

Abstract
Intractable neuropathic pain often results from nerve injury. One immediate event in damaged nerve is a sustained increase in spontaneous afferent activity, which has a well-established role in ongoing pain. Using two rat models of neuropathic pain, the CCI and SNI models, we show that local, temporary nerve blockade of this afferent activity permanently inhibits the subsequent development of both thermal hyperalgesia and mechanical allodynia. Timing is critical—the nerve blockade must last at least 3–5 days and is effective if started immediately after nerve injury, but not if started at 10 days after injury when neuropathic pain is already established. Effective nerve blockade also prevents subsequent development of spontaneous afferent activity measured electrophysiologically. Similar results were obtained in both pain models, and with two blockade methods (200 mg of a depot form bupivacaine at the injury site, or perfusion of the injured nerve just proximal to the injury site with TTX). These results indicate that early spontaneous afferent fiber activity is the key trigger for the development of pain behaviors, and suggest that spontaneous activity may be required for many of the later changes in the sensory neurons, spinal cord, and brain observed in neuropathic pain models. Many pre-clinical and clinical studies of pre-emptive analgesia have used much shorter duration of blockade, or have not started immediately after the injury. Our results suggest that effective pre-emptive analgesia can be achieved only when nerve block is administered early after injury and lasts several days.
Keywords: Neuropathic pain, Chronic constriction injury, Spared nerve injury, Ectopic activity, Allodynia, Hyperalgesia
1. Introduction
Nerve injury caused by trauma, disease or surgery often leads to the development of neuropathic pain, a chronic condition for which effective treatment is lacking. Modifications are observed at several anatomical locations after peripheral nerve injury, including: (1) a large increase in spontaneous (ectopic) activity in dorsal root ganglia (DRG) cell bodies and injured afferent fibers (Wall and Gutnick, 1974); (2) abnormal contacts between sympathetic and sensory nervous systems (McLachlan et al., 1993); and (3) changes in the spinal cord and brain. Although much is known about what molecular and cellular changes occur at these sites in neuropathic pain, including changes in gene expression (Xiao et al., 2002), it is not clear what event(s) and which anatomical site(s) are critical in initially triggering development of neuropathic pain.
In commonly used animal models of neuropathic pain, both spontaneous activity and pain behaviors appear within the first 12 h—2 days post-injury (Bennett and Xie, 1988; Kim and Chung, 1992; Seltzer et al., 1990). Most other pathological changes described above, including spinal cord sensitization and gene expression changes, begin later than this. Spontaneous afferent activity is therefore a likely candidate for initiating chronic pain. A key observation is that temporarily blocking spontaneous activity reduces or eliminates spontaneous pain, hyperalgesia, and allodynia. This has been demonstrated in several different pain models, using methods to suppress spontaneous activity that vary widely in their specific targets (Chaplan et al., 2003; Lai et al., 2002; Lyu et al., 2000; Xiao and Bennett, 1995; Yoon et al., 1996). In these studies, the nerve blocking treatments were started several days after injury, were often brief, and provided only temporary and sometimes incomplete inhibition of pain behaviors.
Other studies indicate that the early period of spontaneous activity may play a special role in neuropathic pain development. Nerve transection or injury induces a rapid 5–10 min injury discharge, followed hours later by onset of spontaneous activity (Govrin-Lippmann and Devor, 1978). Smith et al. provided continual systemic lidocaine for the first 3 post-injury days, and showed this treatment completely reversed thermal hyperalgesia in the rat CCI model (Smith et al., 2002). They did not look after day 3 to determine whether the effect outlasted the lidocaine infusion. Araujo et al. found that brief systemic application of local anesthetics given 2 days after injury reduced mechanical allodynia more effectively than if given at day 7 (Araujo et al., 2003). They did not look beyond day 21.
We were interested in determining whether continuous blockade of spontaneous activity, started soon after nerve injury, could be effective in permanently reducing neuropathic pain behaviors. This idea was supported by the literature, but previous studies either blocked spontaneous activity only briefly, started blockade many days after nerve injury, and/or did not look at long time points after removal of the blockade. We developed methods for applying nerve blockers locally at the site of injury, continually (for 3–7 days) immediately after injury. We then determined the long-term consequences of such blockade on development of neuropathic pain.
2. Materials and methods
2.1. Animals
Adult male Wistar rats (body weight 200–250 g) were housed one or two per cage with free access to water and standard rat chow. Before testing, the animals were allowed to acclimate for 3–5 days under a controlled diurnal cycle of 12 h light and 12 h dark. The ambient environment was maintained at constant temperature (22±0.5 °C) and relative humidity (60–70%). The experimental protocol was approved by the institutional animal care and use committees (IACUC) of the University of Cincinnati and the University of Arkansas for Medical Sciences.
2.2. Bupivacaine and TTX osmotic pump preparation
2.2.1. Bupivacaine OH
The local anesthetic bupivacaine (Cl salt; Sigma, St Louis, MO) was precipitated as bupivacaine OH to make an insoluble depot form. Bupivacaine Cl powder was dissolved in distilled water at concentrations up to 10%. NaOH (1 M) was dripped into the solution with stirring until there was no more precipitate forming. The solid bupivacaine OH was filtered and rinsed with fresh water. The bupivacaine OH was moved to a clean beaker with fresh water (just enough to cover the precipitate), and stirred for 5 h or longer to make it as fine as possible. Then the precipitate was washed carefully with fresh water to remove the extra NaOH. After washing, bupivacaine OH was dried under vacuum and heat (temperature below 80 °C) overnight. Dried bupivacaine OH was a white loose powder and was used in this form.
