

Abstract
We investigated whether co-expression of Neurog1 and Atoh1 in common neurosensory precursors could explain the loss of hair cells in Neurog1 null mice. Analysis of terminal mitosis, using BrdU, supports previous findings regarding timing of exit from cell cycle. Specifically, we show that cell cycle exit occurs in spiral sensory neurons in a base to apex progression followed by cell cycle exit of hair cells in the organ of Corti in an apex to base progression, with some overlap of cell cycle exit in the apex for both hair cells and spiral sensory neurons. Hair cells in Neurog1 null mice show cell cycle exit in an apex to base progression about 1–2 days earlier. Atoh1 is expressed in an apex to base progression rather then a base to apex progression as in wildtype littermates. We tested the possible expression of Atoh1 in neurosensory precursors using two Atoh1-Cre lines. We show Atoh1-Cre mediated β-galactosidase expression in delaminating sensory neuron precursors as well as undifferentiated epithelial cells at E12.5. PCR analysis shows expression of Atoh1 in the otocyst as early as E10.5, prior to any histology based detection techniques. Combined, these data suggest that low levels of Atoh1 exist much earlier in precursors of hair cells and sensory neurons, possibly including neurosensory precursors. Analysis of Atoh1-Cre expression in E18.5 embryos and P31 mice reveal β-galactosidase stain in all hair cells but also in vestibular and cochlear sensory neurons and some supporting cells. A similar expression of Atoh1-LacZ exists in postnatal and adult vestibular and cochlear sensory neurons, and Atoh1 expression in vestibular sensory neurons is confirmed with RT-PCR. We propose that absence of NEUROG1 protein leads to loss of sensory neuron formation through a phenotypic switch of cycling neurosensory precursors from sensory neuron to hair cell fate. Neurog1 null mice show a truncation of clonal expansion of hair cell precursors through temporally altered terminal mitosis, thereby resulting in smaller sensory epithelia.
Keywords: hair cells, sensory neurons, inner ear, neurotrophins, cell fate, terminal mitosis
Introduction:
Atoh1 and Neurog1 belong to an ancient family of basic Helix-Loop-Helix (bHLH) genes that are involved in cell fate determination across phyla and systems, typically being expressed in proliferating precursors. For example, in coelenterates, an ATONAL-like protein determines fate between muscle and neurons (Seipel et al., 2004). In mammals, various bHLH genes determine the relative frequency of distinct retinal cell types (Akagi et al., 2004), regulate the size and progression of precursor fate in the olfactory epithelium (Wu et al., 2003), are involved in cell fate determination of neurons and switch between neuron and glial cell fates in the brain (Anderson et al., 1997; Bertrand et al., 2002), and determine the fate of progenitors of the gut epithelium (Yang et al., 2001). Except for continuously proliferating systems, these bHLH genes seem to be downregulated in neonatal mammals within days after birth (Kageyama, 1995). As in these other neuronal and non-neuronal systems, at least three bHLH transcription factors are involved in the development of hair cells and sensory neurons of the ear: Neurog1 is essential for sensory neuron development (Ma et al., 1998), Atoh1 is essential for hair cell differentiation (Bermingham et al., 1999), and Neurod1 is required for sensory neuron survival (Liu et al., 2000; Kim et al., 2001). In apparent agreement with other developing systems in which cell fate alterations have been reported after one or more of these genes have been mutated (Akagi et al., 2004), Neurog1 null mutants show not only loss of all sensory neuron formation but in addition a severe reduction of hair cells in several sensory epithelia (Ma et al., 2000). As reported for seemingly comparable bHLH gene interactions in the eye, spinal cord and the olfactory system (Gowan et al., 2001), it was hypothesized that this hair cell reduction indicates a molecular interaction of NEUROG1 with undisclosed proteins (Ma et al., 2000) in some common precursors of both cell types (Fritzsch et al., 2000; Fritzsch and Beisel, 2003).
In apparent contradiction to this idea are recent data from an elegant study which suggest that Atoh1 is a differentiation factor of postmitotic hair cells that plays no role in hair cell precursor selection in the cochlea (Chen et al., 2002). These conclusions were obtained using an Atoh1 promoter fragment to drive eGFP (Gowan et al., 2001) as well as immunocytochemistry for ATOH1; both sets of data did not show Atoh1 signal prior to cessation of proliferation of hair cell precursors. However, the transgenic construct used in this study requires the presence of Atoh1 protein to be upregulated and may not show the earliest expression of Atoh1 transcript and low levels of protein may not be detected using immunocytochemistry. Indeed, previous and more recent work has shown somewhat earlier Atoh1 expression using other techniques (Bermingham et al., 1999; Lanford et al., 2000; Zine et al., 2001; Woods et al., 2004). Other data showed an even earlier upregulation of Atoh1-eGFP in some vestibular hair cells at a time when both sensory neurons and hair cells become postmitotic in the vestibular system (Radde-Gallwitz et al., 2004). However, the onset of expression of Atoh1 in the ear has not been investigated using more sensitive techniques such as RT-PCR or Q-PCR. It seems therefore fair to say that at the moment we do not know when Atoh1 is first upregulated and what cell type is initially expressing Atoh1.
While Atoh1 is clearly involved in hair cell differentiation (Izumikawa et al., 2005), it appears that sensory epithelia composed of undifferentiated cells which express Atoh1-LacZ and Bdnf-LacZ form even in Atoh1 null mice and receive afferent and efferent innervation (Fritzsch et al., 2005). In addition, recent data (Radde-Gallwitz et al., 2004) support previous results on expression of the transcription factor Islet1 in differentiating vestibular sensory neurons and hair cells (Adam et al., 1998). These Islet1 data agree with information gathered with the neurotrophin BDNF; Bdnf mRNA is also expressed in differentiating hair cells and sensory neuron precursors that appear to delaminate from the future sensory epithelia (Farinas et al., 2001; Fritzsch et al., 2002). Together these data are compatible with the idea that some sensory neurons and hair cells may be derived from common neurosensory precursors (Fritzsch and Beisel, 2004). Indeed, recent lineage tracing work in chicken has shown some lineage relationships of sensory neurons and hair cells (Satoh and Fekete, 2005).
These data leave several questions unanswered: It remains unclear whether the detection of the earliest appearance of Atoh1 expression is compromised by the sensitivity of the employed technique, leading to false negative results. For example, it is possible that the delay in generating eGFP in the Atoh1 transgenic mice may not show the earliest expression of ATOH1 protein (needed to drive the promoter) and certainly will not show the earliest upregulation of Atoh1 message. Likewise, limited expression of Atoh1 mRNA may be below the detection threshold of in situ based techniques thus may lead to false negative results suggesting, for example, absence of Atoh1 expression in neurosensory precursors.
For this study we had six aims: First, we wanted to determine the earliest expression of Atoh1 as revealed with four different histological techniques and compare those data with RT-PCR data. Second, we wanted to identify the cellular distribution of Atoh1 expression in the progeny of these cells using Atoh1-Cre. Third, we wanted to verify atypical expressions (Lumpkin et al., 2003) in Atoh1 transgenic mice (Atoh1-eGFP, Atoh1 Cre) using Atoh1-LacZ and Q-PCR as truly reflecting Atoh1 expression and not integration artifacts. Fourth, we wanted to determine the significance of this expression in sensory neurons using a biological readout system, the pathfinding properties of sensory neurons, to assess the absence of Atoh1. Fifth, we wanted to develop a model that can explain the Neurog1 absence mediated hair cell reduction by assuming a possible NEUROG1-ATOH1 protein interaction in common precursors of hair cells and sensory neurons (neurosensory precursors). Lastly, we wanted to compare terminal mitosis as well as Atoh1 and Neurod1 upregulation in hair cells of Neurog1 null mice as compared to their littermates to study the effect of Neurog1 absence on cell fate decisions of common neurosensory precursors.
Material and Methods
Mouse lines used to generate single and compound mutant mice.
Two Atoh1 null alleles are used in this study. One allele carries a Hprt cassette (Atoh1-Hprt) and the other one carries the LacZ reporter gene (Atoh1-LacZ) instead of the coding sequence (Ben-Arie et al., 1997; Ben-Arie et al., 2000). To analyze the ATOH1/NEUROG1 interaction further we generated doubly heterozygotic mice that carried the Neurog1 null allele (Ma et al., 1998) in combination with the Atoh1-Hprt allele and crossed this line with a line carrying the Bdnf null allele combined with LacZ replacement (Bdnf-LacZ) previously described (Jones et al., 1994; Fritzsch et al., 2005).
Mice with a LacZ or Hprt marker were bred from heterozygotes as previously described (Bermingham et al., 1999; Fritzsch et al., 2005). Embryos were collected at embryonic day 11.5 (E11.5), E12.5, E16.5 and E18.5 and several heterozygotic animals were fixed at 2, 5 and 9 month of age. Older embryos and adults were perfusion or immersion fixed after appropriate anesthesia with 4% PFA for 30 minutes. Ears were rapidly dissected in PBS, and subsequently reacted for Lac-Z histochemistry overnight at room temperature (Farinas et al., 2001). Some of these ears of heterozygotic and homozygotic Atoh1 mutants were also reacted for nerve fiber stain using tubulin antibodies as previously described (Fritzsch et al., 1997) and anti β-galactosidase. All animal breeding and treatment was approved by an IACUC protocol # 0630.
In older embryos the neurotrophin Bdnf is expressed only in differentiated hair cells (Farinas et al., 2001) but may play a role in bHLH gene upregulation in earlier stages (Ito et al., 2003). We studied the expression of Bdnf-LacZ distribution in control, Atoh1 and Neurog1 null littermates. To achieve this we crossed Atoh1-Hprt/Neurog1 doubly heterozygots with Bdnf heterozygote animals. In the offspring we selected an Atoh1-Hprt/Neurog1/Bdnf-LacZ compound heterozygote male and bred this animal with Atoh1-hprt/Neurog1 heterozygote females. The resulting offspring produced Atoh1-Hprt null mutants with Bdnf-LacZ heterozygosity at the expected frequency of 1 in 8 embryos. Ears of E11.5, E12.5 and E18.5 perfusion fixed animals were reacted for β-galactosidase histochemistry as described above.
In these animals we also investigated the distribution of cells in which the terminal mitosis was visualized with BrdU injections (100 μg/g). Injections were administered for each litter only once, at noon, to a given pregnant mouse of the days E9.5–17.5 (with noon after vaginal plug considered being embryonic day 0.5). Animals were perfusion fixed at E18.5, genotyped and the ears processed for BrdU imaging. Briefly, ears were treated for 1h with 1N HCl, washed, blocked with bovine serum and incubated for one hour with anti-BrdU antibody conjugated to Alexa 647 (Invitrogen). Ear epithelia were mounted flat on a slide in glycerol, coverslipped and viewed with a confocal microscope (Biorad Radiance 2000 or Zeiss LSM). Assessment of labeled cells was achieved by first focusing on the surface of the organ of Corti using the 488 nm laser band. Subsequently stacks of images were collected through the hair cells and supporting cells to include the nucleus of hair cells, which appeared as a black hole against the autofluorescence of the cytoplasm. In a second channel, the emission of the Alexa 647 was recorded, showing the distribution BrdU in the nuclei. Nuclei were scored as labeled when 50% or more of the surface was stained in the Z-axis collapsed stacks. Identification of nuclei was supported by the fact that in most cases at least some minor specks of BrdU labeling appeared. Following this high power analysis using a 40x 1.3 NA lens, we took overview images to illustrate the distribution of BrdU labeled cells for illustration purposes only.
Some wildtype and Atoh1 null mutant embryos were also used for neuronal tract tracing as previously described (Fritzsch et al., 2005) to study the possible effect of Atoh1 absence on the pattern of innervation. In addition, some ears of Atoh1 null and wildtype animals were reacted with acetylated tubulin to reveal the pattern of innervation. Ears were whole mounted in glycerol and viewed with the Zeiss LSM confocal system.
The bHLH gene Neurod1 (formerly NeuroD) is expressed in all sensory neurons and in some differentiated hair cells in embryos older than E14.5 (Kim et al., 2001). We studied the Neurod1-LacZ expression in control and Neurog1 null littermates. For this we crossed Neurog1 heterozygotes with Neurod1-LacZ heterozygotic mice. In the offspring we selected Neurog1/Neurod1-LacZ compound heterozygote males and bred these animals with Neurog1 heterozygote females. We bred two litters of E12.5–E18.5 embryos in which we had two offspring that were Neurog1 null mutants and that were also heterozygous for Neurod1-LacZ. Ears of these E18.5 embryos were perfusion fixed and reacted for β-galactosidase histochemistry as described above.
