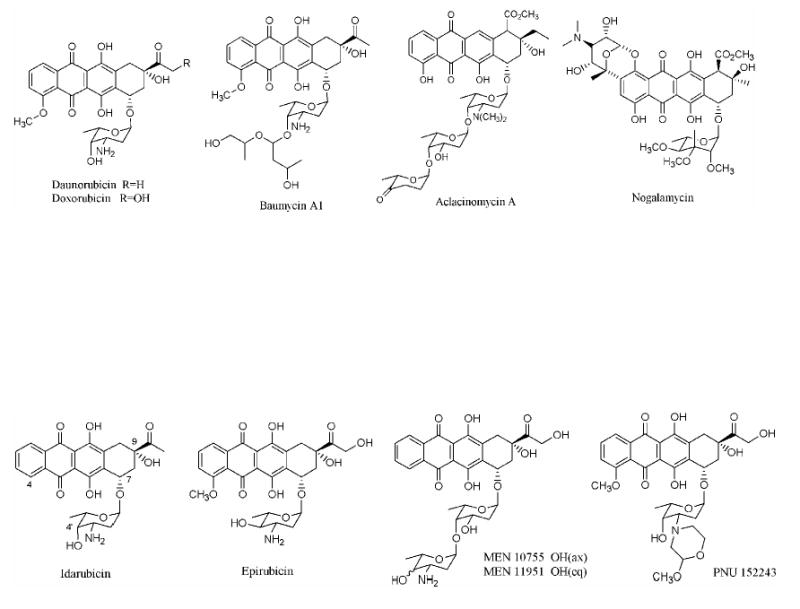
(A) Examples of natural anthracycline polyketides
(B) Examples of clinically useful semisynthetic analogs of daunorubicin and doxorubicin.
Figure 2.
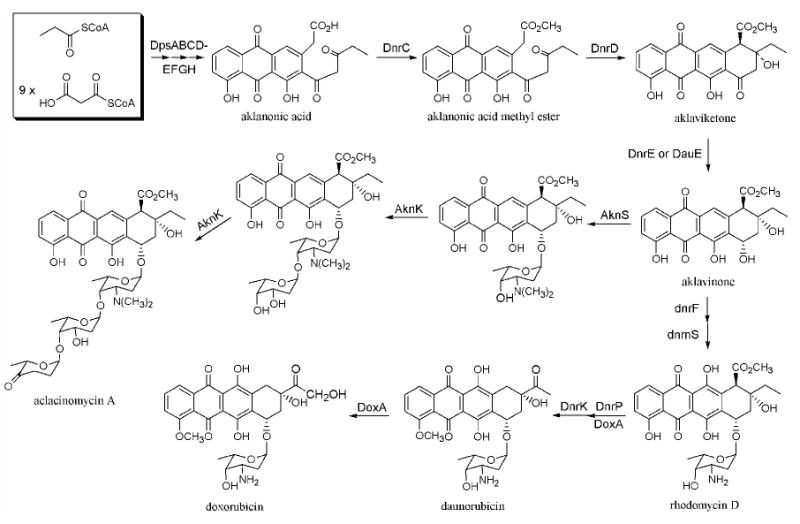
Biosynthesis of daunorubicin, doxorubicin, and aclacinomycin A
Much has been learned within the past two decades about the biosynthesis of anthracycline natural products through the combined exploitation of chemical, biochemical and genetic approaches.4,12,13 These insights have started to yield practical benefits; for example, a bacterium has been engineered to synthesize epirubicin, a clinically important semi-synthetic analog of doxorubicin with reduced side effects.14 (Figure 1(B)) Notwithstanding this progress, an even greater diversity of biosynthetic anthracyclines can be realized when the polyketide backbones of these natural products are systematically modified. Here we describe a new strategy for achieving this goal.
The primer unit in aromatic polyketide biosynthesis is an attractive site for introducing novel chemical functionality into existing natural products.15 In the absence of a dedicated mechanism for introducing non-acetyl primer units, the minimal PKS, comprising a ketosynthase (KS), a chain length factor (CLF), an acyl-carrier protein (ACP) and a malonyl-CoA:ACP transacylase (MAT), decarboxylates one malonyl equivalent and initiates polyketide assembly with an acetyl group.16,17,18 However, certain natural products, such as doxorubicin, wailupemycin and the R1128, estrogen receptor antagonists, can incorporate various non-acetyl primers, such as propionate, benzoate and a range of short-chain fatty acids, into their backbones. (Figure 3)12,19,20,21 In some cases, such as the R1128 PKS and the daunorubicin PKS, the synthase includes a dedicated module of enzymes for chain initiation.22,23 Recent studies have demonstrated that these initiation modules can collaborate with elongation modules from heterologous octaketide synthases and decaketide synthases to produce non-acetyl primed hexaketide and octaketide natural products, respectively.24 In each case, chain length of the polyketide backbone is dictated by the elongation module (i.e., the minimal PKS). Since the anthracycline antitumor antibiotics shown in Figure 1(A) are decaketides, it was therefore hypothesized that primer unit modifications of this class of natural products would require the use of a dodecaketide PKS. Specifically, we targeted the engineering of aklanonic acid and aklanonic acid methyl ester analogs, since these compounds are the first common stable intermediates in the biosynthesis of the antitumor compounds shown in Figure 1(A).
Figure 3.
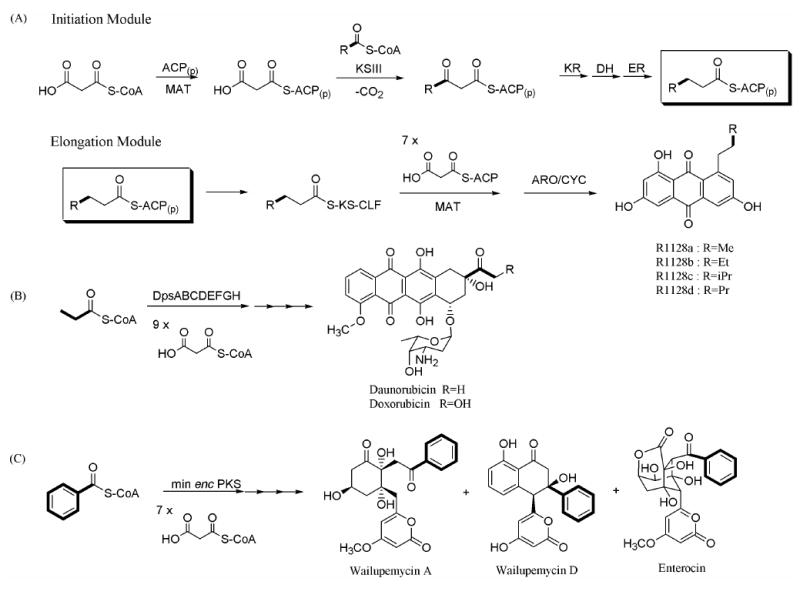
Non-acetate primed polyketide biosynthesis. (A) R1128 biosynthesis (B) doxorubicin biosynthesis (C) wailupemycin A biosynthesis. Primer units are in bold.
Several dodecaketide synthases have been reported in the literature. For example, the spore pigment PKSs from various Streptomyces species, including whiE from S. coelicolor 25, sch from S. halstedii, and cur from S. curacoi, synthesize dodecaketide backbones.26,27 The pradimicin PKS from Actinomadura hibisca has also been identified as a dodecaketide synthase.28 Although our initial efforts to prepare aklanonic acid analogues attempted to harness the spore pigment PKSs, we observed a fundamental incompatibility between the R1128 initiation module and the spore pigment elongation modules. We therefore harnessed the pradimicin KS-CLF to successfully produce a range of aklanonic acid analogs in good yield. In addition to providing a route to new antitumor compounds, our results highlight the biochemical logic of PKS specificity during contemporaneous antibiotic and spore pigment synthesis in the life cycle of an actinomycete bacterium.