2.2.2. TTX osmotic pump
Tetrodotoxin (TTX; Sigma, St Louis, MO) dissolved in saline (780 μM) was used to fill an osmotic pump (Model 2001 or Model 1003D, Durect Corporation, Cupertino, CA) which was attached to silicone tubing (50–60 mm in length). The silicone tubing was slit over the final 15 mm. Before implantation, filled osmotic pumps were incubated 4 h for 3 day pumps or overnight for 7 day pumps in saline at 37 °C. Saline osmotic pumps used in control experiments were prepared identically except for omission of TTX.
2.3. Surgical preparations
2.3.1. Chronic constrictive injury (CCI) model
A peripheral mononeuropathy was produced by loosely ligating the sciatic nerve (chronic constrictive injury, CCI) according to a method previously described (Bennett and Xie, 1988). Briefly, under pentobarbital sodium anesthesia (40 mg/kg, i.p.), the sciatic nerves were exposed bilaterally at mid-thigh level by blunt dissection through the biceps femoris muscle. On one side, four loosely constrictive ligatures (4–0 chromic gut suture) were tied around the nerve at spaces of about 1 mm. Muscle and skin were closed in layers. On the contralateral side, sham operations were performed to expose and mobilize the nerve, but there was no ligation. To minimize differences in technique, all operations were done by the same person. A single dose of antibiotics was administered (Ampicillin 8000 u/rat, Sigma, St Louis, MO). Each animal was allowed to recover for 24 h before any behavioral testing.
2.3.2. Spared nerve injury (SNI) model
A peripheral mononeuropathy was produced by axotomy and ligation of the tibial and common peroneal nerves, leaving the sural nerve intact, as described by Decosterd and Woolf (2000). Briefly, under pentobarbital sodium anesthesia (40 mg/kg, i.p.), the sciatic nerve and its three terminal branches: the sural, common peroneal and tibial nerves were exposed at low-thigh level by blunt dissection through the biceps femoris muscle. The common peroneal and the tibial nerves were tightly ligated with 5–0 silk. The nerve distal to the ligature was sectioned and 2–4 mm of the nerve stump was removed. Great care was taken to avoid any contact with or stretching of the intact sural nerve. Muscle and skin were closed in layers. A single dose of antibiotics was administered (Ampicillin 8000 u/rat, Sigma, St Louis, MO). Each animal was allowed to recover for 24 h before further testing.
2.4. Transient nerve blockade
Two different chemicals, bupivacaine OH and TTX, were used independently to suppress activity in normal and injured sciatic nerve.
2.4.1. Bupivacaine OH
For testing the effects of the drug in normal sciatic nerve, approx. 20 mm of sciatic nerve was freed from surrounding tissue, and bupivacaine OH was placed evenly around the freed sciatic nerve. For suppressing spontaneous afferent activity in injured sciatic nerve, the injury site was completely and evenly covered with 200 mg bupivacaine OH powder immediately (within 10 min) after the sciatic nerve was injured with either the CCI or SNI technique. In the case of the SNI technique, the bupivacaine powder was placed covering both severed nerve branches and the more proximal area before the sciatic nerve divides.
Although previous studies reported that the injected soluble form of bupivacaine can cause cardiotoxicity (Covino, 1986; Graf, 2001; Mather and Chang, 2001) and neurotoxicity (Barsa et al., 1982; Li et al., 1985; Naguib et al., 1998; Sakura and Hashimoto, 1997), in our studies no animal showed cardiac problems. In addition, there was no significant change in the histology of sciatic nerve blocked with bupivacaine OH compared with normal sciatic nerve. Also, on rudimentary histological examination, bupivacaine OH did not alter the appearance of the CCI site.
2.4.2. TTX pump
To test the effects of TTX in uninjured sciatic nerve, either 7-day or 3-day osmotic pumps filled with TTX solution were implanted underneath the skin covering the thigh. Silicon tubing (0.64 mm ID × 1.19 mm OD; Dow Corning Corporation Midland, MI) was led from the pump to the sciatic nerve at high-thigh level. The two halves of the slit end of the tubing were placed surrounding the sciatic nerve and tightened together at the very end with 5–0 silk suture. Similarly, to block injured sciatic nerve, a portion of the sciatic nerve at least 5 mm long and located 10–20 mm proximal to the injury site, was infused with TTX. Osmotic TTX pumps were implanted either immediately following sciatic nerve injury surgery (CCI or SNI) or 10 days after this surgery, as indicated. The method was similar to that previously published (Gardiner et al., 1992) except that we found that implanting a silicone cuff on injured sciatic nerve was extremely difficult to do without further damaging the nerve. Hence instead of a cuff we surrounded the nerve with two soft pieces of silicon formed by cutting a slit in the end of silicon tubing. As a control, in some experiments the sciatic nerve was perfused in an identical fashion from saline-filled osmotic pumps. The pump and tubing alone did not cause any changes in neuropathic pain behavior or in spontaneous activity (see saline controls in Figs. 1–5). Our technique differed from that of Mosconi and Kruger (1996), who placed tubing around the sciatic nerve to produce a pain syndrome, in that we used a softer tubing type with very limited contact to the nerve, and the tubing did not constrict the nerve, due to the presence of the slit.
Fig. 1.