After β-galactosidase reaction, ears were dissected and mounted flat for analysis of the sensory epithelia area. Some ears were embedded in soft epoxy resin, sectioned at 20μm thickness and analyzed in a compound lightmicroscope (Nikon Eclipse 800). Images were captured using a CCD camera and processed using ImagePro. Images were compiled into plates using CorelDraw.
Atoh1-Cre line:
To generate this line we used the JQ2-Atoh1 promoter fragment originally described by (Gowan et al., 2001) and used to analyze Atoh1 upregulation in developing hair cells (Chen et al., 2002) provided by Dr. J. Johnson. This fragment was ligated to the bacteriophage P1 Cre recombinase cDNA and injected into mouse embryo pronuclei to generate two transgenic mouse lines that carried the Atoh1-Cre. Integrity of the construct was checked by Southern blot. Transgenic animals were mated with Rosa 26 lines to demonstrate expression of Cre using the conditional LacZ reporter system of this line. We generated two transgenic strains of mice with this construct. Mice of one strain were fixed in 4% paraformaldehyde at E12.5 and E18.5, mice of the second strain were fixed at 1 month of age after lethal anesthesia by perfusion. Ears were dissected and either reacted as whole mounts for β-galactosidase or sectioned and reacted for β-galactosidase. Ears and sections were viewed in an Olympus EZ dissection scope or a Nikon Eclipse 800 and images were taken using a Coolsnap camera and Metamorph software.
RT-PCR was done on dissected inner ear tissues at E10.5 and E11.5 and Q-PCR was done on vestibular ganglia dissected form P0 and P10 ears using gene specific primers according to a published procedure (Beisel et al., 2000) to determine the absence or presence of Atoh1 transcripts. Care was taken that this tissue was not contaminated with adjacent brain tissue known to express Atoh1. The samples were treated with DNase to eliminate any trace genomic DNA. Afterwards, the quality of the RNA was assessed using the Agilent 2100 Bioanalyzer. Primer/probe sets were designed over introns to eliminate any possible amplification from contaminating genomic DNA. In the reactions themselves, no Template Control samples were used and none of these amplified a PCR product. First strand synthesis was done using a T7-dT(24) primer (Beisel et al., 2000). A primary amplification was done using mAtoh1-896 FOR (5′ CATCACCTTCGCACCGCCTC CTC 3′) and a T7-specific oligomer at standard PCR conditions with 35 cycles. A secondary amplification step was done using the nested primer sets of mAtoh1-1094 FOR (5′ CTAACA GCGATGATGGCACAGAAG 3′) and mAtoh1-1777 REV (5′ AGTGATGAAGTGCGTGTATT CTGG 3′) with 30 cycles. A primary amplification was done using mAtoh1-896 FOR (5′ CATCACCTTCGCACCGCCTCCTC 3′) and a T7-specific oligomer. A secondary amplification step was done using the nested primer sets of mAtoh1-1094 FOR (5′ CTAACAGCGATGATGGCACAGAAG 3′) and mAtoh1-1777 REV (5′ AGTGATGAAGTGCGTGTATTCTGG 3′. We also dissected vestibular ganglia of newborn (PO; N=3) and 7 day old mice (P7; N=2) and processed these ganglia for Q-PCR. We used the primers outlined above and compared the Atoh1 transcripts with Neurod1 (a bHLH gene present in sensory neurons), Neurog1 (a bHLH gene absent in postnatal sensory neurons) and ribosomal RNA.
Results
Comparison of Bdnf expression in Atoh1 and Neurog1 simple and double null mice.
We previously showed that Neurog1 null mice lose not only all sensory neurons but also show loss and reorganization of hair cells (Ma et al., 2000). We also showed that undifferentiated cells that express Atoh1-LacZ develop in Atoh1 null mice and attract afferent fibers likely because many of these cells co-express Bdnf (Fritzsch et al., 2005) a known major attractor for growing ear sensory neuron processes (Tessarollo et al., 2004). Here we wanted to analyze how the temporal expression of Bdnf is altered in the absence of Atoh1, Neurog1 or both bHLH genes using Bdnf-LacZ as a marker for hair cells and sensory neuron precursors, possibly including common neurosensory precursors.
The following differences and similarities in Bdnf-LacZ expression were apparent in the E12.5 and E18.5 ears of these mutants. First, the canal cristae and the apex of the cochlea showed roughly identical expression of Bdnf, regardless of age or genetic background and whether differentiated hair cells existed or not (Fig. 1A–K). Second, delaminating sensory neurons showed expression of Bdnf as they exited from the utricle, saccule and ductus reunions/basal turn of the cochlea of Atoh1 null and wildtype embryos (Fig. 1E,F). As expected, no Bdnf positive sensory neurons were visible outside the otocyst in Neurog1 null mice at any time during development (Fig. 1C, G). Interestingly, delaminating cells were clearly positive for Bdnf-β-galactosidase in Atoh1-Hprt null mice despite the fact that no Bdnf was expressed in the areas they delaminated from (Fig. 1F). This Bdnf positivity suggests that sensory neuron precursors are capable of Bdnf upregulation as they delaminate, but does not exclude the possibility that Bdnf is already present in wildtype neurosensory precursors, giving rise to Bdnf positive hair cells and sensory neurons. Lastly, there was a conspicuous lack of expression of Bdnf in the basal turn, utricle and saccule of Atoh1 null mutants (Fig. 1B, F). This absence of Bdnf expression in Atoh1 null mutants contrasted sharply with the profound expression of Bdnf in distinctly labeled cells of the utricle in Neurog1 null mice (Fig. 1F, G). We show Bdnf-LacZ positive cells in the utricle of E12.5 Neurog1 null mice (Fig. 1G) and Bdnf-LacZ positive cells in the ductus reuniens of E18.5 Neurog1 null mice (Fig. 1C). The frequency of these cells and their distribution was variable and we are currently investigating whether such cells are hair cells by crossing Neurog1 with Atoh1-LacZ.
Fig 1 .
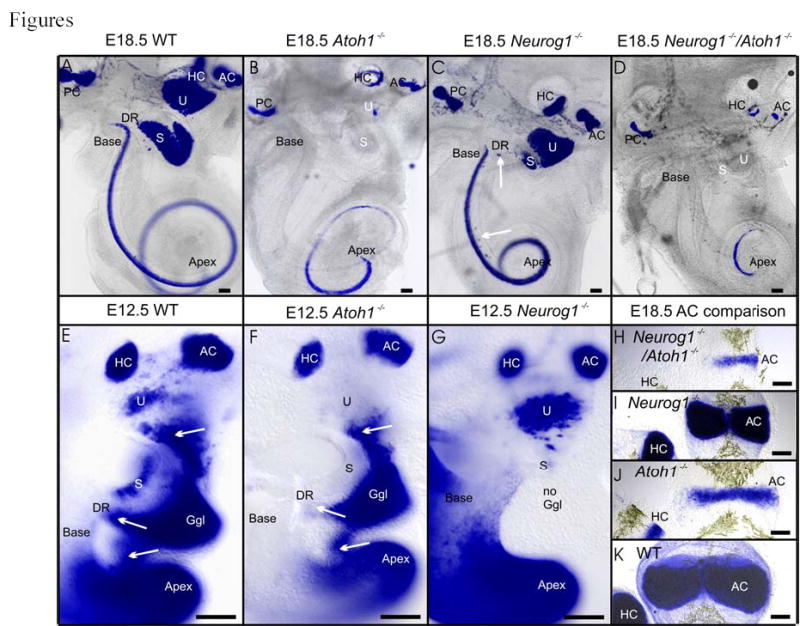
Expression patterns of Bdnf-LacZ in E18.5 (A–D, H–K), E12.5 (E–G) ears of wildtype (A, E, K), Atoh1 null mice (B, F, J), Neurog1 null mice (C, G, I) and Neurog1/Atoh1 double null mice (D, H) is shown. Note that the canal cristae (HC, AC) and the cochlear apex are strongly labeled in all cases. However, the basal turn shows no Bdnf-LacZ staining in Atoh1 null mice (B, F) and Neurog1/Atoh1 double null (D) mice. Neurog1 null and Neurog1/Atoh1 double null mice show decreased length of all sensory epithelia (C, D) with the cochlea showing only one, tightly coiled turn. The saccule (S) is almost lost in Neurog1 null mice (C) and Bdnf-LacZ positive cells appear in the ductus reuniens (DR, vertical arrow) and the GER (horizontal arrow). In E12.5 embryos, sensory neurons delaminate in wildtype (E) and Atoh1 null mice (F) from the utricle (U; arrow), the ductus reuniens (DR) leading from the saccule (S) to the cochlea (arrow), and the upper middle turn of the cochlea (arrow). No Bdnf-LacZ positive sensory neurons and no ganglion exist in Neurog1 null mutants (G; no ggl). Note that delaminating cells may upregulate Bdnf-LacZ in Atoh1 null mice de novo (F) since no Bdnf-LacZ is expressed in the utricle and saccule in Atoh1 null mutants. In contrast to the absence of Bdnf–LacZ positive cells in Atoh1 null mice (F) is the more profound expression of Bdnf-LacZ in the utricle of Neurog1 null mice (G) which have a saccule (S) formed of few cells (out of focus in G). These data suggest a differential effect of Atoh1 and Neurog1 on Bdnf-LacZ expression in hair cells, in particular in the utricle, saccule and basal turn. Direct comparison of the anterior crista in various genetic backgrounds shows the additive effect of Neurog1 and Atoh1 null mutation on the size and degree of differentiation of this sensory epithelium (H–K). AC, anterior crista; DR, ductus reuniens; Ggl, ganglion; HC, horizontal crista; PC, posterior crista; s, saccule; u, utricle. Bar indicates 100 μm.
Since sensory neurons, but not hair cells of utricle and saccule, express Bdnf in Atoh1 null mutants (Fig. 1B, F) we generated mice that were homozygotic null for both Atoh1 and Neurog1 combined with Bdnf-LacZ heterozygosity. These mice showed the combined phenotype of Neurog1 null mice (shortened cochlea, reduction of saccular recess, absence of sensory neurons; Fig. 1D; Table I) and Atoh1 (absence of Bdnf expression in utricle, saccule and basal turn of cochlea; Fig. 1D).
Table 1.
Mutation of Atoh1, Neurog1 and double Atoh1/Neurog1 affect the size and length of cochlea and anterior crista as judged by measuring the area of Bdnf expression or total length.
| Cochlea | WT | Atoh1 null | Atoh1/WT % of length | Neurog1null | Neurog1/WT % of length | Atoh1-Neurog1double null | Atoh1-Neurog1/WT % of length |
|---|---|---|---|---|---|---|---|
| Bdnf (μm) | 5495+/−68 | 3722+/− 37 | 68% | 2356+/−29 | 42% | 1329+/−18 | 24% |
| Total (μm) | 5776+/−86 | 5407+/−78 | 93% | 3083+/−48 | 53% | 3003+/−76 | 52% |
| Anterior crista | |||||||
| Width (μm) | 237+/−21 | 125+/−19 | 53% | 193+/−44 | 81% | 79+/−32 | 33% |
| Length (μm) | 608+/−35 | 451+/−38 | 74% | 413+/−35 | 68% | 225+/−86 | 37% |
| Area (μm2) | 107350+/−193 | 36487+/−283 | 34% | 58003+/−175 | 54% | 12755+/−88 | 12% |
| N=6 | N=6 | N=4 | N=4 | ||||
The data on more prominent expression of Bdnf in cells of the utricle of Neurog1 null mice at E12.5 are compatible with at least three scenarios: A) in the absence of Neurog1 hair cell precursors exit the cell cycle in younger animals and upregulate Bdnf; B) hair cells, instead of sensory neurons, differentiate out of common neurosensory precursors that are positive for Bdnf; C) sensory neurons fail to delaminate and thus remain as Bdnf positive cells, eventually dying and being replaced by hair cells forming later. In either case, more cells should be positive for Bdnf in the utricle of Neurog1 null mice at an earlier stage, precisely what we demonstrated (Fig. 1G). The assumption of earlier exit from the cell cycle of hair cells can be verified by demonstrating alterations in the time of terminal mitosis of hair cells in the Neurog1 and Atoh1 null mice, our next experiment. Such data can also show whether cells that exit the cycle at this early stage remain or die. If such cells would die, BrdU positive hair cells should not be found at later stages, thus allowing us to distinguish between the three possible scenarios.