Experimental Methods
Materials
Silica gel plates Si250F for thin layer chromatography were obtained from J. T. Baker. All other chemicals and solvents were purchased from Sigma, Aldrich, and Fisher Scientific and were of the highest available grade.
Bacterial Strain and General Methods for DNA Manipulation
S. coelicolor strain CH999 was used as the host for transformation by shuttle vectors. Protoplast preparation and PEG-assisted transformation were performed as described by Hopwood et al.29 All cloning steps were performed in Escherichia coli strain XL-1 Blue (Stratagene). PCR was performed using PfuTurbo polymerase (Stratagene). PCR products were first cloned into a pCR-Blunt vector (Invitrogen) followed by DNA sequencing (Stanford PAN Facility). Unmethylated DNA was obtained using the methylase-deficient strain GM2163 (New England Biolabs). T4 DNA ligase (Invitrogen) was used for ligation of restriction fragments.
Construction of Plasmids
The following primers were used to amplify the individual genes: Sch KS: 5′-GATTAATTAAGGAGGAGCCATCATCATGAGCCGCCGGGTCGTCGTCACCGG-3′, 5′-CGATGCATTCATCTCAGCCTCCTCTCCGGTCCGG-3′ (PacI/NsiI); Sch CLF: 5′-GAATGCATGGAGGAGCCATCATGAGCGCACCCCGGCGAGCCGTCG-3′ (NsiI); whiE ACP: 5′-CCTCTAGAGGAGGAGCCCATATGACCGATCAGCAGCTGGAC-3′, 5′-GGACTAGTCCACTCCCGTCTTGAGGGCGCC-3′ (XbaI,NdeI/SpeI); whiE cyclase: 5′-GCACTAGTGCCCTCAAGACGGGAGTGTGACATG-3′ (SpeI); Pms KS: 5′-GATTAATTAAGGAGGAGCCATCATGAGCCGACCGCAGGGGGGCGGG-3′, 5′-GGATGCATCATGCGGCCTCCCGCAGGACGG-3′ (PacI/NsiI); Pms CLF: 5′-GGATGCATGGAGGAGCCATCATGAGCGTCCTGACGGCGGACGCGCCGG-3′, 5′-GGTCTAGAGGTCAGGTGTTCGGGCCGCGCAGCACC-3′ (NsiI/XbaI); Pms ACP: 5′-GGTCTAGAGGAGGAGCCCATATGGCAACCCGCGAACGCACCATCG-3′, 5′-GGACTAGTCATCGCTCGGCCACCGCCCGCCCG-3′ (XbaI,NdeI/SpeI); Pms CYC: 5′-GGACTAGTGGAGGAGCCATCATGACGCAGCTGGCGCACCGACAGCG-3′, 5′-GGTCACCGCGCGCCCCGGGCCGCCCG-3′ (SpeI); gris ARO/CYC: 5′-GGTTTAAACGGAGGAGCCAGCATGTCGCAGCCCGGCCTGCGCGAGG-3′, 5′-GGACTAGTCAGCCCCGCCGGCCCGCGGCACG-3′ (PmeI/SpeI); dpsY CYC: 5′-GGACTAGTGGAGGAGCCAGCATGTGCTGTGGTGGCCCGAGAGGAG-3′, 5′-CATCCTAGGAATTAGCTAGCTCATTCGTCGACGAAGGCGACCG-3′ (SpeI/NheI,AvrII); dnrG oxidase: 5′-GATGCTAGCGGAGGAGCCAGCATGCCACAGCCAGAACCGAACGACG-3′, 5′-CATCCTAGGAATATCACGTCGTCCGCCCTCCGGGCCCG-3′ (NheI/AvrII); dnrC methyl esterase: 5′-GGCCTAGGAGGAGCCAGCATGCAGGATTCGTCCTACAAAGAG-3′, 5′-GGTCAGTGGGCGACCCGGGCGGTGACG-3′ (AvrII); dnrD cyclase: 5′-GGCCTAGGAGGAGCCAGCATGAGCACGCAGATCGATCTGGTCCG-3′, 5′-GGACTAGTCAGGGGCGCCAGCCCTCGGGGGTGGG-3′ (AvrII/SpeI). The introduced restriction sites are shown in italics and indicated in parentheses. Genes encoding sch KS and CLF were amplified from pIJ4301,30 and genes encoding whiE ACP and cyclase were amplified from pIJ4296.30 Genes encoding pms KS, CLF, ACP, and CYC were amplified from the genomic DNA of Actinomadura hibisca P157-2.28 Genes encoding gris ARO/CYC were amplified from pIJ5216.31
Genes encoding dpsY CYC, dnrG oxidase, and dnrC methyl esterase were amplified from the genomic DNA of S. peucetius.32,33 Different combinations of cassettes, as shown in Table 1, were introduced into either pYT127 (KR-R1128 initiation module) or pYT128 (R1128 initiation module without KR) to yield the corresponding constructs.24 The physical maps of plasmids pYT266, pYT292, pYT296 and pYT299 are described in Figure 5.
Table 1.
Plasmids Constructions and Resulting Polyketide Productsa
| Plasmid | Minimal PKSb | KR(actIII) | Aromatase, Cyclase | R1128 LM | Polyketide Products | Ref |
|---|---|---|---|---|---|---|
| pYT227 | sch with whiE ACP | − | − | − | TW93c-h | Ref. 30 |
| pYT226 | sch with whiE ACP | + | − | − | TW94c-d YT231a, b (1, 2) | Ref. 30 This work |
| pYT232 | sch with whiE ACP | − | − | + | TW93c-h | This work |
| pYT231 | sch with whiE ACP | + | − | + | TW94c-d YT231a, b (1, 2) | This work |
| pYT236 | sch with whiE ACP | − | whiE CYC | + | TW95a, b | Ref. 30 This work |
| pYT210 | sch with zhuN | − | zhuI, zhuJ | + | No polyketide | This work |
| pYT209 | sch with zhuN | + | actVII, actIV | + | No polyketide | This work |
| pYT243 | pms with zhuN | − | − | + | YT243a, b (3, 4) (SEK15-like) | This work |
| pYT266 | pms with zhuN | + | − | + | YT266a, b (5, 6) (RM20b-like) | This work |
| pYT253 | pms with zhuN | + | pms CYC | + | TW95a, b | This work |
| pYT259 | pms with pms ACP | + | pms CYC | + | TW95a, b | This work |
| pYT292 | pms with zhuN | + | grisARO/CYC | + | YT292a, b (7, 8) (SEK43-like) | This work |
| pYT296 | pms with zhuN | + | grisARO/CYC dpsY | + | YT296a, b (9, 10) (aklanonic acid-like) | This work |
| pYT299c | pms with zhuN | + | grisARO/CYC dpsY | + | YT299a, b (11, 12) (methyl ester) | This work |
The Streptomyces coelicolor strain CH999 is used as the host for polyketide biosynthesis. Each plasmid is derived from pRM5.16 Each construct contains genes for the minimal PKS, cyclases and the R1128 initiation module, which consists of zhuG, zhuH and zhuC. Products are analyzed by LC/MS and NMR.
The minimal PKS consists of the indicated KS-CLF, ACP and the endogenous MAT.
pYT299 has methyl transferase(dnrC) gene which is different from pYT296.