Sciatic nerve blockade inhibits pain sensitivity in uninjured nerve. (A) The indicated amounts of depot bupivacaine were implanted into uninjured sciatic nerve, and thermal pain sensitivity was measured in hind paws on both the treated side and the control side. The difference in thermal pain threshold between the two paws is shown. Note that behavioral data for some higher doses was not recorded for day 1. In this and subsequent figures, the pain threshold difference scores are plotted such that points below the axis represent hypersensitivity to the pain stimulus on the treated side (neuropathic pain behavior), while those above the axis represent hyposensitivity. (B) Absolute values for the thermal pain threshold for the experiment in panel A. The same symbols are used as in panel A, except that data from the treated side is shown in lighter color. For clarity the 30 mg data are omitted. (C) Area under the curve from data in A, showing a near-linear relationship between the drug dose and the effect. (D) The effect of bupivacaine (300 mg) on thermal latency on both control and drug-treated sides is shown on a more extended time scale. (E) Thermal pain difference scores in rats with sham operation (no pump implanted), or implantation of osmotic pump of infusing saline, 7-day TTX, or 3-day TTX unilaterally onto the sciatic nerve. (F) Absolute pain thresholds on both drug-treated and contralateral sides for the experiment shown in E. (G) Mechanical pain sensitivity was tested on hind paws of both the treated side and the normal control side, using von Frey filaments of varying thickness. These filaments are designed to measure an increase in pain sensitivity from normal, but occasionally in naïve animals, and especially in the presence of nerve blockers, the hind paw was often insensitive even to the strongest force that could be applied (18 g). This decrease in sensitivity was quantified as the percentage of animals in which the paw on the treated side did not respond to even the largest mechanical stimulus available (18 g). Shown is the percentage of rats with a mechanical pain threshold above the maximum force in the paw on the treated side, with treatments of either sham operation, or osmotic pumps of saline, 7-day TTX, or 3-day TTX as indicated. (H) Mechanical thresholds on the untreated side for the experiment in panel G. Threshold was generally 18 g (or, occasionally, 13 g, as indicated by lower average values) for most measurements; except that 3 animals in the 3-day TTX group each had a threshold above the cutoff value 18 g on one day (day 0, 1, and 5, respectively). N = 5–7 animals per group, except for TTX-7 day (N = 12) and 300 mg bupivacaine (n = 9).
Fig. 5.

Once the neuropathic pain is established, TTX nerve blockade is no longer effective in preventing subsequent pain behaviors. Rats with CCI (a–d) or SNI (e–h) were tested for thermal pain (a, b, e, f) and mechanical pain (c, d, g, h). Both difference scores and absolute pain thresholds are presented for each experiment, using the same format as in Fig. 2 Contrary to the results in Figs. 2 and 3, application of TTX starting 10 days after onset of neuropathic pain (shaded areas) was not effective in preventing subsequent thermal hyperalgesia or mechanical allodynia. For all CCI data shown, none of the groups differed significantly from the CCI alone group comparing data obtained after day 21 (One-way ANOVA with Dunnett’s post-test.) N = 6 animals per group except for the CCI-TTX group (n = 12) and CCI-saline group (n = 4).
Note that comparison with the literature indicates that both of our blockade methods would not have blocked the injury discharge, which occurs in the first minutes after injury, but that blockers were in place well before the time when spontaneous afferent activity begins.
2.5. Behavioral tests
All behavioral tests were conducted between 10:00 and 19:00 h daily, every day in the first 6 post-operative (PO) days (with thermal and mechanical pain testing on alternate days), and every 4 days after that until PO day 30. After day 30, all behavioral tests were performed every 7 days until PO day 60. For the SNI model only, after PO day 60, all behavioral tests occurred every 14–21 days until 5 months PO. The tester was blind to the treatment condition of the animals.
2.5.1. Thermal hyperalgesia
The method of Hargreaves et al. (1988), was used to assess paw-withdrawal latency to a thermal nociceptive stimulus. Rats were placed in a Plexiglas enclosure on top of the glass surface of a thermal testing apparatus (Model 336, IITC/Life Science Instruments, Woodland Hills, CA) and allowed to acclimate for 30 min before testing. A mobile radiant heat source (a high-intensity light beam) located under the glass was focused onto the hind paw. The paw-withdrawal latency was recorded by a digital timer. Stimulus intensity was kept constant throughout the entire experiment, and was adjusted to give approximately a 10 s withdrawal latency in the normal or sham operated hind paw. A significant reduction in paw-withdrawal latency compared with normal baseline was interpreted as thermal hyperalgesia. The withdrawal latencies of both hind paws were tested once every 10 min until three consistent thresholds were obtained. The mean value of the three consistent thresholds was used as the thermal threshold. A cutoff time of 20 s was used to prevent potential tissue damage.
2.5.2. Mechanical allodynia
Mechanical allodynia was assayed by using a series of calibrated von Frey filaments (Stoelting Co., Wood Dale, IL) (Chaplan et al., 1994). The rats were placed in individual Plexiglas enclosures on a mesh floor and allowed to acclimate for 30 min. The hairless plantar surface of the hind paw was probed by a series of 16 von Frey filaments (ranging from 0.01 to 18 g force; diameters of 15–150 μm). In the absence of response (indicated by brisk withdrawal or flinching), the filament of next greater force was used. In the presence of response, the filament of next lower force was used. Each monofilament was applied with sufficient force to bend for 1–2 s. Each von-Frey hair was used five times (at five distinct, pre-determined anatomical locations) on each hind paw at 30 s intervals, and the response threshold was defined as the lowest force that caused at least three withdrawals out of the five consecutive applications. In occasional control animals and especially in uninjured animals treated with nerve blockers (see Fig. 1), no withdrawal was observed even with the largest filament used (18 g). Larger diameter filaments could not be used because they caused the paw to be lifted by the filament. Hence 18 g was used as the cutoff value in the mechanical pain tests.