Proliferation as revealed by BrdU in wildtype and Neurog1 null mice.
We injected BrdU once in pregnant mice ranging between E9.5 and E17.5 and analyzed the distribution of cells that showed BrdU-Alexa 647 antibody labeling of at least 50% of the area of a given nucleus at E18.5. We used the simple fact that differentiated hair cells form at this stage one row of inner and three rows of outer hair cells to identify hair cells. As is obvious from sections of the cochlea using our hair cell marker Bdnf-LacZ (Fig. 2A), hair cell nuclei are distinctly distributed from the supporting cell nuclei. Thus careful focusing of a flat mounted cochlea allows clear identification of hair cells and their nuclei using autofluorescence of the cytoplasm in combination with nuclear size and distribution to distinguish between the large hair cell nuclei and the smaller nuclei of cells of the greater epithelial ridge (GER; Fig. 2A, B) and lesser epithelial ridge (LER; Fig. 2A, B). Such a criterion provided information on cells that had their terminal mitosis shortly after the time of injection as clearance of BrdU takes around 2 hours. Analysis in other systems has shown that the cell cycle length changes over the time of our analysis from about 8 hours around E9.5 to about 12 hours around E16.5 (Calegari and Huttner, 2003). Any continued cell cycling would have reduced therefore the BrdU content of the nuclei to less than 50% (Fig. 2B) within 8–12 hours after the injection. Such reduction of BrdU results in specks of BrdU-Alexa 647 labeling that amounted to less than 50% of a given nucleus (Fig. 2B). This technique provides a clear picture of the distribution of cells that underwent terminal mitosis shortly after BrdU injection and remained mitotically quiescent afterwards. This technique will also reveal decreasingly labeled cells derived from precursors labeled at the time of BrdU application but which continue to cycle. Such lesser labeling will allow detailing further the topological progression of cells that undergo terminal mitosis at earlier or later stages. Of course, cell death would eliminate such cells and their BrdU labeling. The whole mount approach used here will allow identification of cells using histological criteria established over 100 years ago (Ruben, 1967). In our hands, some of the commonly used antibodies to highlight hair cells showed artifacts or no staining above background, probably due to the acid treatment necessary to uncover the BrdU epitope for the antibody recognition of BrdU.
Fig 2.
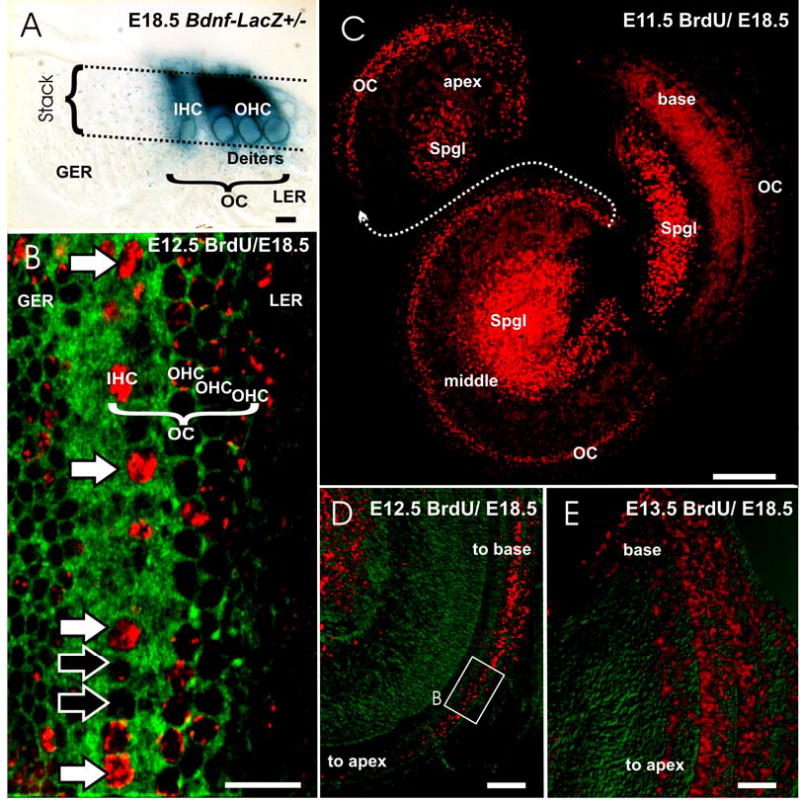
The terminal mitosis of sensory neurons (Spgl) and hair cells (HC) is shown in wild type mice as revealed with BrdU injection followed by delayed recovery at E18.5 (B–E). Differentiated hair cells can be identified at this stage using either sections of Bdnf-LacZ heterozygotic animals (A) or by imaging flat mounted organ of Corti (OC) using a confocal microscope to generate an image stack (stack in A) that extends from the surface to the nuclei of hair cells (A, B). Such a stack of images, if collapsed into a single image (B) will show the cytoplasm as green fluorescence with 488 nm excitation whereas nuclei will be black holes (B). Fluorescent antibody staining of BrdU will reveal labeling (shown in red in B–E) that allows us to asses at higher magnifications the degree of labeling as over 50% (white arrows in B) or less than 50% (black arrows in B). As the organ of Corti is regularly organized into one row of inner hair cells (IHC in A, B) and three rows of outer hair cells (OHC in A, B) with nuclei much large than those in cells of the adjacent greater and lesser epithelial ridge (GER, LER), hair cell identification is reliable with this approach. Low power images (C–E) can be used to assess the overall distribution of labeled cells and thus allow imaging the spatial changes over time following single injections of BrdU at different times in different litters. Injections of BrdU at E11.5 (C) show prominently labeled sensory neuron nuclei in the upper middle turn that were apparently undergoing DNA synthesis for a terminal mitosis at this time as indicated by the almost complete filling of the nucleus of these cell nuclei with anti-BrdU antibody. In contrast, hair cells show only puncta of BrdU staining in the nuclei except for the apex (C), suggesting that most hair cell precursors went through two or more rounds of division before their terminal mitosis. An E12.5 injection of BrdU labels hair cell nuclei in the basal turn and scattered spiral ganglion cells in the apical half (B, D). By E13.5, only few hair cells of the hook region (E) show labeling. Many supporting cell nuclei throughout the cochlea are also positive for BrdU. GER, greater epithelial ridge; IHC, inner hair cell; LER, lesser epithelial ridge; OC, Organ of Corti; OHC, outer hair cell; Spgl, spiral ganglion. Bar in A, B indicates 10 μm, in C–E indicates 100 μm.
Using these criteria in whole mounted ears we detected labeled spiral sensory neurons in a base to apical progression and of hair cells in an apex to base progression as previously described (Ruben, 1967). Specifically, injections of BrdU at E 9.5 and E 10.5 and imaging at E18.5 showed label in only few neurons in the basal turn with only punctate labeling in the apex (data not shown). Injections at E11.5 and imaging the distribution of nuclear staining at E18.5 showed that neurons were labeled in the basal and middle spiral turn (Fig. 2C). In contrast, neurons in the apex had less than 50% filled nuclei suggesting that they had at least one or more additional mitosis after the BrdU application. Using the same criterion we found only a few labeled hair cells in the apical most tip of the organ of Corti. However, a trail of less labeled hair cell nuclei extended throughout the middle turn almost to the basal hook (Fig. 2C).
Injections of BrdU at E12.5 labeled only neurons in the apex of the cochlea and many hair cells in the apical half turn of the organ of Corti (Fig. 2B, D). As in earlier stages, there was a sharp boundary toward the apex of labeled hair cells whereas the boundary toward the basal turn was fuzzy with many cells showing spotty BrdU labeling. Injections at E13.5 labeled very few spiral sensory neurons and showed labeled hair cells extending throughout the middle and basal turn. These injections also showed profound, but spotty labeling of other cell types throughout the cochlea, making it difficult to identify the labeled hair cells at low magnification. However, using the above described procedure of generating a single focal stack through the hair cells alone we were able to identify a few hair cells near the basal tip that showed profound nuclear labeling (Fig. 2E). Injections at E14.5 labeled no sensory neurons and only occasionally a few hair cells in the most basal tip of the organ of Corti where the rows of outer hair cells become reduced (data not shown).
These data suggest that in the apex the first hair cells become postmitotic around the time the last spiral sensory neurons become postmitotic, with an apparent spatial and temporal overlap. In contrast, in the base hair cells become postmitotic up to three days after spiral sensory neurons. These data suggest that overall, sensory neurons and hair cells are the first cells to exit cell cycling in opposing gradients as previously demonstrated (Ruben, 1967).
Neurog1 null mice: We next investigated the effects of Neurog1 on time of hair cell terminal mitosis in Neurog1 null littermates of our wildtype analyses presented above. In animals injected at E9.5 to E11.5 and analyzed at E18.5 we found no labeling in sensory neurons, consistent with previous data that suggests complete abrogation of any sensory neuron formation in Neurog1 null mice (Ma et al., 1998; Ma et al., 2000). However, in the middle to apical turn of the shortened and widened organ of Corti of these mutants we found numerous hair cells in which the nuclei were at least 50% filled with the BrdU antibody fluorescence (Fig. 3B, C). Injections after E13.5 did not reveal any hair cells with labeled nuclei, suggesting that the entire phase of hair cell proliferation was shifted in Neurog1 null mutants to an approximately 1–2 days earlier start of hair cell terminal mitosis compared to wildtype littermates (Fig. 3A). Data in the vestibular endorgans were less obvious, presumably because of the prolonged time over which hair cells are generated in these epithelia (Ruben, 1967) and the lack of a simple linear progression of terminal mitosis along one axis, as in the cochlea. In summary, the data on the cochlea suggest that abrogation of Neurog1 leads to a terminal mitosis of hair cells in the cochlea that temporally overlaps with the time of terminal mitosis of sensory neurons in wildtype littermates.
Fig. 3.

We show comparison of temporal patterns of terminal mitosis in the cochlea of the wild type and Neurog1 null littermates imaged at E18.5 (A, – C), and of Neurod1–LacZ expression at E18.5 in a Neurod1-LacZ heterozygote wild type and Neurog1 null littermate (D–G). In the absence of Neurog1, the hair cells exhibit alteration of the temporal pattern of terminal mitosis. BrdU injection at E9.5 and examination of the cochlea at E18.5 reveals some fully labeled nuclei (i.e. terminal mitotic cells) in the apex of the Neurog1 mutant cochlea (arrows in C; green is autofluorescence of hair cells). BrdU injected at E11.5 labels cells in the middle turn of the organ of Corti (OC) with only specks of BrdU labeling in wildtype (A) and shows prominent nuclear labeling of spiral ganglion neurons (Spgl in A). In Neurog1 null littermates there are no spiral ganglion cells (B). Instead, a prominent labeling of hair cell nuclei of the organ of Corti indicates a premature terminal mitosis of hair cells in the middle turn in a pattern similar to the proliferation of spiral ganglion neurons in wild-type (compare A, B). Similar effects on altered expression patterns are found in Neurod1 expression in Neurog1 null mutants and wildtype littermates (D–G). At E18.5, spiral neurons are strongly labeled for Neurod1 in the wild-type ear (D). In contrast to the very faint labeling of the wild-type organ of Corti, the Neurog1 null ear (E) shows profound Neurod1-LacZ staining in many hair cells. Note the absence of spiral ganglion neurons in Neurog1 null mice (compare D and E) and the multiple rows of outer hair cells (OHC) in the apex of the mutant cochlea (compare F and G). IHC, inner hair cell; OC, Organ of Corti; Spgl, spiral ganglion. Bar scale indicates 100 μm in A, B, D, E and 10μm in C, F, G.