Culture Conditions, Extractions, and Small-scale Analysis
Strains were grown on R5 media plates29 containing 50 mg/L thiostrepton at 30 °C for 7–10 days. For LC/MS and analytical HPLC analysis, a well-pigmented plate was chopped into fine pieces and extracted with 30 mL of ethyl acetate containing 1% acetic acid. The solvent was removed in vacuo and the residue was dissolved in 1 mL of methanol. The polyketide products were separated by analytical reverse-phase HPLC and detected at 280 and 410 nm using the Alltech Econosphere C18 column (250 mm × 4.6 mm); linear gradient: 10% acetonitrile(MeCN) in water (0.1% TFA) to 60% MeCN in water (0.1% TFA) over 30 min with a flow rate of 1 mL/min. HPLC retention times were as follows; 1: 25.3 min; 2: 21.8 min; 3: 24.1 min; 4: 25.6 min; 5: 30.5 min; 6: 32.6 min; 7: 24.6 min; 8: 27.7 min; 9: 38.2 min; 10: 38.7 min; 11: 40.5 min; 12: 40.8 min; linear gradient: 40% MeCN in water (0.1% TFA) to 80% MeCN in water (0.1% TFA) over 30 min with the flow rate of 1 mL/min; 9: 22.3 min; 10: 25.3 min; 11: 33.8 min; 12: 35.1 min(data not shown). LC/MS was performed at the Vincent Coates Foundation Mass Spectrometry Laboratory at Stanford University using a ThermoFinnigan quadrupole ion trap LC/MS system either by electrospray ionization or by atmospheric pressure chemical ionization (both positive and negative ionization).
Large-scale Production and Isolation
A sufficient number of R5 plates (40–120 plates depending on the yield of the product) streaked with the desired CH999 strains were incubated at 30 °C or 7–10 days. The plates were chopped into fine pieces and extracted with 1 to 3 L of ethyl acetate containing 1% acetic acid according to the number of plates. The extract was dried over sodium sulfate, and the solvent removed in vacuo. The residue was purified by short path silica gel column chromatography (eluted with 50% ethyl acetate in hexane with 1% acetic acid), and the product containing fractions were collected for preparative reverse-phase HPLC. The solvent was removed in vacuo, and the residue was dissolved in 5 mL of methanol and filtered for injection in preparative reverse-phase HPLC column (250 mm × 22.5 mm, C18 column; Alltech Econosil). A 10%–60% MeCN in water (0.1% TFA) gradient was used over 50 min with a flow rate of 3 mL/min. The solvent was removed in vacuo from the collected fractions containing the expected biosynthetic product, and the residue was dissolved in 1 mL of acetone to be loaded onto a preparative TLC plate. Preparative TLC’s were developed in various compositions of ethyl acetate in hexane with or without 1% acetic acid, and the desired bands were excised from the TLC plate and washed with ethyl acetate for the recovery of the compound.
Spectroscopic Analysis
The 1H and 13C NMR spectra were obtained on Varian Inova-500 or Mercury-400 spectrometers. The 1H NMR spectra were referenced at 0 ppm with internal tetramethylsilane in CDCl3 and at 3.31 ppm in methanol-d 4. The 13C NMR spectra were referenced at 77.1 ppm in CDCl3 and at 49.1 ppm in methanol-d 4. Multiple bond 1H–13C connectivity was determined by proton-detected HMBC on Varian Inova-600 NMR instruments. The 1H and 13C NMR assignments are summarized in Tables 2–4. Long-range 13C–1H HMBC correlation data for the compounds: 1: H4 (C2, C3, C5, C6), H8 (C6, C7, C9, C10, C12), H10 (C8, C9, C11), H14 (C6, C12, C13, C16), H20 (C18, C22), H22 (C18, C20, C24), H24 (C18, C22, C23); 2: H4 (C1, C2, C3, C6), H8 (C6, C10, C12), H9 (C7, C11), H10 (C11, C12), H14 (C6, C12, C13, C16), H20 (C18, C22), H22 (C18, C20, C24), H24 (C18, C22, C23); 10: H2 (C1, C3, C4, C16), H4 (C2, C5, C6, C14), H8 (C6, C10), H9 (C7, C11), H10 (C8, C12), H20 (C18, C19, C21, C22, C23), H21 (C19, C20, C22, C23), H22 (C20, C21, C23), H23 (C21, C22, C23); 12: H2 (C1, C3, C4, C16), H4 (C2, C5, C6, C14), H8 (C6, C10), H9 (C7, C11), H10 (C8, C12), H20 (C17, C18, C19, C22), H21 (C19, C20, C22, C23), H22 (C20, C21, C23), H23 (C21, C22, C23), H25 (C1). LR-ESIMS and HR-ESIMS were collected under positive or negative ionization mode as follows: (+)-ESI mass spectra are obtained as [M+H]+ or [M+Na]+ and all (−)-ESI mass spectra are obtained as [M−H]−. LR-ESIMS m/z: 1: 451.2 ([M−H]−, C24H19O9); 2: 433.0 ([M−H]−, C24H17O8); 3: 426.9 ([M+H]+, C23H23O8); 4: 440.9 ([M+H]+, C24H25O8). HR-ESIMS m/z: 5: 427.1389 (calculated for C23H23O8: 427.1393); 6: 441.1549 (calculated for C24H25O8: 441.1549); 7: 409.1293 (calculated for C23H21O7: 409.1287); 8: 423.1442 (calculated for C24H23O7: 423.1444); 9: 419.1125 (calculated for C22H20O7Na: 419.1107); 10: 461.1225 (calculated for C24H22O8Na: 461.1212); 11: 433.1255 (calculated for C23H22O7Na: 433.1263); 12: 475.1364 (calculated for C25H24O8Na: 475.1369). HR-ESIMS was performed at the Vincent Coates Foundation Mass Spectrometry Laboratory at Stanford University using a Micromass QTof API US (Manchester, United Kingdom) quadrupole-time of flight mass spectrometer, which was operated in MS-only mode with an internal standard (poly-alanine, Sigma P9003).
Table 2.
1H and 13C NMR assignments for YT231a (1) and YT231b (2)a
| YT231a
|
YT231b
|
|||
|---|---|---|---|---|
| Nob | 13C δ (ppm) | 1H δ (ppm) (m, area, JHH (Hz)) | 13C δ (ppm) | 1H δ (ppm) (m, area, JHH (Hz)) |
| 1 | 164.3 | 162.6 | ||
| 2 | 90 | 5.44 (d, 1H, 3) | 96 | 5.46 (d, 1H, 2) |
| 3 | 172.8 | 173.5 | ||
| 4 | 107.5 | 6.11 (s, 1H) | 108.0 | 6.09 (d, 1H, 2) |
| 5 | 160.5 | 157.3 | ||
| 6 | 125.8 | 122.8 | ||
| 7 | 141.1 | 136.1 | ||
| 8 | 37 | 3.09 (dd, 1H, 4, 17) | 117.4 | 7.05 (dd, 1H, 1, 8) |
| 2.86 (dd, 1H, 6, 17) | ||||
| 9 | 66.6 | 4.32 (m, 1H) | 129.2 | 7.28 (t, 1H, 8) |
| 10 | 30.8 | 2.97 (dd, 1H, 4, 17) | 109.9 | 6.75 (dd, 1H, 1, 8) |
| 2.75 (dd, 1H, 6, 17) | ||||
| 11 | 203.9 | 155.8 | ||
| 12 | 116.7 | 114.9 | ||
| 13 | 161.9 | 159.9 | ||
| 14 | 119.2 | 6.80 (s, 1H) | 111.9 | 6.71 (s, 1H) |
| 15 | 143.1 | 141.7 | ||
| 16 | 47.3 | 4.25 (d, 2H, 19) | 49.9 | 4.30 (s, 2H) |
| 17 | 204.9 | 205.4 | ||
| 18 | 120.0 | 120.6 | ||
| 19 | 161.2 | 161.5 | ||
| 20 | 101.4 | 6.14 (s, 1H) | 101.3 | 6.13 (d, 1H, 2) |
| 21 | 168.1 | 168.9 | ||
| 22 | 111.1 | 6.14 (s, 1H) | 110.8 | 6.15 (d, 1H, 2) |
| 23 | 145.8 | 140.5 | ||
| 24 | 21.4 | 2.16 (s, 3H) | 21.0 | 2.08 (s, 3H) |
Spectra were obtained at 500 MHz for proton and 125 MHz for carbon and were recorded in CD3OD.
Carbons are labeled as shown in Figure 4.
Table 4.