2.6. Determination of the incidence of spontaneous afferent activity by in vivo electrophysiological recording
Rats were anesthetized with pentobarbital sodium (40 mg/kg) at the indicated times after the initial CCI operation. The rat was put on a warm blanket to keep the body temperature around 37 °C. Supplemental pentobarbital sodium was provided during the entire experiment through a saphenous vein cannula. Six to seven centimeter of the sciatic nerve was exposed at two different sites. One was the injury site, where stimulation electrodes were placed. The second site, where microfilament recordings were made, was exposed just distal to the ischium, about 30–40 mm proximal to injury site. Skin edges of these two sites were used to form two surrounding pools of fluid electrically isolated from each other. The ligated portion of the nerve was freed of adherent tissue. A pair of silver electrodes was placed just proximal to the injury site. The proximal exposed sciatic nerve, the recording site, was bathed in warm paraffin oil. Size 5 tweezers were used to tear off the perineurium of the sciatic nerve at the site of recording. A bundle of fibers, which contained about 1/6–1/5 of the whole sciatic nerve, was teased from the opened nerve and disconnected centrally. This bundle was further separated into microfilaments with approximately equal diameter (30–50 μm). An individual microfilament was placed on a silver wire recording electrode. For each microfilament the following characteristics were checked:
The microfilament was examined for 3 min to see whether any action potentials were present and how many fibers were firing in each strand. The number of fibers with ongoing activity was estimated by the different spike height and frequency of the action potentials. Spike heights were typically 2–10 times the noise level.
We determined whether the recorded ongoing activities represented spontaneous activity originating in the injured axons. This was based on the responses to stimulating the receptive field and the shape and amplitude of the action potentials. The receptive field of the firing fiber was determined by applying gentle mechanical stimuli to the skin and muscle which sciatic nerve innervates. The ongoing activity was considered as spontaneous afferent activity if the mechanical stimuli failed to alter the discharge patterns. Note that this method may slightly overestimate the number of spontaneously active fields due to axons that innervated receptive fields that were difficult to access. However, this caveat applies to animals in all the experimental and control groups.
The total number of conducting axons in each strand was counted. A reported method (Goldstein et al., 1988) was used in this measurement. Briefly, the sciatic nerve was stimulated with a gradually increasing strength of current (0.2–0.5 ms square-wave pulses, 1 Hz) up to 5 mA. The number of all-or-none contributions to the compound action potential on the strand was counted and considered as the total number of conducting myelinated afferents in the strand. Then a 5-mA current was used to stimulate 5 times at 2 s intervals to count unmyelinated fibers.
Finally, the types of axons contained in each recorded strand was determined by their conduction velocities. The conduction velocity was calculated by dividing propagation distance by the response latency to electrical stimulus pulses delivered just proximal to the injury site through a pair of silver electrodes. Fibers with conduction velocity ≥20 m/s were counted as Aβ, those with intermediate velocity as Aδ, and those with conduction velocity ≥2 m/s were counted as C. Six microfilaments were randomly sampled from each bundle of fibers and the rest discarded.
This entire procedure was repeated 5–6 times until the whole sciatic nerve was teased out. The incidence of spontaneous activity was defined as the number of fibers (waveforms) that were spontaneously active divided by the total number of conducting afferents sampled from the sciatic nerve. Data presented are summaries of recordings from 200 to 1200 conducting fibers per experimental group.
2.7. Statistical analyses
Behavioral data from different experimental groups was analyzed using ANOVA, with Dunnett’s post-test, unless otherwise indicated in the text or figure legends. Significance was ascribed for P<0.05. The number of animals per experimental group is given in the figure legends. Data are presented as means±SEM.
3. Results
3.1. Effects of transiently blocking nerve activity
Before attempting to block nerve activity in injured nerve, we first estimated the approximate duration of the blockers action by testing them in intact animals. The first blocking method used a depot form of the local anesthetic bupivacaine, which was imbedded next to sciatic nerve. In these experiments, the nerve was uninjured, though the bupivacaine was imbedded at the same anatomical location that would be injured in later experiments. This relatively insoluble hydroxide form of bupivacaine has a slower rate of absorption, and we found that the duration of action, as estimated by the time course of decreased thermal pain sensitivity, could be adjusted by changing the amount of imbedded bupivacaine (Fig. 1). A second method of blocking nerve activity was to superfuse the sciatic nerve proximal to this site with the sodium channel blocker TTX using either a 3-day or 7-day osmotic pump. We then measured thermal and mechanical pain sensitivities, since thermal hyperalgesia and mechanical allodynia are major behavioral signs of neuropathic pain.
The effects of these two blocking methods on uninjured nerve are shown in Fig. 1. For simplicity, thermal pain behaviors are presented as difference scores—comparing the response to thermal or mechanical stimulation of the hind paw on the injured side with that of the hind paw on the uninjured or sham-operated side of the same animal (Sheen and Chung, 1993; Yamamoto et al., 1993). Hence, in difference score graphs presented, points below the x-axis represent hypersensitivity to the pain stimulus (neuropathic pain behavior), while those above the axis represent hyposensitivity. However, the difference scores observed primarily reflected changes occurring on the injured or drug-treated side, as can be observed by examining the absolute thresholds in experimental vs. control side in each animal. In Fig. 1A,B,E–G and in all subsequent figures presenting behavioral data, the difference scores are presented on the left hand side while the corresponding absolute thresholds from the same experiment are presented on the right.
As shown in Fig. 1, reduction of pain sensitivity by superfused TTX had a duration as expected from the rating of the pump (3-day or 7-day). Bupivacaine depot applied to normal, uninjured sciatic nerves resulted in a reduction in pain sensitivity whose duration depended upon the amount used, lasting 7–9 days at the highest dose used (300 mg). In subsequent experiments using bupivacaine in injured nerve, a dose of 200 mg was selected. As shown in Fig. 1, this dose affects pain behavior for 3–4 days. The 300 mg dose was not used because it was difficult to fit this amount of powder into the space around the nerve, and in some animals swelling around the wound area was observed at this dose. As the data in Fig. 1 indicate, neither blockade method completely blocks the ability to observe pain behaviors. Rather, the data in Fig. 1 were used as an estimate of the time the drugs were present.