We next wanted to confirm and extend this finding with other markers that show a delayed expression in hair cells compared to sensory neurons. Neurod1 is upregulated in sensory neurons immediately after Neurog1 (Ma et al., 1998) but appears only late in development in some hair cells, progressing from base to apex (Kim et al., 2001). We therefore bred mice that were null for Neurog1 and heterozygotic for Neurod1. Wild type E18.5 littermates showed no Neurod1 positive hair cells in the apex, and had only few inner hair cells displaying Neurod1 mediated β-galactosidase staining (Fig.3D, F). In contrast, Neurog1 null mice showed numerous Neurod1-β-galactosidase positive hair cells in the apex that were scattered across the multiple rows of hair cells of these mutants (Fig. 3E, G). As previously shown (Fig. 1A, C), the absence of Neurod1 positive sensory neurons (Fig. 3D) results in the Neurog1 null mice in a much tighter coiling of the cochlea. At the same magnification we can therefore show the entire apex of the Neurog1 null mice but not of the wildtype littermate (Fig. 3D, E). These data show upregulation of Neurod1 in many more hair cells throughout the organ of Corti of E18.5 Neurog1 null mice (Fig. 3F, G). Such data are consistent with the idea that at least some of the hair cells that undergo 1–2 days earlier terminal mitosis are derived from neurosensory precursors that can not differentiate into neurons in the absence of Neurog1 but become hair cells instead. Other genes with similar expression profiles are needed to verify whether this is peculiar to Neurod1 or is a general effect of NEUROG1 protein abrogation.
In conclusion, these data confirm previous findings that absence of Neurog1 affects not only sensory neuron formation but also hair cell formation (Ma et al., 2000). We now show that absence of Neurog1 leads to a terminal mitosis of hair cells up to two days earlier and results in upregulation of Neurod1 in many more hair cells of E18.5 embryos throughout the cochlea. These data are consistent with the notion of a Neurog1/Atoh1 interaction in the common neurosensory (hair cell/sensory neuron) precursors to regulate the terminal mitosis and cell fate of cell types derived from these proliferating precursors. A simple way to explain these effects of Neurog1 null mutation would be that ATOH1 protein mediated onset of differentiation interferes with the cell cycle progression thus resulting in premature terminal mitosis of hair cells and truncation of the proliferative capacity of those sensory neuron/hair cell precursors.
We therefore next tested whether Atoh1 is possibly expressed in mitotically active neurosensory precursors and whether this expression changes in the Neurog1 null mice thus supporting the idea that some sensory neurons of the mammalian ear show coexpression of Neurog1 and Atoh1 which interact either directly or through yet to be determined intermediaries to allow full clonal expansion of their common and/or separate precursors.
Expression of Atoh1 in control and Neurog1 null mice as revealed by various techniques.
We investigated whether the temporally shifted terminal mitosis of hair cells of Neurog1 null mice also lead to an upregulation of Atoh1. This would indicate not only that the onset of terminal mitosis of hair cells is accelerated but also that ATOH1 mediated hair cell differentiation in Neurog1 null mice is accelerated compared to wildtype mice. We studied the expression of Atoh1 in the ear of E12 to E13.5 old mice. In the wildtype littermates, the E12.5 in situ data showed a very faint staining near the forming vestibular epithelia. No differences other than a reduction in size of vestibular epithelia were observed in the Neurog1 null littermate and wildtype control animals. However, a difference was observed in the cochlea of E13 Neurog1 null mice compared to age matched littermates. In the Neurog1 null mice we obtained an Atoh1 signal in the cochlear duct that was strongest near the upper middle turn, fell of steeply near the apex and tapered off in the lower basal turn (Fig. 4C). In contrast, the wildtype littermates showed a fainter in situ signal exclusively in the basal turn (Fig. 4B). Our Atoh1-LacZ mediated β-galactosidase stain in Atoh1 heterozygotic animals showed an identical onset of Atoh1 expression in our mouse lines with a spatially comparable distribution (Fig. 4E). We also obtained some Atoh1-eGFP expressing embryos (courtesy of Dr. P. Chen and N. Segil) and could not detect any eGFP signal in the cochlea of mice that were at least one day older than the mice of our lines (Fig. 4A) thus confirming their reported observations (Chen et al., 2002).
Fig. 4.

The earliest expression of Atoh1 as revealed by different techniques (A, B, D–E) in wildtype and Neurog1 null mice (C) is compared. At E13.5, Atoh1-eGFP shows a profound signal in the vestibular sensory epithelia and in some delaminating cells (arrows in A). However, there is no detectable Atoh1-eGFP signal in the cochlea. In contrast, in situ hybridization (B) and Atoh1-LacZ (E) show Atoh1 expression in the basal turn of E13 and E13.5 animals (B, F). Comparison of Atoh1-LacZ (F) and Atoh1-Cre/Rosa26 (D) shows that more cells are Atoh1 positive in the vestibular sensory epithelia of Atoh1-LacZ animals at an earlier stage (F). In contrast, Atoh1-Cre shows additional expression in delaminating spiral ganglion neurons (Spgl) near the base and vestibular ganglion neurons (Vgl) near the utricle and saccule (D). Of all the techniques employed here, only Atoh1-eGFP shows some of these delaminating cells as well (compare A, D). Atoh1 in situ hybridization in Neurog1 null mutants shows a more apical expression in the E13 cochlea (C). This expression suggests a differential upregulation in the upper middle turn of the cochlea instead of the basal turn as in wildtype littermates (B, C). Note the small size of vestibular sensory epithelia (compare B and C) in Neurog1 mutants. AC, anterior crista; HC, horizontal crista; PC, posterior crista; S, saccule, Spgl, spiral ganglion; U, utricle; Vgl, vestibular ganglion. Bar indicates 100 um.
We next checked whether absence of eGFP signal in these lines is due to a different timetable of Atoh1 upregulation or due to delayed eGFP upregulation owing to the construct. Our Atoh1-LacZ ears showed a weak signal in the basal turn of the cochlea (Fig. 4E) in an animal in which we could not detect any eGFP, suggesting that this technique causes a delay in detectability of Atoh1 upregulation compared to the other two techniques employed here. Most importantly, these data suggest that in our mouse lines there is a potential spatio-temporal overlap of detectable Atoh1 expression in proliferating precursors only in the base of the cochlea, but not in the middle turn and apex, supporting previous suggestions (Chen et al., 2002).
We also tested whether we could detect Atoh1 signal using PCR technique. All three samples of E11.5 embryonic ears exhibited Atoh1 expression (data not shown), while 1 out of two samples from E10.5 embryos were positive for Atoh1 (Fig. 5A). The RNAse treated controls did not have any amplification products. These data suggest that the appearance of Atoh1 transcripts in the developing inner ear is at or around E10.5. This is the earliest time Atoh1 is expressed in detectable amounts in the developing ear.
Fig 5.
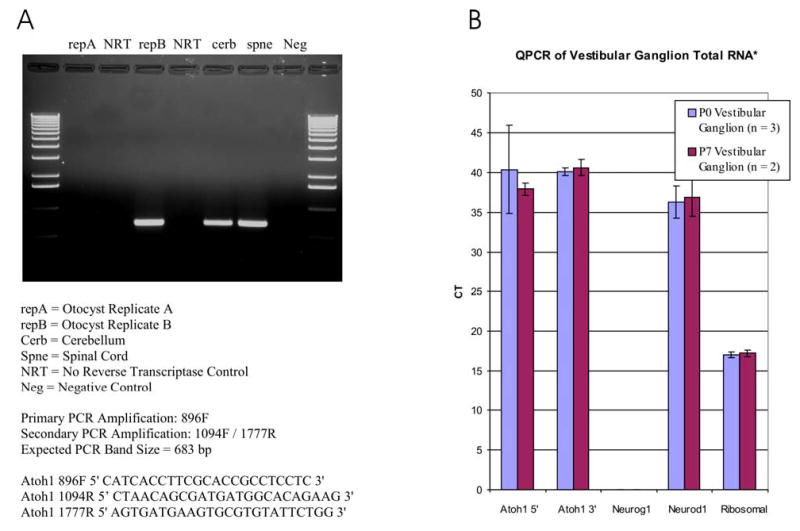
Atoh1 expression as revealed in the embryonic ear with PCR (35 cycles; A) and in the vestibular ganglion of postnatal animals with Q-PCR (B). Atoh1 presence was evaluated using PCR in two ears of E10.5 mice. Note that only one ear is positive for the PCR product, suggesting that Atoh1 expression starts in the ear around E10.5. All three ears of E11.5 old mice were Atoh1 positive (data not shown), suggesting that Atoh1 expression in the ear starts around E10.5. Q-PCR of vestibular ganglia show expression of Atoh1 after 37 cycles at P0 and P7 using both 3′ and 5′ primers. No Neurog1 was detectable in these cells, but Neurod1, known to be expressed in these cells, was detectable at about the same level as Atoh1. These data show the presence of Atoh1 mRNA in the vestibular ganglion in postnatal animals.
In summary, these data imply that Neurog1 expression affects upregulation of Atoh1 in apical hair cells that exit the cell cycle about two days earlier. In the absence of Neurog1 the hair cells that show this earlier terminal mitosis also show upregulation of Atoh1. However, the gradient does not follow the previously reported pattern of upregulation of Atoh1 in wildtype mice and progresses instead from near the apex toward the base. Interestingly, Atoh1 expression shows little temporal alteration (only about ½ day in the apex) compared to terminal mitosis (two days), suggesting that those two events are not tightly coupled. We have recently identified several other bHLH genes in the ear using PCR and their expression profiles could show a more profound upregulation in Neurog1 null mice, something we are actively investigating. Compared to PCR data, our histological techniques do reveal expression of Atoh1 at least 24 hours later (E10.5 versus E11.5) suggesting that such techniques are less than appropriate to exclude expression of low levels of Atoh1 in common neurosensory precursors. We therefore attempted to show such expression using yet another, more sensitive technique, Atoh1-Cre expression in Rosa 26 mice.
Expression of Atoh1 as revealed by Atoh-Cre expression in Rosa 26 mice.
Cre mediated expression of the LacZ reporter results in the Rosa 26 line in β-galactosidase staining wherever the CRE-recombinase was and is active (Ohyama and Groves, 2004). Recently, an Atoh1 promoter fragment used to drive eGFP (Gowan et al., 2001) was engineered by us to drive Cre. If Atoh1 is expressed in neurosensory precursors, the Atoh1-Cre should show β-galactosidase staining in sensory neurons as well as hair cells. We investigated the ears of E 12.5 and E18.5 and P7 of two Atoh1-Cre/Rosa 26 mice. In E12.5 embryos we found expression of β-galactosidase only in single cells in what appeared to be several vestibular sensory epithelia patches of the utricle, saccule, anterior, horizontal and posterior crista. In the cochlea, staining was in the medial wall of the basal and middle turn (Fig. 4D). In addition to these intraepithelial cells, β-galactosidase positive cells were found outside the inner ear epithelia in the vestibular ganglion and the forming spiral ganglion (Fig. 4D). In the hindbrain and midbrain Atoh1-Cre mediated β-galactosidase stain displayed a pattern consistent with the expression of Atoh1 as revealed in same aged Atoh1-LacZ brains (data not shown). This expression was verified in a total of 4 ears and 2 brains of E12.5 mice. Some positive cells were also found outside the sensory epithelia in Atoh1-eGFP ears (Fig. 4A) suggesting that the expression of Atoh1 is possibly transient and thus can not be detected with in situ hybridization or Atoh1-LacZ. Whether the Atoh1 fragment mediated signal is due to an expression in common neurosensory precursors or reflects an independent and transient upregulation in delaminating sensory neurons in addition to hair cell precursors remains unclear as we could not detect it proliferating cells.
At E18.5, β-galactosidase was found in almost all hair cells of all sensory epithelia, with the exception of the most apical tip of the cochlea (Fig. 6A, B). In addition, Atoh1-Cre mediated LacZ expression was found in spiral ganglion neurons, vestibular ganglion neurons, pillar cells and other supporting cells (Fig. 6B). We also investigated this expression of Atoh1-LacZ in hair cells of E18.5 Neurog1 null mice combined with Atoh1-LacZ heterozygosity and obtained comparable results. Combined, these data suggest that the most apical tip of the cochlea, the area in which the first hair cells become postmitotic, has a profound delay of Atoh1 upregulation of several days, irrespective of the genetic background and the technique used. Cells other than hair cells were also Atoh1-Cre-β-galactosidase positive. These cells were particularly frequent in the forming spiral prominence (Fig. 6A, B). In addition, few supporting cells of the cochlea and the vestibular epithelia were also β-galactosidase positive. Interestingly, some cells of the non-sensory epithelium of the ear were also β-galactosidase positive. These cells were particularly prominent in the extension of the saccule into the endolymphatic duct (Fig. 6A, B) but also as extension of the cruciate eminence of the canal cristae into the canals, in particular the anterior and posterior canal.
Fig. 6.