1H and 13C NMR assignments for YT296a, b (9, 10), YT299a, b (11, 12)a
| YT296a
|
YT296b
|
YT299a
|
YT299b
|
|||||
|---|---|---|---|---|---|---|---|---|
| Nob | 13Cδ (ppm) | 1H δ (ppm) (m, area JHH (Hz)) | 13Cδ (ppm) | 1H δ (ppm) (m, area JHH (Hz)) | 13Cδ (ppm) | 1H δ (ppm) (m, area JHH (Hz)) | 13Cδ (ppm) | 1H δ (ppm) (m, area JHH (Hz)) |
| 1 | 172.4 | 172.6 | 170.3 | 170.3 | ||||
| 2 | 39.4 | 3.80 (s, 2H) | 40.7 | 3.86 (s, 2H) | 38.9 | 3.791 (s, 2H) | 39.5 | 3.86 (s, 2H) |
| 3 | 141.1 | 141.3 | 141.5 | 142.7 | ||||
| 4 | 122.5 | 7.78 (s, 1H) | 122.3 | 7.85 (s, 1H) | 122.6 | 7.78 (s, 1H) | 122.4 | 7.77 (s, 1H) |
| 5 | 133.9 | 134.2 | 133.5 | 133.7 | ||||
| 6 | 181.0 | 179.6 | 192.8 | 181.2 | ||||
| 7 | 133.3 | 132.8 | 133.4 | 132.5 | ||||
| 8 | 125.2 | 7.86 | 120.6 | 7.86 | 120.4 | 7.86 | 120.4 | 7.86 |
| (dd, 1H, 8, 1.2) | (dd, 1H, 8, 1.2) | (dd, 1H, 8, 1.2) | (dd, 1H, 8, 1.2) | |||||
| 9 | 137.8 | 7.73 (t, 1H, 8) | 137.9 | 7.73 (t, 1H, 8) | 137.7 | 7.73 (t, 1H, 8) | 137.7 | 7.72 (t, 1H, 8) |
| 10 | 136.0 | 7.35 | 125.2 | 7.34 | 125.1 | 7.35 | 125.1 | 7.33 |
| (dd, 1H, 8, 1.2) | (dd, 1H, 8, 1.2) | (dd, 1H, 8, 1.2) | (dd, 1H, 8, 1.2) | |||||
| 11 | 160.3 | 160.6 | 160.0 | 160.3 | ||||
| 12 | 115.5 | 115.6 | 115.3 | 115.8 | ||||
| 13 | 192.8 | 193.6 | 181.2 | 192.7 | ||||
| 14 | 115.8 | 115.8 | 115.8 | 115.8 | ||||
| 15 | 162.8 | 162.8 | 162.8 | 162.8 | ||||
| 16 | 130.4 | 133.0 | 136.6 | 132.5 | ||||
| 17 | 206.7 | 184.4 | 205.8 | 184.2 | ||||
| 18 | 42.4 | 3.04 (t, 2H, 7) | 103.4 | 6.06 (s, 1H) | 42.3 | 3.04 (t, 2H, 7) | 103.2 | 5.92 (s, 1H) |
| 19 | 32.5 | 1.65-1.60 (m, 2H) | 196.0 | 32.3 | 1.67-1.58 (m, 2H) | 195.3 | ||
| 20 | 27.8 | 1.78-1.70 (m, 1H) | 37.4 | 2.44 (t, 2H, 8) | 27.8 | 1.78-1.69 (m, 1H) | 36.6 | 2.41 (t, 2H, 8) |
| 21 | 22.5 | 0.94 (d, 6H, 6) | 34.8 | 1.60-1.56 (m, 2H) | 22.5 | 0.94 (d, 6H, 6) | 33.6 | 1.59-1.53 (m, 2H) |
| 22 | 28.1 | 1.70-1.62 (m, 1H) | 52.5 | 3.714 (s, 3H) | 28.1 | 1.66-1.61 (m, 1H) | ||
| 23 | 22.4 | 0.94 (d, 6H, 6) | 22.8 | 0.93 (d, 6H, 6.5) | ||||
| 25 | 52.5 | 3.71 (s, 3H) | ||||||
Spectra were obtained at 500 MHz for proton and 125 MHz for carbon and were recorded in CDCl3.
Carbons are labeled as shown in Scheme 1.
Results
The plasmids constructed for this study are shown in Table 1. The plasmids were derived from the E. coli/S. coelicolor shuttle vector pRM5.16 (Figure 1 in Supporting Information) Each of the plasmids was transformed into the engineered S. coelicolor host CH99916 and the production of polyketides was initially assayed by using LC-MS.
Supporting Information Figure 1.
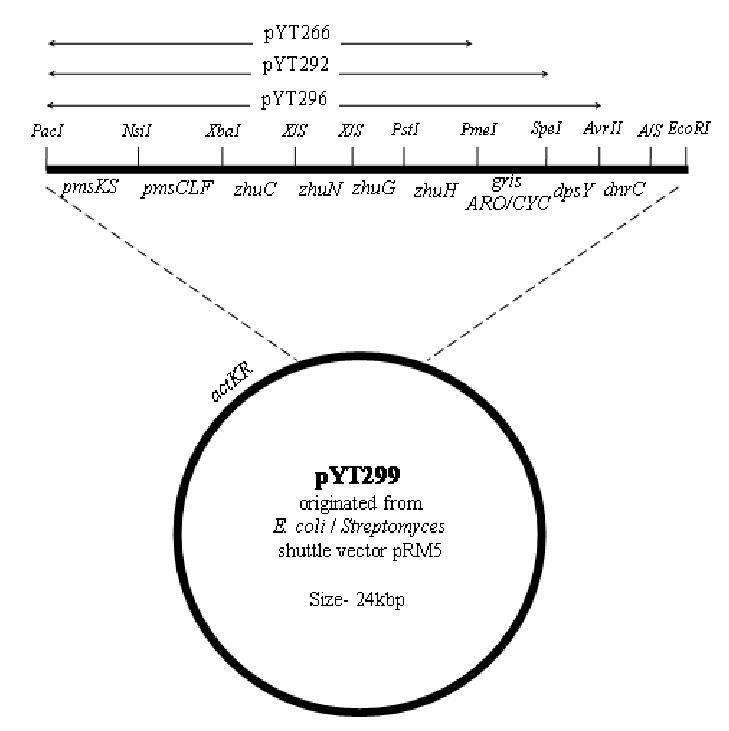
Physical map of pYT299. The recombinant gene cassettes present in pYT266, pYT292 and pYT296 are also indicated. All biosynthetic operons have PacI and EcoRI restriction sites at their ends. X/S is made by ligating the compatible ends of XbaI and SpeI restriction sites. A/S is made by ligating the compatible ends of AvrII and SpeI restriction sites.
Evaluation of KS-CLF enzymes from spore pigment biosynthetic pathways as catalysts for novel decaketide biosynthesis
For the biosynthesis of new aklanonic acid derivatives, we first examined the ability of dodecaketide-specific Sch KS-CLF from the spore pigment biosynthetic pathway of S. halstedii 26 to productively interact with the R1128 initiation PKS module. The biosynthetic products of both the Sch minimal PKS alone and the Sch minimal PKS with actIII KR have been characterized.30 (Figure 4, and Figure 2 in Supporting Information) When the Sch KS-CLF was recombined with the WhiE ACP (pYT227), we were able to observe the unreduced dodecaketides (TW93c-d). Similarly, addition of KR to pYT227 (resulting in pYT226) gave rise to the reduced dodecaketides TW94c-d. The genes encoding the R1128 initiation module were therefore added to pYT227 and pYT226 to yield pYT232 and pYT231, respectively. Constructs pYT232 and pYT231 were introduced into S. coelicolor CH999 via transformation; however LC/MS analysis of their biosynthetic products did not reveal the presence of isobutyryl or propionyl primed polyketides, as observed earlier for minimal PKSs with shorter chain length specificity.24 Instead, the transformants only produced acetyl-primed products such as TW93c-d (CH999/pYT232) and TW94c-d (CH999/pYT231). Two new acetyl-primer derived biosynthetic products were isolated from CH999/pYT231, with masses of 452 and 434. The yields of these two compounds were comparable to TW94c-d. The two compounds were characterized using 1H and 13C NMR (Table 2) along with COSY and HMBC analysis. We anticipated that the compound with mass 434 (YT231b, 2) may be a dehydration product of the compound with mass 452 (YT231a, 1); this was confirmed by 1H NMR spectroscopic analysis which shows that the two pairs of dd peaks of H-8 and H-10 in the latter compound were replaced by aromatic proton peaks in the former. The two compounds were eventually characterized as having distinct cyclization patterns compared to TW94d (Figure 4 and Table 2).