3.2. Transient nerve blockade in the CCI model
We then examined the effect of nerve blockade in a rodent model of neuropathic pain, the CCI model (Bennett and Xie, 1988). This model produces robust thermal hyperalgesia that starts within 3–5 days of the injury and that lasts for about 40–50 days before progressing into hypoalgesia (Bennett and Xie, 1988), as shown in Fig. 2A and B (open symbols). The mechanical allodynia produced by this model (Fig. 2C and D, open symbols) lasts even longer, and does not seem to progress to a hyposensitive state. Nerve blockade, when applied during the initial stage of nerve injury (shaded areas in Fig. 2), inhibited both types of neuropathic pain behavior (Fig. 2, closed symbols). In fact, the observed thermal pain behavior was hypoalgesic, similar in magnitude to that observed in untreated CCI rats after 40 days. Note that the hypersensitivity to thermal pain is eliminated for the entire duration of the experiment, well beyond the initial several days duration of nerve blockade. Blockade of initial nerve activity also significantly reduced mechanical allodynia, though it was not completely eliminated (Fig. 2C and D). Similar results were obtained with either method of transient nerve blockade—TTX (7-day pump) or depot bupivacaine (200 mg). The differences between thermal hyperalgesia and mechanical allodynia in response to nerve blockade may reflect the fact that they are mediated by different sensory fibers—unmyelinated C-fibers and large diameter, myelinated A-fibers, respectively (Field et al., 1999; Ossipov et al., 2000), and/or that mechanical allodynia may more rapidly develop a prominent central component (Chen et al., 2004). Only partial reduction of allodynia was obtained with the 3-day TTX pump or with doses of bupivacaine (30 or 120 mg) that blocked nerve activity for less than 3 days (data not shown).
Fig. 2.
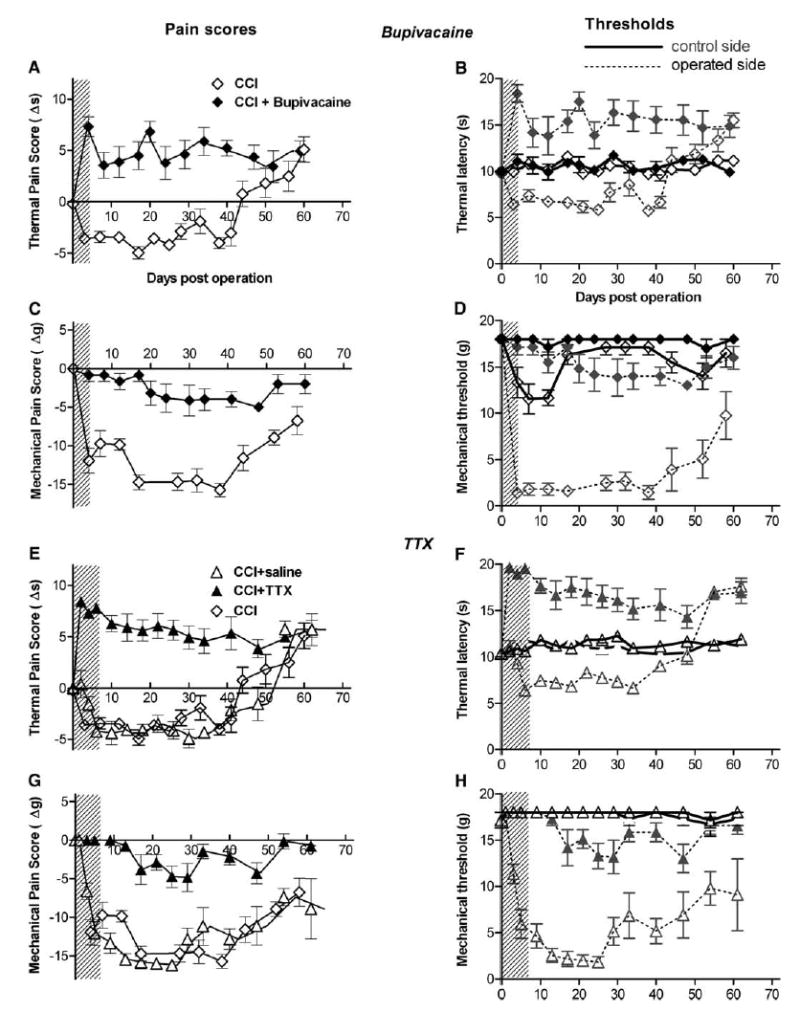
Local application of nerve blockers during the initial stage of neuropathic pain is sufficient to inhibit the subsequent development of neuropathic pain. Rats with chronic constriction injury (CCI) of the sciatic nerve were tested for thermal pain (a, b, e, f) and mechanical pain (c, d, g, h). Thermal hyperalgesia lasted over 30 days, and mechanical allodynia lasted over 60 days. Nerve blockade applied at the initial stage of neuropathic pain (shaded areas) by 200 mg bupivacaine (a–d) or by TTX (e–h) prevented the development of neuropathic pain. Duration of blocker activity (shaded areas) was estimated by the experiment shown in Fig. 1. Mechanical pain threshold was defined as the lowest applied force that caused at least three withdrawals out of the five consecutive applications, with a cutoff value of 18 g. As in a and b, difference scores are presented on the left, with negative values corresponding to increased pain sensitivity, while absolute thresholds on both operated and contralateral sides are shown on the right for each experiment, using the same symbols as in the difference data, with lighter shading and dotted lines used to distinguish the operated side from the control side. For all data shown, the groups with nerve blockade (N = 6–7) differed significantly from the group with CCI only (N = 6; one-way ANOVA with Dunnett’s post-test, P<0.01). The CCI plus saline pump group (N = 6) did not differ significantly from CCI only.