Expression of Atoh-Cre as revealed with a Rosa 26 line is shown for E18.5 embryo ears (A–D). Hair cells are most profoundly positive for the β-galactosidase reaction product (A, B, C) and can be seen in whole ears (A), flat mounted cochlea (B) or sections imaged with confocal microscope (C, level of section is indicated by dotted line in B). However, additional cells in the cochlea are also positive (A, B). Note that Atoh1-Cre staining can be shown in some spiral ganglion neurons (Spgl in A, B; SpglN in D) at any level of magnification (A, B, D). Note that the X-Gal reaction product deposited at places of β-galactosidase activity tends to form small deposits outside of cells (small blue specks in C) but is most prominently deposited in the cytoplasm of Atoh1-Cre positive cells like inner and outer hair cells and spiral ganglion neurons (C, D). Dotted line in B indicates the section plane with C, D indicated as squares. IHC, inner hair cell; OC, Organ of Corti; OHC, outer hair cell; S, saccule; Spgl, spiral ganglion; SpglN, spiral ganglion neuron; U, utricle Bar indicates 100 μm in A, B, 20 μm in C, D.
The ears of the one month old second Atoh1-Cre-Rosa 26 line (P31) also showed expression other than the hair cells of the sensory epithelia (Fig. 7B, C, G). This expression showed three prominent distribution patterns, sensory neurons, supporting cells in the sensory epithelia (in particular pillar cells; Fig. 7G) and non-sensory cells of the ear outside the sensory epithelia such as a few cells in the stria vascularis (Fig. 7B). Almost all sensory neurons were positive in both the spiral ganglion and the vestibular ganglion in this line with little staining elsewhere (Fig. 7B, C) with labeling being so prominent that the nerve fibers showed faint β-galactosidase reaction product (Fig. 7C).
Fig. 7.
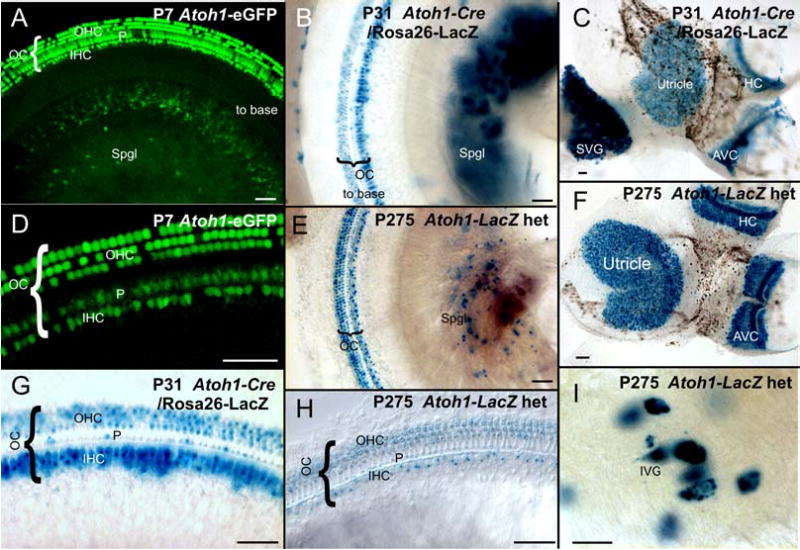
Expression of Atoh1 (AI) in hair cells and sensory neurons of juveniles and adults is shown as revealed with different techniques. With all these techniques, hair cells are most profoundly positive (D, C, F, G, H). However, additional cells in the cochlea are also positive for Atoh1 using either Atoh1-eGFP (A, D), Atoh1-Cre (B, C, G) or Atoh1-LacZ (H) and can be identified as pillar cells. Additional label is found in cells of the stria vascularis (near B). Note that Atoh1-LacZ staining in spiral or vestibular sensory neurons can be readily demonstrated in postnatal animals of all ages with any of these techniques (A, B, C, E, I). High power images show that the vestibular ganglion neurons (I) are approximately 50 μm in diameter and thus can not be confused with any other cell type in the vestibular ganglion. The similarities in detail of the sensory neuron expression obtained with all three techniques supports the idea that at least some vestibular and spiral neurons express Atoh1. However, the expression of β-galactosidase is particularly profound in spiral and vestibular neurons of Atoh1-Cre mice, suggesting that there may be a build up β-galactosidase over time, leading to a more obvious reaction in these large cells including a faint labeling in the nerve leading to the sensory epithelia. IVG, inferior vestibular ganglion; OC, Organ of Corti; S, saccule; Spgl, spiral ganglion; SVG, superior vestibular ganglion; U, utricle Bar indicates 50 μm in D, G, H, I and 100 μm in A, B, C, E, F.
These data obtained from two Atoh1-Cre transgenic lines suggest that Atoh1-Cre expressing precursors may not only give rise to hair cells but also to sensory neurons. Other cells found positive in both lines are some supporting cells and non-sensory epithelia of the ear. We will next focus on the possibility that Atoh1 is indeed expressed in sensory neurons where our two Atoh1-Cre lines showed strong expression (Fig. 6A, B; 7B, C).
Expression of Atoh1 in vestibular and spiral sensory neurons as revealed by Atoh1-eGFP, Atoh1-LacZ staining and Q-PCR.
Atoh1-Cre mediated β-galactosidase could provide a false positive signal that is related to the genomic integration of the promoter fragment. To test for this possibility, we investigated the ears of E12.5 and P7 Atoh1-eGFP mice (courtesy of Drs. Chen and Segil). Initially we could not detect any staining outside the sensory epithelia. However, using the Zeiss LSM confocal system color unmixing algorithm we were able to extract a weak eGFP signal form the background in scattered spiral sensory neurons of P7 (Fig. 7A) mice. We also found few eGFP positive cells delaminating in E12.5 Atoh1-eGFP mice (Fig. 4A). Detailed comparison of Atoh1-Cre and Atoh1-eGFP thus confirm staining in hair cells and some sensory neurons, but also show some differences in these three transgenic lines using the same Atoh1 promoter fragment in additional staining of cells in the cochlea. Most importantly, the signal in delaminating cells of E12.5 Atoh1-eGFP mice was stronger than in sensory neurons at P7 (Figs. 4A, 7A), suggesting that Atoh1-might be transiently upregulated. Moreover, only one of our two Atoh1-Cre transgenic lines showed labeling in a large fraction of sensory neurons, suggesting that the possibly transient expression of Atoh1 in these cells might be more widespread. While it is virtually inconceivable that this expression of Atoh1 in sensory neurons presents an integration artifact that would need to be similar in all three transgenic lines, we nevertheless wanted to obtain independent confirmation of the validity of the staining in sensory neurons indicated by the Atoh1 promoter fragment to demonstrate that this expression in sensory neurons is truly not an aberration of Atoh1 promoter integration.
We reasoned that if Atoh1 staining is indeed in sensory neurons and not an artifact of the Atoh1 promoter fragment, than we should be able to detect Atoh1-LacZ expression in the spiral and vestibular sensory neurons of juvenile and adult mice and should also detect it with Q-PCR. We investigated this possibility by staining the ears of 9 day old, 2 month, 5 month and 9 month old Atoh1-LacZ heterozygotic mice for β-galactosidase. Our data demonstrated that indeed there is an expression of β-galactosidase not only in hair cells but also in sensory neurons. Moreover, the overall distribution was not unlike that found in E18.5 Atoh1-Cre mice and showed only a small fraction of cells to be positive for Atoh1-LacZ (Fig. 7E, I). The distribution of these cells in the cochlea showed a more profound cellular staining in the apex, the area in which Neurog1 null mutants show the most profound effect on the spatial expression of Atoh1 and on temporal changes of hair cell terminal mitoses. Vestibular ganglion neurons are among the largest cells in the ear with 40–50 μm in size and can not be mistaken for any other cell type in the vestibular ganglia (Fig. 7I). Interestingly, all transgenic lines as well as adult Atoh1-LacZ lines showed prominent expression in virtually all hair cells and most pillar cells (Fig. 6B; 7D, G, H) implying that these cells should also be considered as Atoh1 positive cells.
We next tested whether spiral sensory neurons express Atoh1 and if so how this expression changes quantitatively over time. Previous work had shown that Atoh1 is sharply downregulated in the brain (Akazawa et al., 1995) and it could therefore be possible that the Atoh1-LacZ expression is unrelated to actual Atoh1 protein and gene expression. We therefore investigated the expression of Atoh1 using in situ hybridization and Q-PCR at P0 and P10. With both techniques we confirmed expression of Atoh1 in neonatal and older mice in the sensory neurons of the cochlea at virtually identical levels compared to newborns (Fig. 5B). Together these data firmly establish the presence of Atoh1 in spiral and vestibular ganglion neurons.
Absence of Atoh1 may affect pathfinding properties of sensory neurons.
We next wanted to understand whether Atoh1 presence in sensory neurons is biologically meaningful. We reasoned that pathfinding properties of sensory neurons are most likely to be disturbed and thus investigated this issue using tract tracing and immunocytochemistry for tubulin. The fibers projecting to the posterior crista have previously been demonstrated to provide a good readout for molecular perturbation of pathfinding properties such as neurotrophin misexpression (Tessarollo et al., 2004) or semaphorin signaling (Gu et al., 2003). We could not detect any obvious alterations in pathfinding at E11.5 (Fig. 8A, B), but found aberrations at E12.5 (Fig. 8C, D) and E13.5 (Fig. 8F, G). Specifically, fibers tended to branch profusely off the main branchlet to the posterior crista with only few fibers entering the epithelium (Fig. 8D, E) in Atoh1 null mice. The aberrations observed resembled those previously reported (Gu et al., 2003) in targeted disruption of semaphorin signaling through its neuropilin 1 receptor (Fig. 8E). In contrast, wildtype mice showed bifurcation of the posterior crista branchlet to enter the two hemicristae of the posterior cristae epithelium. Similar data were obtained for the anterior and horizontal crista and suggest that around the onset of Bdnf mediated rerouting of sensory processes (Tessarollo et al., 2004), Atoh1 null affects targeting of afferents near the sensory epithelia in a matter reminiscent of that in Sema3a signaling disrupted mutants (Gu et al., 2003).
Fig. 8.

The effect of Atoh1 null mutation (B, D, G) on the pattern of innervation of the posterior crista is compared with a wildtype (A, C, F) and a npn1-sema3a- mutant (E) as revealed with lipophilic tracers (A–F) and acetylated tubulin immunocytochemistry (G). The initial growth toward the posterior crista is unaffected in E11.5 embryos (A, B). However, afferent fibers continue to branch outside the sensory epithelium in both Atoh1 null and npn1-sema3a- mutant embryos (D, E) whereas fibers have entered and branch inside the sensory epithelium in wildtype littermates (C). By E13.5 afferents have entered the epithelium and branched to reach the Bdnf positive undifferentiated hair cells of the Atoh1 null mutant. This branching is more profuse in wildtype animals and the fibers have split into two areas, each supplying the Bdnf positive hair cells of the hemichristae (F). These data suggest that Atoh1 absence interferes with homing of afferent fibers near the target sensory epithelium. Bar indicates 100 μm.
In summary, we show here that Neurog1 null mice have a reduction in the number of hair cells and a reduced longitudinal growth of the cochlea. This is in part mediated by a terminal mitosis of hair cells of up to two days earlier and, to a lesser extent, by topographically altered upregulation of Atoh1 in the apical turn of the cochlea and a more prominent expression of Neurod1 in Neurog1 null mice. We demonstrate presence of Atoh1 in sensory neurons using three Atoh1 transgenic lines, Atoh1-LacZ heterozygotes and Q-PCR of vestibular ganglion neurons and show a possible effect of Atoh1 absence on neuronal pathfinding properties. Combined, these data support the notion that Atoh1 is expressed in some common hair cell and sensory neuron precursors, in particular in the cochlea and saccule. What remains to be shown is that Neurog1 is also co-expressed with Atoh1 in neurosensory precursors giving rise to hair cells and neurons, a possibility that was recently suggested (Raft, to be submitted).