Supporting Information Figure 2.
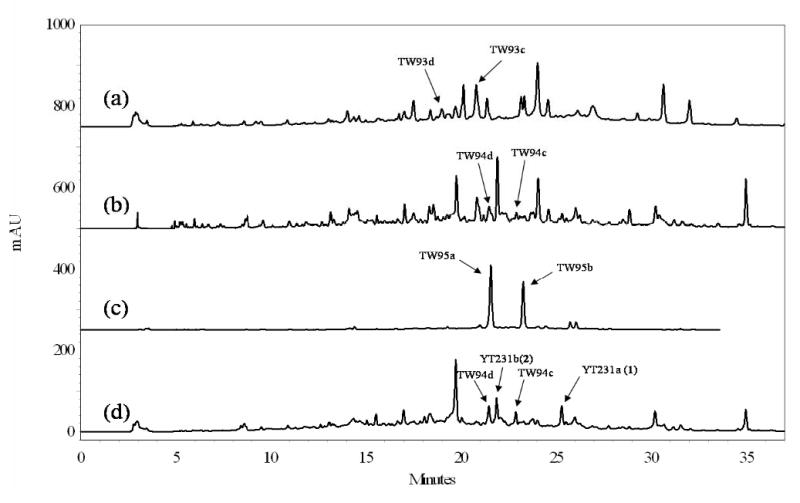
HPLC trace of extracts from (a) CH999/YT227, (b) CH999/YT226, (c) CH999/YT236, and (d) CH999/YT231. A linear gradient of 10%–60% CH3CN in water was used and the trace was monitored by UV detector at 280 nm. Peaks were assigned by LC/MS analysis. The indicated peaks were compared against authentic standards from extracts of the reported strains (Ref 30). The unassigned peaks were not further characterized, since they had a mass lower than 200 via LC/MS analysis.
Figure 4.
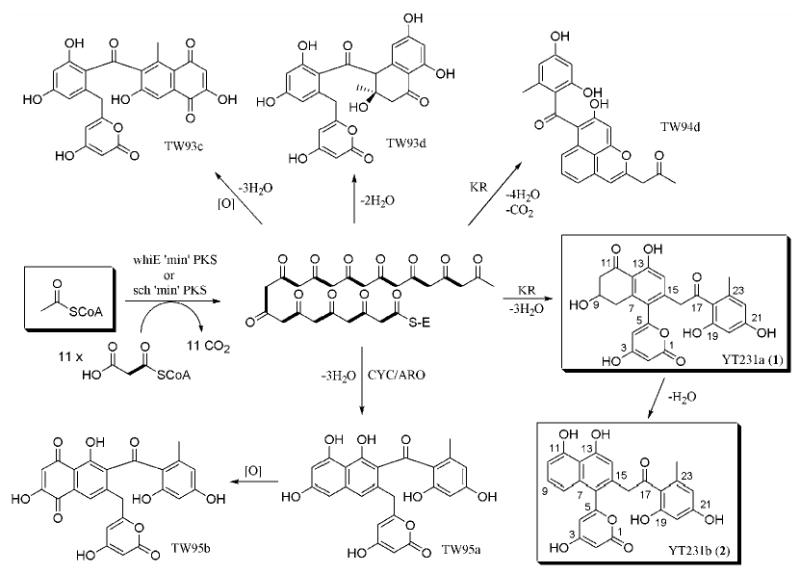
Biosynthesis by spore pigment producing dodecaketide synthases
We further examined the biosynthetic capacity of Sch KS-CLF by addition of the WhiE cyclase to pYT232 (resulting in pYT236). As we expected from the previous study by Moore et al.30, this construct resulted in abundant production of TW95a and TW95b (the isolated yields were 50 mg/L and 8 mg/L, respectively), but it still did not produce any non-acetyl primed polyketide.
To attempt improving crosstalk between the R1128 initiation module and the Sch KS-CLF, we replaced the WhiE ACP with ZhuN, the ACP from the elongation module of the R1128 PKS.24 This modified bimodular PKS was constructed in the absence (pYT210) and presence (pYT209) of the ketoreductase gene. However, neither construct yielded any polyketide, not even the dodecaketides observed with CH999/pYT231 and CH999/pYT232. Thus there appears to be a fundamental incompatibility between initiation PKS modules from antibiotic pathways and elongation PKS modules from spore pigment pathways.
Biosynthesis of non-acetyl primed decaketide backbones using the pradimicin KS-CLF
To find a dodecaketide synthase that could communicate successfully with R1128 initiation module, we selected the antifungal pradimicin polyketide synthase (pms) as a possible partner for the R1128 initiation module. The pms KS and CLF genes were amplified from genomic DNA of Actinomadura hibisca P157-2 by PCR.28 We replaced tcm KS and tcm CLF cassettes in pYT128 with the 1.3 kb PacI-NsiI cassette of pms KS and 1.3 kb NsiI-XbaI cassette of pms CLF to yield pYT243 (Table 1). This construct was introduced into S. coelicolor CH999 by PEG-mediated transformation. LC/MS analysis of the crude extract from the resulting transformant showed two peaks with strong absorption at 280 nm and masses of 426 and 440, corresponding to the expected masses of SEK15-analogs primed by propionyl and isobutyryl groups, respectively (Scheme 1). The more abundant isobutyryl-primed compound was purified by preparative HPLC and TLC. 1H NMR analysis of this compound showed similar peak assignments for most backbone protons to SEK1534. (Table 3) This compound, however, had 3 more sets of peaks in aliphatic proton region than SEK15 had, and the most characteristic peak was a doublet with 6 protons at δ = 0.8 ppm which corresponds to two terminal methyl groups in isobutyryl primed compound. Further spectroscopic analysis with 13C NMR and COSY confirmed the structure of this compound as the expected SEK15-analog (YT243b, 4) along with the comparison with spectroscopic data for SEK1534.
Scheme 1.
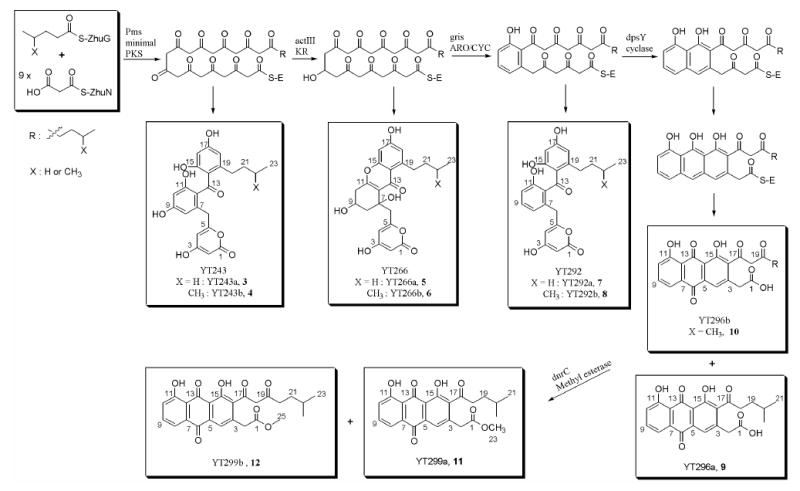
Biosynthesis of non-acetate primed decaketides by the bimodular dodecaketide polyketide synthases.
Table 3.