3.3. Transient nerve blockade in the SNI model
Although CCI is considered a useful neuropathic pain model because of the robust neuropathic pain behaviors that develop, it differs from clinical situations in humans because in this model the pain behaviors last no longer than 2 months. In the more recently developed SNI model (Decosterd and Woolf, 2000), neuropathic pain behavior was reported to last almost 6 months. In order to examine the effects of transient nerve blockade over a longer term, we used the SNI model, and performed nerve blockade experiments similar to those shown in Fig. 2. We observed very similar results: both TTX and bupivacaine depot, when applied at the time of nerve injury so as to suppress nerve activity for 4–7 days starting immediately after nerve injury, inhibited both mechanical allodynia and thermal hyperalgesia (Fig. 3). As in the original study describing the SNI model (Decosterd and Woolf, 2000), we found that the mechanical allodynia and thermal hyperalgesia were longer-lasting in this model than in the CCI model, although in our hands the thermal hyperalgesia lasted 3–4 months rather than 6 months (Fig. 3A and B), and eventually progressed to thermal hypoalgesia much as the CCI model did. In the original study, significant changes in thermal withdrawal latency were not observed following SNI; hence, a change in the duration of paw withdrawal was measured (Decosterd and Woolf, 2000). It is not clear why we were able to see significant differences in withdrawal latency in the SNI model; we did use a different strain of rat.
Fig. 3.

Local application of nerve blockers during the initial stage of neuropathic pain blocks the subsequent development of neuropathic pain in spared nerve injury (SNI) model. Rats with SNI were tested for thermal pain (a, b, e, f) and mechanical pain (c, d, g, h). Both thermal hyperalgesia and mechanical allodynia lasted well over 60 days. Both difference scores and absolute pain thresholds are presented for each experiment, using the same format as in Fig. 2. Nerve blockade at the initial stage of neuropathic pain (shaded areas) by 200 mg bupivacaine (a–d) or by TTX (e–h) inhibited the development of neuropathic pain. For all data shown, the groups with nerve blockade (N = 12–13) differed significantly from the group with SNI only (N = 6; one-way ANOVA with Dunnett’s post-test, P<0.01). The SNI plus saline group (N = 6) did not differ significantly from SNI only.
3.4. Effect of transient nerve blockade on subsequent spontaneous activity
The association between spontaneous afferent activity in the injured nerve and neuropathic pain is well established—temporarily blocking ongoing spontaneous afferent activity can temporarily block pain behaviors in animal models (Eglen et al., 1999; Lyu et al., 2000; Xiao and Bennett, 1995; Zimmermann, 2001). In contrast, our results have shown that temporarily blocking the early spontaneous afferent activity permanently eliminates neuropathic pain behaviors. This raises the question: after the nerve blockade effect of bupivacaine or TTX wears off, does the spontaneous afferent activity return even though the neuropathic pain is absent, or is the spontaneous afferent activity also permanently eliminated by the initial transient nerve blockade? To address this question, we used nerve fibers dissected out from the sciatic nerve, and recorded electrical activity of A- (Fig. 4, left) and C-fibers (Fig. 4, right). In animals with CCI, spontaneous afferent activity appeared in both A- and C-fibers one day after the nerve injury, lasted for at least a month, and subsided at later time points (Fig. 4B and E) concomitant with the disappearance of neuropathic pain behaviors in this model. Since spontaneous afferent activity remains robust during the period of 19–27 days post-CCI, we examined animals with various treatments during this time period. As shown in Fig. 4C and F, sham-operated animals did not show elevated spontaneous afferent activity, while CCI animals without any drug treatment (‘naïve’) or with only saline perfusion (‘saline’) displayed robust spontaneous afferent activity as expected based on the time-course data (Fig. 4B and E, marked by bars above the 19–27 days). Remarkably, blockers (bupivacaine or TTX) applied only during the initial stage after injury reduced spontaneous afferent activity at PO day 19–27 to levels similar to controls. These results indicate that initial nerve blockade prevented subsequent spontaneous afferent activity in the injured sciatic nerve, long after the acute effects of the blockers had worn off (see Fig. 1). These data confirm and extend the association between spontaneous afferent activity and neuropathic pain behavior, and suggest that events during the initial period after nerve injury are critical for subsequent maintained spontaneous afferent activity in neuropathic pain.
Fig. 4.
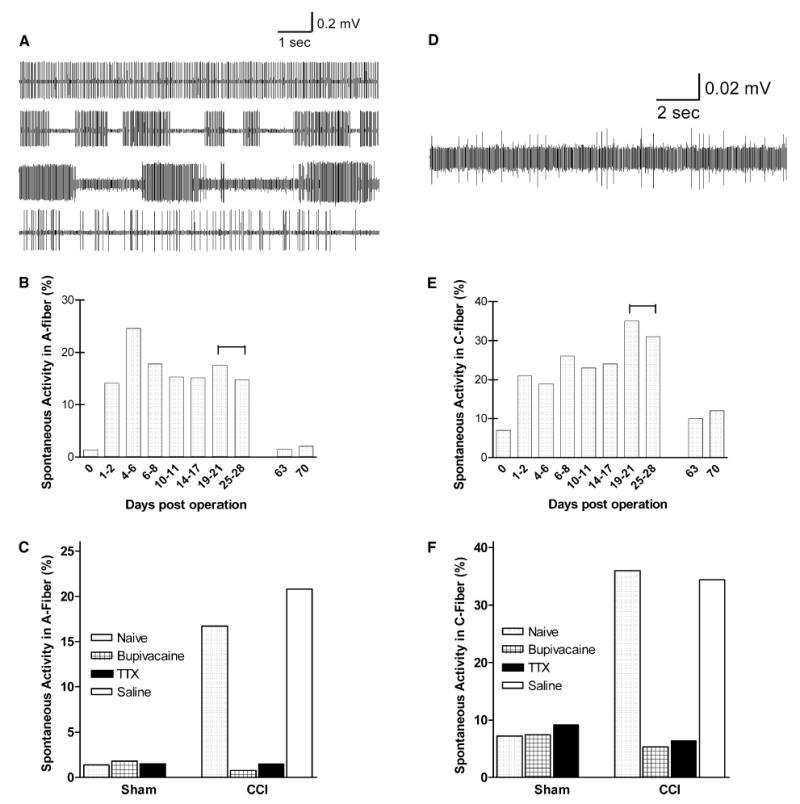
Nerve blockade during the initial stage of neuropathic pain inhibits subsequent appearance of spontaneous afferent activity in injured sciatic nerve. Examples shown are of spontaneous electrical activity recorded in afferent nerve fibers in A-fibers (a) and C-fibers (d). Time course of spontaneous activity after sciatic nerve injury was determined in A-fibers (b) and C-fibers (e), showing the percentages of activatable fibers that displayed spontaneous (ectopic) firing. High levels of spontaneous activity lasted for over 4 weeks after injury. The bar above the 19–28 days in (b) and (e) indicates the time period for recordings of (c) and (f), where rats with various treatments were analyzed for the level of spontaneous activity in sciatic nerve. After CCI nerve injury, both A-fibers (c) and C-fibers (f) displayed high levels of spontaneous activity in the CCI-rats that were untreated (‘naïve’) or perfused with saline, but treatment with either bupivacaine or TTX during the first 4–7 days post-injury inhibited this elevation in spontaneous activity. For both C and A fiber data, the reduction in percentage of spontaneously active fibers by 200 mg bupivacaine or by TTX was significant (P<0.0003), while animals receiving CCI plus saline did not differ significantly those receiving CCI alone (Fisher’s exact with Bonferroni correction for multiple comparisons).