Discussion
Past research in the ear has demonstrated that three bHLH genes are essential for the formation and/or differentiation of the two types of neurosensory cells of the ear, the hair cells and the sensory neurons (Ma et al., 1998; Bermingham et al., 1999; Liu et al., 2000; Kim et al., 2001). Additional information on Neurog1 and Atoh1 null mutations have shown that loss of sensory neuron formation in Neurog1 null mice affects hair cell formation (Ma et al., 2000). In Atoh1 null mutants, undifferentiated and/or dying cells still form in the topology normally occupied by hair cells (Chen et al., 2002; Fritzsch et al., 2005). It still is unclear how the interaction between Neurog1 and Atoh1, resulting in overlapping effects upon the ear neurosensory epithelium, could be reconciled, especially in light of the complex interactions of bHLH genes known in other developing sensory systems (Bertrand et al., 2002; Wu et al., 2003; Akagi et al., 2004). The Neurog1 and Atoh1 interaction also suggests a possible clonal relationship between sensory neurons and hair cells in mammals (Fritzsch and Beisel, 2004), which was recently demonstrated in chickens (Satoh and Fekete, 2005). As previously pointed out, our data and those of others (Bermingham et al., 1999; Chen et al., 2002; Fritzsch et al., 2005) do not support the recent idea that Atoh1 is not only essential for hair cell differentiation but also for sensory epithelia formation (Woods et al., 2004). In fact, our data show that even in combined Atoh1/Neurog1 double null mutants there is formation of Bdnf-LacZ positive cells in a topology that closely corresponds to some of the sensory epithelia found in wildtype littermates, suggesting that neither of these two bHLH genes is essential for the formation of undifferentiated sensory epithelia precursors and their maintenance up to E18.5 (Fig. 1D). The observed reduction in total length of the cochlea in Neurog1 null mice exceeds the length of Bdnf-LacZ expressing cells in Atoh1 null mice. The data on length and area changes in Neurog1 null, Atoh1 null and double null mice suggest a simple additive effect, implying that Neurog1 affects both hair cell and non-sensory epithelium formation (Fig. 1; Table I). These data are more in line with the idea that other genes are responsible for setting up the neurosensory precursor domains of the ear (Zou et al., 2004; Daudet and Lewis, 2005; Kiernan et al., 2005).
Atoh1 in sensory neurons:
With almost any technique employed to reveal Atoh1 in the ear we are able to demonstrate Atoh1 presence not only in the hair cells but also in some spiral and vestibular sensory neurons (Figs. 4–7). In the spiral ganglion we find labeled cells in particular in the apex, an area were past research suggests a more prominent presence of type II spiral neurons that project to the outer hair cells (Ryugo, 1992; Rubel and Fritzsch, 2002). The reported lack of projection deeper into the undifferentiated organ of Corti of the remaining sensory neuron afferent fibers of Atoh1 null mutants at E18.5 (Fritzsch et al., 2005) could indicate lack of projection of type II neurons. Alternatively, it could relate to the limited attraction provided by the undifferentiated cells that express Atoh1-LacZ and Bdnf-LacZ (Fritzsch et al., 2005) as these cells are apparently negative for any other hair cell or supporting cell protein marker tested thus far (Chen et al., 2002; Woods et al., 2004).
It remains somewhat unclear why none of the previous publications has indicated such an expression of Atoh1 in sensory neurons. In large part this may be related to the more prominent expression of Atoh1 in hair cells combined with the known effects of Atoh1 on hair cell maturation (Bermingham et al., 1999; Izumikawa et al., 2005) that diverted a complete inner ear expression analyses. In part this appears to be inherent to some of the techniques employed, like the Atoh1-eGFP reporter, which requires additional techniques to extract the rather faint signal from the background. A compounding factor is likely the early formation of bone around the sensory neurons which preclude proper penetration of detection solutions, thus leading to false negatives as we can detect Atoh1 in spiral and vestibular sensory neurons in older animals in which bone encloses these neurons. Lastly, without reason to suspect expression of Atoh1, one would not dissect the vestibular ganglia and process them for PCR to detect the clearly present Atoh1 signal (Fig. 5B).
In summary, Atoh1 expression is not only in hair cells but also in some sensory neurons and may be functionally meaningful for the development of specific projection patterns (Fig. 8).
Neurog1 and hair cell terminal mitosis and differentiation:
Our past work has shown that many hair cells do not form in Atoh1 null mice (Ma et al., 2000), in particular in the saccule and the cochlea. We now show that combined mutation of Atoh1 and Neurog1 leads to a simple additive effect of the Atoh1 phenotype on the Neurog1 phenotype (Fig. 1; Table 1). Our data confirm previous reports on a base to apex progression of sensory neuron and apex to base progression of hair cell terminal mitosis (Ruben, 1967). Minor differences in the timing of these events relate to differences in staging the mice and differences in gestational length of the mouse strains used. Most importantly, this past work has indicated that apical hair cells may become postmitotic before apical sensory neurons. We now show that there is a widespread overlap of timing of terminal mitosis in apical sensory neurons and hair cells around E11.5–E12.5 (Fig. 2). On closer examination of terminal mitosis, Neurod1 and Atoh1 expression in Neurog1 null embryos shows both terminal mitosis and Neurod1 and Atoh1 expressions are temporally and/or spatially altered in Neurog1 null mice. In fact, terminal mitosis of hair cells occurs up to two days earlier in Neurog1 null mice. This temporal acceleration of hair cell terminal mitosis, combined with our inability to demonstrate earlier expression of Atoh1 implies that other genes are responsible for the cell fate switch from sensory neurons to hair cells in the Neurog1 null mice: instead of being able to differentiate as sensory neurons, postmitotic cells now become hair cells. The altered expression of Neurod1 and Atoh1 in the cochlea with a spatial shift toward the apex suggest that Neurog1 is not only affecting cell fate selection but also expression of other bHLH genes.
The simplest way of explaining these data as well as the expression of Atoh1 in sensory neurons would be the assumption that neurosensory precursors co-express Atoh1 and Neurog1 and switch their cell fate to hair cell differentiation in Neurog1 null mice. (Fig. 9). However, we can not exclude other models, namely that Neurog1 absence disrupts co-ordinated cell cycling of nearby cells and affects their gene expression profiles, thus causing Neurod1 upregulation in hair cells. This alternative scenario was previously raised in developing dorsal root ganglion (Ma et al., 1999). Clearly, the truncation of hair cell formation may relate to either direct effects in neurosensory precursors through interactions of these bHLH genes as previously reported (Gowan et al., 2001) or may interfere with the coordinated inhibitory processes or collaboration with other factors that set up sensory epithelia (Adam et al., 1998; Daudet and Lewis, 2005).
Fig. 9.
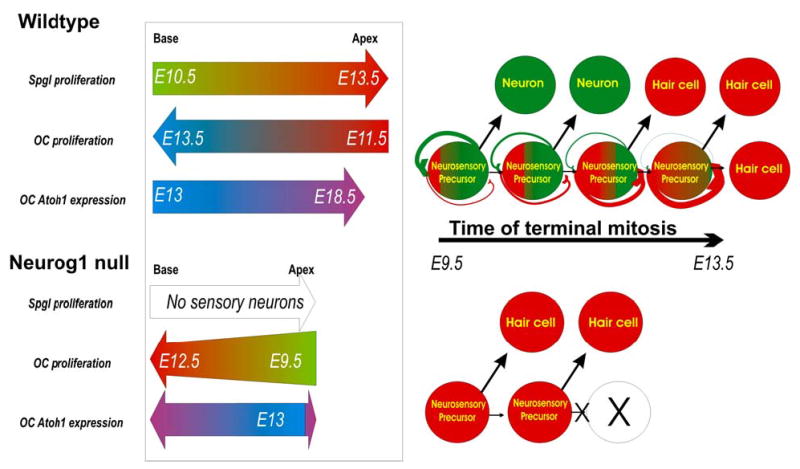
This summary diagram shows the spatiotemporal progression of sensory neuron and hair cell terminal mitosis, and Atoh1 expression in wildtype (top) and Neurog1 null mice (bottom). Our data and those of Ruben (1967) suggest that in wildtype the spiral ganglion terminal mitosis (Spgl) progresses from the base of the cochlea to the apex whereas the proliferation of hair cells of the organ of Corti progresses from apex to base with a broad overlap of terminal mitosis of both cell types in the apex around E11.5–12.5. The Neurog1 null ear has no spiral neurons, and shows a premature onset and cessation of terminal mitosis in hair cells in an apex to base gradient, along a time line comparable to sensory neurons in wildtype mice. Neurog1 null mice have fewer hair cells in a shortened and widened cochlea. Neurog1 null mice also show some alteration in the topology of Atoh1 expression. The right half of the scheme presents a hypothetical relationship between neurosensory precursors that give rise to both hair cells and sensory neurons. The model assumes that some neurosensory precursors will undergo a progressive up-regulation of Atoh1 in Neurog1 positive precursors until eventually they will turn these cells into hair cell producing precursors. It also assumes a mutual inhibition of Neurog1 and Atoh1 in the same precursor. According to this model, absence of NEUROG1 protein in Neurog1 null mutants causes premature commitment to hair cell fate and thus a terminal mitosis of hair cells at earlier developmental stages as well as truncation of the progenitor pool expansion, resulting in a shortened cochlea.
[modified after Ma et al., 1999; Gowan et al., 2001; Bertrand et al., 2002]
Similar effects on reduced clonal expansion and changes in cell fate assignment have been reported for null mice of the winged helix gene Bf1 (now Foxg1). In these mutants the clonal expansion of cortical precursors is truncated through premature terminal mitosis and cell fate is limited to only one cortical neuron type (Hanashima et al., 2002; Hanashima et al., 2004). It appears that Neurog1 null mutation has comparable effects on neurosensory precursors of the ear, limiting the fate choice to one (hair cells) and initiating an earlier terminal mitosis of these cells resulting in clonal compression. Clearly, not only are fewer hair cells formed in the smaller epithelia of Neurog1 null mice, but these mutants also miss all sensory neurons, suggesting that NEUROG1 protein is essential for the complete clonal expansion of the entire neurosensory population of the ear, possibly including some hair cell precursors.
In summary, much like in the spinal cord (Gowan et al., 2001), these data could be most easily explained by assuming an inhibitory interaction of Neurog1 and Atoh1 in a set of neurosensory precursor cells of the ear (Fig. 9). Absence of this interaction in Neurog1 null mice leads to temporal shifting of hair cell terminal mitosis through a phenotype switch of postmitotic cells. Recent work in chicken has demonstrated such a clonal relationship (Satoh and Fekete, 2005) and our data are compatible with such a relationship for mice but do not prove. Direct evidence is still needed.
Atoh1 and hair cell terminal mitosis
Obviously, our Atoh1 in situ hybridization data as well as our Atoh1-LacZ data strongly suggest that Atoh1 expression is at best in a small set of cycling hair cell precursors in the basal turn, the last hair cells to become postmitotic in the cochlea. These data are in agreement with almost all other data on Atoh1 expression in the cochlea published thus far (Bermingham et al., 1999; Zine et al., 2001; Chen et al., 2002). Only some recent Atoh1-LacZ data suggest suggest a somewhat more profound expression of Atoh1 throughout the cochlea at E13.5 (Woods et al., 2004) which partially agrees with our Atoh1-LacZ data (Fig. 4E). Combined, our data confirm the previous conclusion based on Atoh1-eGFP expression that Atoh1 is not expressed in detectable levels in cycling precursors (Chen et al., 2002), except for a few cells at the basal tip of the organ of Corti.
A low level of Atoh1 may be present but this possibility can not be ruled out with in situ hybridization, Atoh1-LacZ or Atoh1-eGFP. Indeed, our Atoh1-Cre data show somewhat earlier upregulation of Atoh1 expression in the cochlea basal turn at E12.5 as well as β-galactosidase labeling in sensory neurons at this developmental stage. These data imply that some Atoh1 expression in amounts not detectable by current histological techniques exist in the growing organ of Corti several hours or days prior to E12.5. Recent data on another bHLH gene, MyoD showed that more sensitive in situ techniques can demonstrate cellular distribution prior to detection with more established in situ techniques, providing cellular distribution information for already existing RT-PCR data (Gerhart et al., 2004). This Atoh1 expression revealed with the Cre line is particularly remarkable as it is so early despite that fact that this reporter must be delayed through the multiple steps required for expression and eventual detection. This is apparent by the earlier Atoh1-Cre activation and detection in sensory neurons in Atoh1-Cre/Rosa 26 lines. Indeed, using PCR we find Atoh1 signal as early as E10.5. In the light of these data, combined with the clear effect of Neurog1 on changes in timing of terminal mitosis of hair cells, we suggest that the spatio-temporal overlap of Atoh1 expression with proliferating hair cell precursors might be more profound than our data and those of others suggest (Chen et al., 2002) as they may be biased toward detection only of larger amounts of Atoh1 transcript or ATOH1 protein, rather than the earliest onset of expression.