1H and 13C NMR assignments for YT243b (4) and YT292b (8)a
| YT243b
|
YT292b
|
|||
|---|---|---|---|---|
| Nob | 13Cδ (ppm) | 1H δ (ppm) (m, area JHH (Hz)) | 13Cδ (ppm) | 1H δ (ppm) (m, area, JHH (Hz)) |
| 1 | 164.0 | 166.8 | ||
| 2 | 86.6 | 4.90 (s, 1H) | 88.0 | 4.96 (s, 1H) |
| 3 | 172.8 | 172.3 | ||
| 4 | 101.8 | 5.49 (s, 1H) | 101.8 | 5.51 (s, 1H) |
| 5 | 163.1 | 162.3 | ||
| 6 | 39.0 | 3.53 (s, 2H) | 36.6 | 3.53 (s, 2H) |
| 7 | 135.4 | 133.6 | ||
| 8 | 109.1 | 6.27 (s, 1H) | 130.7 | 6.76 (d, 1H, 8) |
| 9 | 159.8 | 120.8 | 7.18 (t, 1H, 8) | |
| 10 | 101.6 | 6.16 (s, 1H) | 114.7 | 6.72 (d, 1H, 8) |
| 11 | 157.4 | 154.5 | ||
| 12 | 121.1 | 130.0 | ||
| 13 | 200.1 | 199.4 | ||
| 14 | 117.2 | 116.8 | ||
| 15 | 163.6 | 163.0 | ||
| 16 | 100.6 | 6.16 (s, 1H) | 100.4 | 6.09 (s, 1H) |
| 17 | 162.2 | 162.7 | ||
| 18 | 111.0 | 6.30 (s, 1H) | 109.3 | 6.11 (s, 1H) |
| 19 | 141.9 | 147.4 | ||
| 20 | 36.8 | 2.23 (t, 2H, 8) | 32.3 | 2.27 (t, 2H, 8) |
| 21 | 27.7 | 1.37-1.32 (m, 2H) | 27.8 | 1.06-1.01 (m, 2H) |
| 22 | 31.0 | 1.37-1.32 (m, 1H) | 39.4 | 1.28-1.24 (m, 1H) |
| 23 | 22.2 | 0.78 (d, 6H, 6.5) | 22.2 | 0.70 (d, 6H, 6.5) |
Spectra were obtained at 500 MHz for proton and 125 MHz for carbon and were recorded in CD3OD.
Carbons are labeled as shown in Scheme 1.
To produce decaketide analogs reduced at C-9, we inserted the act KR gene into pYT243, yielding pYT266 (Table 1). The expected biosynthetic products were the propionyl- and isobutyryl-primed analogs of RM20b/c35 (Scheme 1). Indeed, LC/MS analysis revealed two peaks with the expected masses of propionyl (YT266a, 5) and isobutyryl (YT266b, 6) primed RM20b (m/z = 427 and 441 by (−)-ESIMS, respectively), both of which were confirmed by HR-ESIMS. The above results demonstrated that the compatible interactions between the R1128 initiation module and the elongation module from the pms PKS can indeed produce non-acetyl primed decaketides.
In a further step toward engineered biosynthesis of aklanonic acid analogs, we inserted the gene encoding the pradimicin cyclase into plasmids carrying the R1128 initiation module, the act KR, pms KS-CLF and either zhuN or pms ACP, yielding plasmids pYT253 and pYT259, respectively. However, neither construct produced the expected C-7/C-12 cyclized polyketide. Instead, these constructs produced only the acetyl-primed compounds TW95a and TW95b, as evidenced by LC/MS analysis (data not shown). This result suggests that Pms CYC is a C-9 unreduced polyketide-specific cyclase like ZhuI and ZhuJ in R1128 PKS24 and the cyclases in WhiE PKS30. We could not observe YT266 compounds peaks in LC/MS analysis and this suggests that Pms CYC works faster than act KR. We therefore replaced the pms CYC with another bifunctional aromatase/cyclase, the gris ARO/CYC from the griseusin gene cluster, which has been previously used for synthesizing SEK4336. The resulting construct (pYT292) was transformed into S. coelicolor CH999. The recombinant strain appeared to produce both the propionyl (YT292a, 7) and the isobutyryl (YT292b, 8) primed analogs of SEK43, as confirmed by LC/MS (m/z = 411 and 425, respectively, by (+)-ESIMS; 409 and 423, respectively, by (−)-ESIMS) with a combined yield over 30 mg/L. (Figure 4) The compound with m/z = 425 from LC/MS, which was more abundant, was purified by successive purification steps and identified as the expected isobutyryl primed SEK43 (YT292b, 8) by 1H and 13C NMR, (Table 3) COSY, and comparison with SEK43.17 These results provide a basis for attempting the engineered biosynthesis of new aklanonic acid analogs.
Biosynthesis of aklanonic acid analogs
For the synthesis of aklanonic acid analogs with non-acetyl primer units, the monocyclic polyketide precursors of YT292a and YT292b must be cyclized in a controlled fashion into a tricyclic anthraquinone. For this we selected the dpsY cyclase from the daunorubicin biosynthetic pathway,12,37 The 820bp dpsY gene was amplified from genomic DNA of S. peucetius by PCR, and inserted as an SpeI-AvrII cassette into pYT292 to yield the construct pYT296. Based on earlier precedents, we anticipated that an endogenous oxygenase in S. coelicolor CH999 would be able to oxidize the tricyclic core into anthracyclic aklanonic acid analogs.17,24 The HPLC trace of the crude extract of CH999/pYT296 showed several characteristic anthraquinone-type peaks with absorption at 280 nm and 410 nm. LC/MS analysis demonstrated that the two most abundant anthraquinone-type peaks have masses of 396 and 438, corresponding to the expected isobutyryl-primed aklanonic acid analog and perhaps a related compound that lacks one acetyl elongation unit. (Figure 3 in Supporting Information) Production of these metabolites was scaled up. Preparative TLC yielded three major yellow products when developed with 25% ethyl acetate in hexane plus 1% acetic acid. 1H NMR analysis of one of the products revealed peaks to the aromatic hydrogen atoms and the C-2 benzylic hydrogen of aklanonic acid, but this compound lacked the enolizable methylene in the aklanonic acid side chain (δ = 6.0 ppm). (Table 4) MS analysis of this compound was also consistent with its identity as an isobutyryl primed nonaketide. Further spectroscopic analysis of the compound via 13C NMR, COSY, and HMBC confirmed its structure as shown in Scheme 2 (YT296a, 9, 2 mg/L). The 1H and 13C NMR spectra of the second compound suggested it was more closely related to aklanonic acid38, with additional peaks corresponding to the alkyl side chain, including a doublet corresponding to terminal methyl groups, analogous to YT243b (4) and YT292b (8). COSY and HMBC analyses confirmed the incorporation of isobutyryl primer into this polyketide backbone, and the compound was identified as the isobutyryl primed decaketide (YT296b, 10, 3 mg/L), expected analog of aklanonic acid. (Scheme 1) These findings demonstrate the feasibility of engineered biosynthesis of aklanonic acid analogs by harnessing the initiation module from the R1128 PKS, the elongation module from the pradimicin PKS, the act ketoreductase, the bifunctional gris aromatase/cyclase, and the DpsY cyclase from the daunorubicin biosynthetic pathway.
Supporting Information Figure 3.