The electrophysiological experiments also included measurement of the total number of activatable (i.e. both silent and spontaneously active) fibers in each microfilament; roughly comparable numbers of microfilaments were recorded in each animal (see Methods). These data indicated that there was no gross loss of fibers due to the treatment with nerve blockers: for the extensive set of measurements conducted 19–27 days after injury, the average number of activatable myelinated and unmyelinated fibers per animal, respectively, was 249 and 34 in CCI rats, 259 and 37 in CCI rats treated with TTX, 244 and 34 in CCI rats treated with bupivacaine. In uninjured rats the numbers were 266 and 49 in naïve rats, 259 and 31 in rats treated with bupivacaine, and 196 and 54 in rats treated with TTX. It should be noted that these measurements count axons stimulated just proximal to the injury site and measured more proximally; for TTX-treated animals this included conduction through the site at which the TTX-tubing had been placed.
3.5. Transient nerve blockade after neuropathic pain is well established
To determine whether the timing of nerve blockade is critical, we applied nerve blockade as before, except that the nerve blockers were applied starting at 10 days after the nerve injury, a time point at which robust thermal hyperalgesia (Fig. 5A and B, open symbols) and mechanical allodynia (Fig. 5C and D, open symbols) had already developed. Nerve blockade by TTX pump (filled symbols in Fig. 5) only temporarily reduced pain sensitivity during the nerve blockade (shaded areas), but did not affect the subsequent neuropathic pain behaviors once the blockade had worn off, in either the CCI (Fig. 5A and C) or the SNI model (Fig. 5B and D). Similar results were obtained using late application of bupivacaine rather than TTX: applying 200 mg to the injury site 10 days after injury was ineffective in providing prolonged reduction of neuropathic pain behaviors in either the CCI or SNI model (Fig. 6). In fact, there was a small, transient, but significant increase in mechanical pain in the SNI model following application of bupivacaine at day 10 (Fig. 6G) and delay in development of the late thermal hypoalgesia phase (Fig. 6E).
Fig. 6.

Once the neuropathic pain is established, bupivacaine nerve blockade is no longer effective in preventing subsequent pain behaviors. Rats with CCI (a–d) or SNI (e–h) were tested for thermal pain (a, b, e, f) and mechanical pain (c, d, g, h). Both difference scores and absolute pain thresholds are presented for each experiment, using the same format as in Fig. 2 Contrary to the results in Figs. 2 and 3, local application of 200 mg bupivacaine starting 10 days after onset of neuropathic pain (shaded areas) was not effective in preventing subsequent thermal hyperalgesia or mechanical allodynia. N = 6 animals per group. Omitting data from the blockade period (days 10–17), there were no significant differences between drug-treated and untreated groups in either of the CCI experiments shown (Mann–Whitney test).
4. Discussion
4.1. Key role of initial activity in neuropathic pain
The present study indicates that the development of neuropathic pain after peripheral nerve injury is permanently inhibited by transient local application of nerve blockers early after injury. Our interpretation is that these agents worked by reducing spontaneous activity at the injury site (bupivacaine depot), or preventing this activity from reaching the DRG cell bodies (TTX). Additional possibilities must be considered. We estimated the duration of action of the local blocking agents by testing them in noninjured animals. However, nerve injury induces wide-ranging changes in the function, expression and localization of sodium channels in sensory nerves. In particular, neuropathic pain can be relieved by lower doses of systemic local anesthetics than are required to block normal conduction (Mao and Chen, 2000). This raises the possibility that the local bupivacaine depot in our experiments actually worked systemically. It is difficult to quantitatively evaluate this possibility since most studies on systemic local anesthetics in chronic pain models are done with lidocaine. Arguments against this possibility include our finding that local bupivacaine was equally effective in the SNI and CCI models, although the latter is much less sensitive to systemic local anesthetics (Erichsen et al., 2003). In addition, we obtained very similar results with local TTX application, requiring a very similar time window, despite its very different pharmacological profile. Finally, although the profile of ion channels present in DRG cell bodies and axons changes over the first 2 weeks post-injury, many of the early injury-induced changes in ion channels are long-lasting (Waxman et al., 1999; Xiao et al., 2002), yet we found locally applied blockers were completely ineffectual when applied at day 10 after injury.