As outlined above, it is possible that none of the employed histological techniques may detect the earliest onset of Atoh1 expression. Such early upregulation of Atoh1-Cre/Rosa 26 signal indicates that Atoh1 transcription may exist in cochlea precursor cells as early as E10.5, a time in which the majority of hair cells are not yet postmitotic. We assume that this early expression is in common neurosensory precursors of sensory neurons and hair cells, leading to the formation of enough ATOH1 protein to activate the Atoh1 promoter fragment that will drive the Cre expression which will eventually activate the LacZ reporter system of the Rosa 26 line. However, we can not exclude that most hair cells and sensory neurons arise from discrete progenitors and that the early Atoh1-Cre expression reflects an early upregulation of Atoh1 in some sensory neurons.
In many other systems, quantitative relationships within clones are determined by one or more bHLH genes through regulation of both cell fate and numbers of cells of a given fate (Bertrand et al., 2002; Akagi et al., 2004). Elimination of one bHLH gene my lead to replacement of a given cell type through clonal expansion and regulation of cell fate using another bHLH gene (Ma et al., 1999). In the ear, there appears to be a dichotomy of bHLH gene channeling towards either hair cell or sensory neuron fate. Thus, abrogation of Neurog1 may cause premature upregulation of Neurod1 and possibly other genes which can, however, in the context of the ear only cooperate in hair cell differentiation (Fig. 9). The temporal acceleration of terminal mitosis of hair cells will move these cells out of synchrony with other developing gene expression patterns, thus leading to the formation of multiple rows of hair cells and disorganization of apical specializations (Ma et al., 2000) presumably by moving hair cell differentiation out of synchrony with polarity determining gene expressions such as Vangl (Amonlirdviman et al., 2005). Closer examination of Vangl upregulation (Montcouquiol et al., 2003) in Neurog1 null mice and control littermates is needed to support this notion. Alternatively, the more profound upregulation of Neurod1 driven LacZ reporter in these hair cells of Neurog1 null mice may affect cell polarity organization more directly.
Interestingly, our data on Atoh1 expression, regardless of any genetic background, show that apical hair cells express Atoh1 only in early neonates, but become postmitotic between E11.5 and E12.5. In contrast to Atoh1 null mice, where Atoh1 absence apparently leads to rapid degeneration of many of the undifferentiated Atoh1-lacZ positive precursors found in these mutants (Chen et al., 2002; Fritzsch et al., 2005), Atoh1 negative postmitotic hair cells of the apex in wildtype mice seem to do just fine for several days, even in Atoh1 null mice (Fritzsch et al., 2005). Several explanations for this conflicting data are apparent. It is possible that cells require ATOH1 protein once the upstream regulators have initiated Atoh1 expression and failure of ATOH1 protein formation leads to cell death within several hours. Available data on base to apex progression of apoptosis would be compatible with such an idea (Chen et al., 2002). Since lack of ATOH1 protein will invariably lead to apoptosis, the reported base to apex progression of apoptosis in Atoh1 null mutants might also relate to inherited specific maximal times postmitotic hair cells can survive without a specified amount of ATOH1 protein.
Atoh1 expression in sensory neurons may be biologically meaningful.
The expression of Atoh1 in the vestibular ganglion neurons, sensory neurons known to project to the saccule and the posterior crista (Maklad and Fritzsch, 1999), appears to relate to a possible effect of Atoh1 null mutation on targeted afferent fiber growth (Fig. 8). Examination of initial vestibular fiber growth has shown unusual deviations of fibers near the target epithelia that could not be explained by lack of neurotrophins in the target epithelia (Fritzsch et al., 2005). Clearly, Bdnf, the most important neurotrophin to guide and maintain afferent fibers to the crista organs (Fritzsch et al., 2004; Tessarollo et al., 2004), is expressed in these epithelia even in mutants lacking both Neurog1 and Atoh1 (Fig. 1H–J). Moreover, Atoh1 null mice show a prominent innervation of the posterior crista as late as E18.5 (Fritzsch et al., 2005) thus ruling out that Bdnf expression might be the problem as it would have resulted in loss of all crista innervation by E14.5 (Fritzsch et al., 2004; Tessarollo et al., 2004). We now have extended this initial finding and show aberrations that resemble those previously reported for Npn1 sema3a- mutant mice (Gu et al., 2003). We suggest that the expression of Atoh1 in vestibular sensory neurons mediates certain pathfinding properties of sensory neurons either by regulating neuropilin expression (Gu et al., 2003) or by affecting the BDNF/TRPC pathway for guiding growth cones of vestibular ganglion neurons (Li et al., 2005; Wang and Poo, 2005). Such pathfinding properties are essential for the connection of insect mechanosensory neurons with the CNS and may even be mediated by Atoh1 knocked into atonal in flies (Ben-Arie et al., 2000; Wang et al., 2002). Governing pathfinding may also be a property of Atoh1 in CNS neurons in which it is expressed (Ben-Arie et al., 1997; Bermingham et al., 1999; Bermingham et al., 2001). Such involvement of Atoh1 in regulating the molecular basis of pathfinding may directly relate to the ancestry of the atonal gene family and their association with the sensory ciliated neuron (Fritzsch and Beisel, 2003) and the sensory system development and evolution (Fritzsch and Beisel, 2003; Fritzsch et al., 2005). Testing this suggestion requires conditional null mutation of Atoh1 only in sensory neurons using a Neurog1 mediated Cre expression. Alternatively, analyzing the expression of Neuropilin1, TrkB and TrpC in wildtype and Atoh1 null ears could reveal differential regulation of these genes known to be involved in pathfinding.
In summary, our data support the notion that some sensory neurons express low levels of Atoh1, particularly those of the apex of the cochlea and the inferior vestibular ganglion known to innervate the saccule and posterior crista. Neurog1 abrogation leads to earlier terminal mitosis of hair cells, somewhat displaced upregulation of Atoh1 and Neurod1 and downregulation of clonal expansion of hair cell precursors (Fig. 9). Consistent with recent data in chicken (Satoh and Fekete, 2005), our data are compatible with a clonal relationship of some sensory neurons and hair cells in mice, being derived from common neurosensory precursors. For translational research, it would be extremely helpful if the molecular switches from sensory neuron precursor to hair cell precursors are better understood and can be molecularly manipulated. Ultimately this may lead to implantation of a single neurosensory precursor population that can give rise to both sensory neurons and hair cells. Indeed, sensory neuron precursors that have the capacity to proliferate in vitro have recently been isolated from human cochlea (Rask-Andersen et al., 2005) and thus offer a possible therapeutic approach in combination with the above outlined insights.
Acknowledgments
This work was supported by grants from NIH (RO1 DC005590, BF; R01 DC04279 KWB). We wish to acknowledge the help of Dr. P. Chen and Q. Ma to improve the manuscript and Drs. P. Chen, H. Zoghbi, N. Segil, D. Ginty and Q. Ma for providing their mutant mouse lines. This investigation was conducted in a facility constructed with support from Research Facilities Improvement Program Grant Number 1 C06 RR17417-01 from the National Center for Research Resources, National Institutes of Health. We acknowledge the use of the confocal microscope facility of the NCCB, supported by EPSCoR EPS-0346476 (CFD 47.076).
References
- Adam J, Myat A, Le Roux I, Eddison M, Henrique D, Ish-Horowicz D, Lewis J. Cell fate choices and the expression of Notch, Delta and Serrate homologues in the chick inner ear: parallels with Drosophila sense-organ development. Development. 1998;125:4645–4654. doi: 10.1242/dev.125.23.4645. [DOI] [PubMed] [Google Scholar]
- Akagi T, Inoue T, Miyoshi G, Bessho Y, Takahashi M, Lee JE, Guillemot F, Kageyama R. Requirement of multiple basic helix-loop-helix genes for retinal neuronal subtype specification. J Biol Chem. 2004;279:28492–28498. doi: 10.1074/jbc.M400871200. [DOI] [PubMed] [Google Scholar]
- Akazawa C, Ishibashi M, Shimizu C, Nakanishi S, Kageyama R. A mammalian helix-loop-helix factor structurally related to the product of Drosophila proneural gene atonal is a positive transcriptional regulator expressed in the developing nervous system. J Biol Chem. 1995;270:8730–8738. doi: 10.1074/jbc.270.15.8730. [DOI] [PubMed] [Google Scholar]
- Amonlirdviman K, Khare NA, Tree DR, Chen WS, Axelrod JD, Tomlin CJ. Mathematical modeling of planar cell polarity to understand domineering nonautonomy. Science. 2005;307:423–426. doi: 10.1126/science.1105471. [DOI] [PubMed] [Google Scholar]
- Anderson DJ, Groves A, Lo L, Ma Q, Rao M, Shah NM, Sommer L. Cell lineage determination and the control of neuronal identity in the neural crest. Cold Spring Harb Symp Quant Biol. 1997;62:493–504. [PubMed] [Google Scholar]
- Beisel KW, Nelson NC, Delimont DC, Fritzsch B. Longitudinal gradients of KCNQ4 expression in spiral ganglion and cochlear hair cells correlate with progressive hearing loss in DFNA2. Brain Res Mol Brain Res. 2000;82:137–149. doi: 10.1016/s0169-328x(00)00204-7. [DOI] [PubMed] [Google Scholar]
- Ben-Arie N, Bellen HJ, Armstrong DL, McCall AE, Gordadze PR, Guo Q, Matzuk MM, Zoghbi HY. Math1 is essential for genesis of cerebellar granule neurons. Nature. 1997;390:169–172. doi: 10.1038/36579. [DOI] [PubMed] [Google Scholar]
- Ben-Arie N, Hassan BA, Bermingham NA, Malicki DM, Armstrong D, Matzuk M, Bellen HJ, Zoghbi HY. Functional conservation of atonal and Math1 in the CNS and PNS. Development. 2000;127:1039–1048. doi: 10.1242/dev.127.5.1039. [DOI] [PubMed] [Google Scholar]
- Bermingham NA, Hassan BA, Price SD, Vollrath MA, Ben-Arie N, Eatock RA, Bellen HJ, Lysakowski A, Zoghbi HY. Math1: an essential gene for the generation of inner ear hair cells. Science. 1999;284:1837–1841. doi: 10.1126/science.284.5421.1837. [DOI] [PubMed] [Google Scholar]
- Bermingham NA, Hassan BA, Wang VY, Fernandez M, Banfi S, Bellen HJ, Fritzsch B, Zoghbi HY. Proprioceptor pathway development is dependent on Math1. Neuron. 2001;30:411–422. doi: 10.1016/s0896-6273(01)00305-1. [DOI] [PubMed] [Google Scholar]
- Bertrand N, Castro DS, Guillemot F. Proneural genes and the specification of neural cell types. Nat Rev Neurosci. 2002;3:517–530. doi: 10.1038/nrn874. [DOI] [PubMed] [Google Scholar]
- Calegari F, Huttner WB. An inhibition of cyclin-dependent kinases that lengthens, but does not arrest, neuroepithelial cell cycle induces premature neurogenesis. J Cell Sci. 2003;116:4947–4955. doi: 10.1242/jcs.00825. [DOI] [PubMed] [Google Scholar]
- Chen P, Johnson JE, Zoghbi HY, Segil N. The role of Math1 in inner ear development: Uncoupling the establishment of the sensory primordium from hair cell fate determination. Development. 2002;129:2495–2505. doi: 10.1242/dev.129.10.2495. [DOI] [PubMed] [Google Scholar]