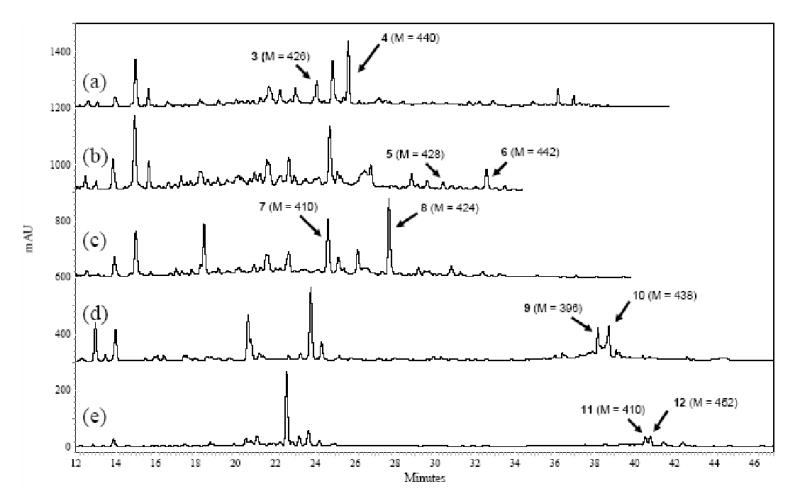
HPLC trace of extracts from (a) CH999/YT243, (b) CH999/YT266, (c) CH999/YT292, (d) CH999/YT296, and (e) CH999/YT299. A linear gradient of 10%–60% CH3CN in water was used and the trace was monitored by UV detector at 280 nm. Peaks were assigned by LC/MS analysis and confirmed by NMR and/or HR-ESIMS analysis. None of major unassigned peaks show a mass higher than 200. In particular, the major peaks in (d) and (e) at 23.8 min and at 22.5 min, respectively, were purified and analyzed. The peak at 23.8 min has a mass of 196, and the peak at 22.5 min shows a mass of 182. This, together with their 1H NMR spectra (single aromatic peak), suggests that these compounds are either truncated polyketides (e.g., see ref. 30) or not polyketides at all.
Biosynthesis of aklanonic acid methyl ester analogs
In daunorubicin biosynthesis (and presumably in the biosynthesis of other anthracyclines as well), the carboxylic acid of aklanonic acid is converted into a methyl ester, presumably by the DnrC protein.33 Therefore, we cloned the 850bp dnrC gene from genomic DNA of S. peucetius by PCR, and inserted the corresponding SpeI-AvrII cassette into pYT296, yielding pYT299. The recombinant S. coelicolor strain CH999/pYT299 was found to produce anthraquinone-like compounds, with expectedly longer retention times via reverse phase HPLC. LC/MS analysis of crude extracts from this culture identified the two most abundant anthraquinone-type products as compounds with masses 410 and 452. Corresponding [M−OMe]+ peaks (379 and 421, respectively) were also observed, suggesting that these compounds were the isobutyryl-primed nonaketide and decaketide anthraquinones, respectively. Large scale production led to their isolation and identification as methyl esters of YT296a (YT299a, 11, 2 mg/L) and YT296b (YT299b, 12, 1 mg/L) by spectroscopic comparison with authentic aklanonic acid methyl ester.38
Discussion
Polyketides are a pharmacologically important class of natural products, having yielded many clinically useful drugs. A number of improved analogs of therapeutically promising polyketides have been prepared by synthetic and, more recently, biosynthetic means. In particular, engineering biosynthetic pathways of polyketide antibiotics to incorporate novel primer units is an effective way to generate analogs of existing natural products.15,39,24
In recent reports, we and others have shown that bacterial aromatic PKSs can be engineered to synthesize rationally modified polycyclic aromatic polyketides.24,40,41,42 Specifically, we have harnessed bimodular PKSs to produce engineered hexaketides and octaketides.24 Since some of the most pharmacologically interesting polyketides, including important anticancer drugs such as doxorubicin and aclacinomycin, are decaketides, we wished to extend our methodology for primer unit modification to decaketide natural products. In doing so, however, our attempt to harness the dodecaketide PKS from spore pigment pathways failed. Analysis of this failure has highlighted the possible existence of a fundamental incompatibility between initiation modules from antibiotic pathways and elongation modules from spore pigment pathways. Fortunately, the dodecaketide synthase from the pradimicin pathway was able to productively interact with the R1128 initiation module to produce non-acetyl primed decaketides, including novel aklanonic acid analogs. Aklanonic acid is a key biosynthetic intermediate in the formation of several pharmacologically intriguing anthracyclines. The 4-methylpentanoyl primed aklanonic acid analog (YT296b, 10) and its methyl ester (YT299b, 12) were biosynthesized by an engineered bimodular aromatic polyketide synthase, together with a nonaketide product (YT296a, 9) and its methyl ester (YT299a, 11).
As has been recently demonstrated, the biosynthetic potential of this new source of anthracycline antibiotics can be considerably expanded by feeding natural and unnatural amino acids to the producer organisms.43 For example, we recently produced the 2-cyclopropylacetyl primed aromatic octaketide, 3,8-dihydroxy-1-(3-cyclopropylpropyl)-anthraquinone-2-carboxylic acid, from a culture of CH999/pYT12724 by feeding 1 g/L of cyclopropyl alanine to this organism. The amino acid is converted into 2-cyclopropylacetyl-CoA through amino acid catabolism, which in turn is incorporated into the polyketide backbone.44 The cyclopropyl functionality is useful for a variety of synthetic modifications, highlighting the potential for chemo-biosynthetic expansion of the aglycone library.45
Further biosynthetic elaboration of these new aklanonic acid analogs can be achieved by reconstituting in S. coelicolor CH999 a variety of downstream enzymes such as the fourth ring cyclase (dnrD), the ketoreductase (dnrE), and the glycosyltransferases from the daunorubicin pathway (Figure 2).33,46,47 Analogous glycosyltransferases have been identified in the daunorubicin48 and aclacinomycin A49 biosynthetic gene clusters. Although the tolerance of these tailoring enzymes for our new aklanonic acid analogs remains to be established, recent studies suggest that the l-2-deoxyfucosyltransferase (AknK) from the aclacinomycin A pathway has broad substrate specificity.49
In conclusion, engineering of bimodular aromatic polyketide synthases has opened the door to qualitatively novel anthracycline antibiotics through modifications at a position not readily accessible by synthetic methods alone.
Acknowledgments
We thank Dr. Allis Chien at the Stanford Mass Spectrometry Facility and Dr. Stephen Lynch at the Stanford Chemistry Department NMR Facility for providing helps with the mass spectrometric and the NMR spectroscopic analysis of the compounds. This work was supported by a grant from the National Institutes of Health (NIH) (CA 77248 to CK).
Footnotes
Supporting Information Available. Physical maps of plasmids pYT266, pYT292, pYT296 and pYT299. HPLC traces for CH999/pYT227, CH999/pYT226, CH999/pYT236, CH999/pYT231, CH999/pYT243, CH999/pYT266, CH999/pYT292, CH999/pYT296, CH999/pYT299 with peak assignment by LC/MS.