- Daudet N, Lewis J. Two contrasting roles for Notch activity in chick inner ear development: specification of prosensory patches and lateral inhibition of hair-cell differentiation. Development. 2005;132:541–551. doi: 10.1242/dev.01589. [DOI] [PubMed] [Google Scholar]
- Farinas I, Jones KR, Tessarollo L, Vigers AJ, Huang E, Kirstein M, de Caprona DC, Coppola V, Backus C, Reichardt LF, Fritzsch B. Spatial shaping of cochlear innervation by temporally regulated neurotrophin expression. J Neurosci. 2001;21:6170–6180. doi: 10.1523/JNEUROSCI.21-16-06170.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsch B, Beisel KW. Molecular conservation and novelties in vertebrate ear development. Curr Top Dev Biol. 2003;57:1–44. doi: 10.1016/s0070-2153(03)57001-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsch B, Beisel KW. Keeping sensory cells and evolving neurons to connect them to the brain: molecular conservation and novelties in vertebrate ear development. Brain Behav Evol. 2004;64:182–197. doi: 10.1159/000079746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsch B, Beisel KW, Bermingham NA. Developmental evolutionary biology of the vertebrate ear: conserving mechanoelectric transduction and developmental pathways in diverging morphologies. Neuroreport. 2000;11:R35–44. doi: 10.1097/00001756-200011270-00013. [DOI] [PubMed] [Google Scholar]
- Fritzsch B, Beisel KW, Jones K, Farinas I, Maklad A, Lee J, Reichardt LF. Development and evolution of inner ear sensory epithelia and their innervation. J Neurobiol. 2002;53:143–156. doi: 10.1002/neu.10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsch B, Farinas I, Reichardt LF. Lack of neurotrophin 3 causes losses of both classes of spiral ganglion neurons in the cochlea in a region-specific fashion. J Neurosci. 1997;17:6213–6225. doi: 10.1523/JNEUROSCI.17-16-06213.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsch B, Matei VA, Nichols DH, Bermingham N, Jones K, Beisel KW, Wang VY. Atoh1 null mice show directed afferent fiber growth to undifferentiated ear sensory epithelia followed by incomplete fiber retention. Dev Dyn. 2005;233:570–583. doi: 10.1002/dvdy.20370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsch B, Matei VA, Nichols DH, Bermingham N, Jones K, Beisel KW, Wang VY. 2005. Atoh1 null mutants show directed afferent fiber growth to undifferentiated ear sensory epithelia followed by incomplete fiber retention. Dev Dyn in press. [DOI] [PMC free article] [PubMed]
- Fritzsch B, Piatigorsky J, Tessmar-Raible K, Jekely G, Guy K, Raible F, Wittbrodt J, Arendt D. Ancestry of Photic and Mechanic Sensation? Science. 2005;308:1113–1114. doi: 10.1126/science.308.5725.1113. [DOI] [PubMed] [Google Scholar]
- Fritzsch B, Tessarollo L, Coppola E, Reichardt LF. Neurotrophins in the ear: their roles in sensory neuron survival and fiber guidance. Prog Brain Res. 2004;146:265–278. doi: 10.1016/S0079-6123(03)46017-2. [DOI] [PubMed] [Google Scholar]
- Gerhart J, Baytion M, Perlman J, Neely C, Hearon B, Nilsen T, Getts R, Kadushin J, George-Weinstein M. Visualizing the Needle in the Haystack: In Situ Hybridization With Fluorescent Dendrimers. Biol Proced Online. 2004;6:149–156. doi: 10.1251/bpo84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowan K, Helms AW, Hunsaker TL, Collisson T, Ebert PJ, Odom R, Johnson JE. Crossinhibitory activities of Ngn1 and Math1 allow specification of distinct dorsal interneurons. Neuron. 2001;31:219–232. doi: 10.1016/s0896-6273(01)00367-1. [DOI] [PubMed] [Google Scholar]
- Gu C, Rodriguez ER, Reimert DV, Shu T, Fritzsch B, Richards LJ, Kolodkin AL, Ginty DD. Neuropilin-1 Conveys Semaphorin and VEGF Signaling during Neural and Cardiovascular Development. Dev Cell. 2003;5:45–57. doi: 10.1016/s1534-5807(03)00169-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanashima C, Li SC, Shen L, Lai E, Fishell G. Foxg1 suppresses early cortical cell fate. Science. 2004;303:56–59. doi: 10.1126/science.1090674. [DOI] [PubMed] [Google Scholar]
- Hanashima C, Shen L, Li SC, Lai E. Brain factor-1 controls the proliferation and differentiation of neocortical progenitor cells through independent mechanisms. J Neurosci. 2002;22:6526–6536. doi: 10.1523/JNEUROSCI.22-15-06526.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Nakajima A, Nomoto H, Furukawa S. Neurotrophins facilitate neuronal differentiation of cultured neural stem cells via induction of mRNA expression of basic helix-loop-helix transcription factors Mash1 and Math1. J Neurosci Res. 2003;71:648–658. doi: 10.1002/jnr.10532. [DOI] [PubMed] [Google Scholar]
- Izumikawa M, Minoda R, Kawamoto K, Abrashkin KA, Swiderski DL, Dolan DF, Brough DE, Raphael Y. 2005. Auditory hair cell replacement and hearing improvement by Atoh1 gene therapy in deaf mammals. Nat Med. [DOI] [PubMed]
- Jones KR, Farinas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan AE, Pelling AL, Leung KK, Tang AS, Bell DM, Tease C, Lovell-Badge R, Steel KP, Cheah KS. Sox2 is required for sensory organ development in the mammalian inner ear. Nature. 2005;434:1031–1035. doi: 10.1038/nature03487. [DOI] [PubMed] [Google Scholar]
- Kim WY, Fritzsch B, Serls A, Bakel LA, Huang EJ, Reichardt LF, Barth DS, Lee JE. NeuroD-null mice are deaf due to a severe loss of the inner ear sensory neurons during development. Development. 2001;128:417–426. doi: 10.1242/dev.128.3.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanford PJ, Shailam R, Norton CR, Gridley T, Kelley MW. Expression of Math1 and HES5 in the cochleae of wildtype and Jag2 mutant mice. J Assoc Res Otolaryngol. 2000;1:161–171. doi: 10.1007/s101620010023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Jia YC, Cui K, Li N, Zheng ZY, Wang YZ, Yuan XB. Essential role of TRPC channels in the guidance of nerve growth cones by brain-derived neurotrophic factor. Nature. 2005;434:894–898. doi: 10.1038/nature03477. [DOI] [PubMed] [Google Scholar]
- Liu M, Pereira FA, Price SD, Chu MJ, Shope C, Himes D, Eatock RA, Brownell WE, Lysakowski A, Tsai MJ. Essential role of BETA2/NeuroD1 in development of the vestibular and auditory systems. Genes Dev. 2000;14:2839–2854. doi: 10.1101/gad.840500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumpkin EA, Collisson T, Parab P, Omer-Abdalla A, Haeberle H, Chen P, Doetzlhofer A, White P, Groves A, Segil N, Johnson JE. Math1-driven GFP expression in the developing nervous system of transgenic mice. Gene Expr Patterns. 2003;3:389–395. doi: 10.1016/s1567-133x(03)00089-9. [DOI] [PubMed] [Google Scholar]
- Ma Q, Anderson DJ, Fritzsch B. Neurogenin 1 null mutant ears develop fewer, morphologically normal hair cells in smaller sensory epithelia devoid of innervation. J Assoc Res Otolaryngol. 2000;1:129–143. doi: 10.1007/s101620010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Chen Z, del Barco Barrantes I, de la Pompa JL, Anderson DJ. neurogenin1 is essential for the determination of neuronal precursors for proximal cranial sensory ganglia. Neuron. 1998;20:469–482. doi: 10.1016/s0896-6273(00)80988-5. [DOI] [PubMed] [Google Scholar]
- Ma Q, Fode C, Guillemot F, Anderson DJ. Neurogenin1 and neurogenin2 control two distinct waves of neurogenesis in developing dorsal root ganglia. Genes Dev. 1999;13:1717–1728. doi: 10.1101/gad.13.13.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maklad A, Fritzsch B. Incomplete segregation of endorgan-specific vestibular ganglion cells in mice and rats. J Vestib Res. 1999;9:387–399. [PubMed] [Google Scholar]
- Montcouquiol M, Rachel RA, Lanford PJ, Copeland NG, Jenkins NA, Kelley MW. Identification of Vangl2 and Scrb1 as planar polarity genes in mammals. Nature. 2003;423:173–177. doi: 10.1038/nature01618. [DOI] [PubMed] [Google Scholar]
- Ohyama T, Groves AK. Generation of Pax2-Cre mice by modification of a Pax2 bacterial artificial chromosome. Genesis. 2004;38:195–199. doi: 10.1002/gene.20017. [DOI] [PubMed] [Google Scholar]
- Radde-Gallwitz K, Pan L, Gan L, Lin X, Segil N, Chen P. Expression of Islet1 marks the sensory and neuronal lineages in the mammalian inner ear. J Comp Neurol. 2004;477:412–421. doi: 10.1002/cne.20257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rask-Andersen H, Bostrom M, Gerdin B, Kinnefors A, Nyberg G, Engstrand T, Miller JM, Lindholm D. Regeneration of human auditory nerve. In vitro/in video demonstration of neural progenitor cells in adult human and guinea pig spiral ganglion. Hear Res. 2005;203:180–191. doi: 10.1016/j.heares.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Rubel EW, Fritzsch B. AUDITORY SYSTEM DEVELOPMENT: Primary Auditory Neurons and Their Targets. Annu Rev Neurosci. 2002;25:51–101. doi: 10.1146/annurev.neuro.25.112701.142849. [DOI] [PubMed] [Google Scholar]
- Ruben RJ. Development of the inner ear of the mouse: a radioautographic study of terminal mitoses. Acta Otolaryngol:Suppl. 1967;220:221–244. [PubMed] [Google Scholar]
- Ryugo DK. 1992. The auditory nerve; peripheral innervation, cell body morphology, and central projections. In: Webster DB, Popper AN, Fay RR, editors. The Mammalian Auditory Pathway: Neuroanatomy. New York: Springer-Verlag. pp 23–65.
- Satoh T, Fekete DM. 2005. Clonal analysis of the relationships between mechansosensory cells and the neurons that innervate them in the chicken ear. Development in press. [DOI] [PubMed]
- Seipel K, Yanze N, Schmid V. Developmental and evolutionary aspects of the basic helix-loop-helix transcription factors Atonal-like 1 and Achaete-scute homolog 2 in the jellyfish. Dev Biol. 2004;269:331–345. doi: 10.1016/j.ydbio.2004.01.035. [DOI] [PubMed] [Google Scholar]
- Tessarollo L, Coppola V, Fritzsch B. NT-3 replacement with brain-derived neurotrophic factor redirects vestibular nerve fibers to the cochlea. J Neurosci. 2004;24:2575–2584. doi: 10.1523/JNEUROSCI.5514-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GX, Poo MM. Requirement of TRPC channels in netrin-1-induced chemotropic turning of nerve growth cones. Nature. 2005;434:898–904. doi: 10.1038/nature03478. [DOI] [PubMed] [Google Scholar]
- Wang VY, Hassan BA, Bellen HJ, Zoghbi HY. Drosophila atonal fully rescues the phenotype of Math1 null mice: new functions evolve in new cellular contexts. Curr Biol. 2002;12:1611–1616. doi: 10.1016/s0960-9822(02)01144-2. [DOI] [PubMed] [Google Scholar]
- Woods C, Montcouquiol M, Kelley MW. 2004. Math1 regulates development of the sensory epithelium in the mammalian cochlea. Nat Neurosci. [DOI] [PubMed]
- Woods C, Montcouquiol M, Kelley MW. Math1 regulates development of the sensory epithelium in the mammalian cochlea. Nat Neurosci. 2004;7:1310–1318. doi: 10.1038/nn1349. [DOI] [PubMed] [Google Scholar]
- Wu HH, Ivkovic S, Murray RC, Jaramillo S, Lyons KM, Johnson JE, Calof AL. Autoregulation of neurogenesis by GDF11. Neuron. 2003;37:197–207. doi: 10.1016/s0896-6273(02)01172-8. [DOI] [PubMed] [Google Scholar]
- Yang Q, Bermingham NA, Finegold MJ, Zoghbi HY. Requirement of Math1 for secretory cell lineage commitment in the mouse intestine. Science. 2001;294:2155–2158. doi: 10.1126/science.1065718. [DOI] [PubMed] [Google Scholar]
- Zine A, Aubert A, Qiu J, Therianos S, Guillemot F, Kageyama R, de Ribaupierre F. Hes1 and Hes5 activities are required for the normal development of the hair cells in the mammalian inner ear. J Neurosci. 2001;21:4712–4720. doi: 10.1523/JNEUROSCI.21-13-04712.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou D, Silvius D, Fritzsch B, Xu PX. Eya1 and Six1 are essential for early steps of sensory neurogenesis in mammalian cranial placodes. Development. 2004;131:5561–5572. doi: 10.1242/dev.01437. [DOI] [PMC free article] [PubMed] [Google Scholar]