References
- 1.O’Hagan, D., The Polyketide Metabolites. Chichester, UK, 1991
- 2.Hopwood DA. Chem Rev. 1997;97:2465. doi: 10.1021/cr960034i. [DOI] [PubMed] [Google Scholar]
- 3.Binaschi M, Bigioni M, Cipollone A, Rossi C, Goso C, Maggi CA, Capranico G, Animati F. Curr Med Chem-Anti-Cancer Agents. 2001;1:113. doi: 10.2174/1568011013354723. [DOI] [PubMed] [Google Scholar]
- 4.Arcamone F, Cassinelli G. Curr Med Chem. 1998;5:391. [PubMed] [Google Scholar]
- 5.Arcamone FM. Biochimie. 1998;80:201. doi: 10.1016/s0300-9084(98)80003-5. [DOI] [PubMed] [Google Scholar]
- 6.Grandi M, Pezzoni G, Ballinari D, Capolongo L, Suarato A, Bargiotti A, Faiardi D, Spreafico F. Cancer Treat Rev. 1990;17:133. doi: 10.1016/0305-7372(90)90037-g. [DOI] [PubMed] [Google Scholar]
- 7.Capranico G, Supino R, Binaschi M, Capolongo L, Grandi M, Suarato A, Zunino F. Mol Pharmacol. 1994;45:908. [PubMed] [Google Scholar]
- 8.Capranico G, Zunino F, Kohn KW, Pommier Y. Biochemistry. 1990;29:562. doi: 10.1021/bi00454a033. [DOI] [PubMed] [Google Scholar]
- 9.Bachur NR. J Pharmacol Exp Ther. 1971;177:573. [PubMed] [Google Scholar]
- 10.Cassinelli G, Grein A, Masi P, Suarato A, Bernardi L, Arcamone F, Di Marco A, Casazza AM, Pratesi G, Soranzo C. J Antibiot. 1978;31:178. doi: 10.7164/antibiotics.31.178. [DOI] [PubMed] [Google Scholar]
- 11.Acton EM, Tong GL, Mosher CW, Smith TH, Henry DW. J Med Chem. 1979;22:922. doi: 10.1021/jm00194a007. [DOI] [PubMed] [Google Scholar]
- 12.Hutchinson CR. Chem Rev. 1997;97:2525. doi: 10.1021/cr960022x. [DOI] [PubMed] [Google Scholar]
- 13.Kantola J, Kunnari T, Hautala A, Hakala J, Ylihonko K, Mantsala P. Microbiology. 2000;146(Pt 1):155. doi: 10.1099/00221287-146-1-155. [DOI] [PubMed] [Google Scholar]
- 14.Madduri K, Kennedy J, Rivola G, Inventi-Solari A, Filippini S, Zanuso G, Colombo AL, Gewain KM, Occi JL, MacNeil DJ, Hutchinson CR. Nat Biotechnol. 1998;16:69. doi: 10.1038/nbt0198-69. [DOI] [PubMed] [Google Scholar]
- 15.Moore BS, Hertweck C. Nat Prod Rep. 2002;19:70. doi: 10.1039/b003939j. [DOI] [PubMed] [Google Scholar]
- 16.McDaniel R, Ebert-Khosla S, Hopwood DA, Khosla C. Science. 1993;262:1546. doi: 10.1126/science.8248802. [DOI] [PubMed] [Google Scholar]
- 17.McDaniel R, Ebert-Khosla S, Hopwood DA, Khosla C. Nature. 1995;375:549. doi: 10.1038/375549a0. [DOI] [PubMed] [Google Scholar]
- 18.Hopwood DA, Sherman DH. Annu Rev Genet. 1990;24:37. doi: 10.1146/annurev.ge.24.120190.000345. [DOI] [PubMed] [Google Scholar]
- 19.Hori Y, Abe Y, Ezaki M, Goto T, Okuhara M, Kohsaka M. J Antibiot. 1993;46:1055. doi: 10.7164/antibiotics.46.1055. [DOI] [PubMed] [Google Scholar]
- 20.Sitachitta N, Gadepalli M, Davison BS. Tetrahedron. 1996;52:8073. [Google Scholar]
- 21.Piel J, Hertweck C, Shipley PR, Hunt DM, Newman MS, Moore BS. Chem Biol. 2000;7:943. doi: 10.1016/s1074-5521(00)00044-2. [DOI] [PubMed] [Google Scholar]
- 22.Meadows ES, Khosla C. Biochemistry. 2001;40:14855. doi: 10.1021/bi0113723. [DOI] [PubMed] [Google Scholar]
- 23.Bao W, Sheldon PJ, Wendt-Pienkowski E, Hutchinson CR. J Bacteriol. 1999;181:4690. doi: 10.1128/jb.181.15.4690-4695.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang Y, Lee TS, Khosla C. PLoS Biol. 2004;2:E31. doi: 10.1371/journal.pbio.0020031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davis NK, Chater KF. Mol Microbiol. 1990;4:1679. doi: 10.1111/j.1365-2958.1990.tb00545.x. [DOI] [PubMed] [Google Scholar]
- 26.Blanco G, Pereda A, Mendez C, Salas JA. Gene. 1992;112:59. doi: 10.1016/0378-1119(92)90303-7. [DOI] [PubMed] [Google Scholar]
- 27.Bergh S, Uhlen M. Gene. 1992;117:131. doi: 10.1016/0378-1119(92)90501-f. [DOI] [PubMed] [Google Scholar]
- 28.Dairi T, Hamano Y, Igarashi Y, Furumai T, Oki T. Biosci Biotechnol Biochem. 1997;61:1445. doi: 10.1271/bbb.61.1445. [DOI] [PubMed] [Google Scholar]
- 29.Kieser, T.; Bibb, M. J.; Buttner, M. J.; Charter, K. F.; Hopwood, D. A., Practical Streptomyces Genetics. The John Innes Foundation: Norwich, 2000
- 30.Yu TW, Shen Y, McDaniel R, Floss HG, Khosla C, Hopwood DA, Moore BS. J Am Chem Soc. 1998;120:7749. [Google Scholar]
- 31.Yu TW, Bibb MJ, Revill WP, Hopwood DA. J Bacteriol. 1994;176:2627. doi: 10.1128/jb.176.9.2627-2634.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lomovskaya N, Doi-Katayama Y, Filippini S, Nastro C, Fonstein L, Gallo M, Colombo AL, Hutchinson CR. J Bacteriol. 1998;180:2379. doi: 10.1128/jb.180.9.2379-2386.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Madduri K, Hutchinson CR. J Bacteriol. 1995;177:3879. doi: 10.1128/jb.177.13.3879-3884.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu H, Ebert-Khosla S, Hopwood D, Khosla C. J Am Chem Soc. 1994;116:4166. [Google Scholar]
- 35.Fu H, McDaniel R, Hopwood DA, Khosla C. Biochemistry. 1994;33:9321. doi: 10.1021/bi00197a036. [DOI] [PubMed] [Google Scholar]
- 36.Zawada RJ, Khosla C. J Biol Chem. 1997;272:16184. doi: 10.1074/jbc.272.26.16184. [DOI] [PubMed] [Google Scholar]
- 37.Hautala A, Torkkell S, Raty K, Kunnari T, Kantola J, Mantsala P, Hakala J, Ylihonko K. J Antibiot. 2003;56:143. doi: 10.7164/antibiotics.56.143. [DOI] [PubMed] [Google Scholar]
- 38.Eckardt K, Tresselt D, Schumann G, Ihn W, Wagner C. J Antibiot. 1985;38:1034. doi: 10.7164/antibiotics.38.1034. [DOI] [PubMed] [Google Scholar]
- 39.Denoya CD, Fedechko RW, Hafner EW, McArthur HA, Morgenstern MR, Skinner DD, Stutzman-Engwall K, Wax RG, Wernau WC. J Bacteriol. 1995;177:3504. doi: 10.1128/jb.177.12.3504-3511.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hutchinson CR, Colombo AL. J Ind Microbiol Biotechnol. 1999;23:647. doi: 10.1038/sj.jim.2900673. [DOI] [PubMed] [Google Scholar]
- 41.Metsa-Ketela M, Palmu K, Kunnari T, Ylihonko K, Mantsala P. Antimicrob Agents Chemother. 2003;47:1291. doi: 10.1128/AAC.47.4.1291-1296.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shen Y, Yoon P, Yu TW, Floss HG, Hopwood D, Moore BS. Proc Natl Acad Sci U S A. 1999;96:3622. doi: 10.1073/pnas.96.7.3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang Y, Lee TS, Lee HY, Khosla C. Tetrahedron. 2004;60:7659. [Google Scholar]
- 44.The manuscript is prepared.
- 45.Wessjohann LA, Brandt W. Chem Rev. 2003;103:1625. doi: 10.1021/cr0100188. [DOI] [PubMed] [Google Scholar]
- 46.Dickens ML, Ye J, Strohl WR. J Bacteriol. 1995;177:536. doi: 10.1128/jb.177.3.536-543.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kendrew SG, Katayama K, Deutsch E, Madduri K, Hutchinson CR. Biochemistry. 1999;38:4794. doi: 10.1021/bi9827924. [DOI] [PubMed] [Google Scholar]
- 48.Otten SL, Liu X, Ferguson J, Hutchinson CR. J Bacteriol. 1995;177:6688. doi: 10.1128/jb.177.22.6688-6692.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu W, Leimkuhler C, Oberthur M, Kahne D, Walsh CT. Biochemistry. 2004;43:4548. doi: 10.1021/bi035945i. [DOI] [PubMed] [Google Scholar]