Similar arguments apply to other possible mechanisms of the bupivacaine and TTX effects: We cannot completely eliminate a possible role for known effects of local anesthetics or TTX on axonal transport, vascularity, inflammation, or sprouting (Kalichman, 1993; Radwan et al., 2002), nor can we completely eliminate the possibility that our results can be explained by neurotoxicity of TTX and bupivicaine that is specific to injured fibers. However, the complete lack of long-lasting behavioral effects of bupivacaine or TTX in uninjured animals, the lack of effect of bupivacaine or TTX when applied at day 10 after injury, and the striking similarity in magnitude of effect and time course between local bupivacaine and local TTX, all suggest that the most parsimonious interpretation of the data is that both agents inhibited neuropathic pain by reducing spontaneous activity or preventing it from reaching the DRG. Future studies including careful histology will be needed to determine whether local blockade exacerbates nerve damage in injured nerve, although fiber counts gave no evidence for such an effect.
In our experiments in which nerve blockade was applied for 4–7 days starting immediately after SNI or CCI, subsequent pain behavior was eliminated (thermal pain) or substantially reduced (mechanical pain) for the rest of the experiment, up to 5 months. Instead, we observed thermal hypoalgesia on the injured blocker-treated side. We interpret this as a more rapid progression to the hypoalgesia normally seen much later in the CCI model. This hypoalgesia was noted in the original description of the CCI method (Bennett and Xie, 1988), though it occurs somewhat earlier in our hands (40–50 days vs. 80 days). It is unlikely that this is due to the different strain of rat used (Sprague Dawley vs. Wistar), as another group using CCI in Sprague Dawley reports an even earlier progression to hypoalgesia (Jasmin et al., 1998). It seems more likely that this is due to differences in technique between labs. The SNI model also eventually progressed to thermal hypoalgesia in our study. It is unlikely that the hypoalgesia observed in experiments with nerve blockers reflects persistence (local or systemic) of low concentrations of TTX or bupivacaine. Although injury will alter the pharmacology of sensory neurons and axons, these alterations are probably not large enough to account for the blockers affecting channels for several months even though uninjured nerve is affected for only 4–7 days in behavioral experiments in uninjured animals. For example, TTX-resistant sodium channels are only 2½ times more sensitive to bupivacaine than TTX-sensitive channels (Scholz et al., 1998); use-dependent block by bupivacaine, which might account for some of its higher potency in spontaneously active fibers, makes it only 10–50% more effective (Gerner et al., 2001; Scholz et al., 1998). In addition, it would be difficult to account for our observation that the hypoalgesia observed early in injured, blockade treated animals was always quantitatively the same as that observed much later in untreated animals with the same injury, regardless of which blocker was used and regardless of which pain model was used.
Spontaneous activity originating in the injured afferent fibers and sensory neurons is proposed to be involved in both triggering and maintaining central sensitization following peripheral nerve injury (Ji and Woolf, 2001). Many studies have demonstrated that blocking spontaneous activity reduces spontaneous pain, allodynia, and hyperalgesia in several neuropathic pain models (Eglen et al., 1999; Zimmermann, 2001). As noted in the Introduction, most of these studies either used very brief blockade, started blockade many days after the injury, or did not observe pain behaviors over a long period of time. The new finding in our study is that timely application of drugs designed to reduce or eliminate spontaneous activity can reverse neuropathic pain behavior on a permanent basis, ‘permanent’ being at minimum the duration of pain behaviors in these models (i.e. at least 3–5 months in the case of the SNI model). The transient nerve blockade also prevented the subsequent development of spontaneous activity, corroborating previous findings showing that spontaneous activity and increased pain sensitivity are tightly associated. Our results do conflict with those of Suter et al. (2003), who recently reported that transient nerve blockade by placing bupivacaine-loaded microspheres around the sciatic nerve failed to prevent development of neuropathic pain in the SNI model. There are some differences between the two studies in: pain measurement methods; use of dexamethasone; doses of bupivacaine used; and delivery method. However, it is not clear why their results differed so dramatically from ours.
Our results are most readily interpreted as showing that initiation of the neuropathic pain state is a separate, distinct process from maintenance of that state. In particular, our results strongly suggest that the initial 4–5 day period of spontaneous afferent activity immediately following nerve injury plays a special role in initiating neuropathic pain. It will be interesting to determine whether blocking the initial phase of spontaneous afferent activity can also eliminate some of the other anatomical changes that occur later in the DRG or spinal cord in neuropathic pain models. A recent study found that injury-induced sympathetic fiber sprouting within the axotomized DRG is inhibited by blocking activity originating from the neuroma during the first week after nerve injury (Zhang et al., 2004). Surprisingly, sympathetic sprouting was inhibited for at least one additional week after the removal of the nerve blockade (longer times were not examined). If the results reported in the current study are found to be valid in the chronic pain model used in the Zhang et al. study (Zhang et al., 2004), a simple explanation for the prolonged inhibition of spouting may be that nerve blockade during the first week is sufficient to prevent any subsequent development of spontaneous afferent activity and hence of sprouting.
4.2. Implications for pre-emptive analgesia
Preventing the development of peripheral and central sensitization, particularly after surgical treatment, is the main objective of pre-emptive analgesia (Katz, 2001). A variety of methods and analgesics have been studied for their long-term effect on post-operative pain, yet the results remain inconsistent (Kelly et al., 2001b; Moiniche et al., 2002). The results presented in this report validate the principle of pre-emptive analgesia. Furthermore, our findings demonstrate that the critical time window is immediately post-operative and may need to be 4–5 days long. In a recent review of human trials of pre-emptive analgesia (Kelly et al., 2001a), it was noted that many of the studies that failed to show an effect of pre-emptive regional nerve blockade were those that used a treatment that was not long enough to block the post-surgical nociceptive barrage (see also Table 7 in Moiniche et al., 2002). This supports the hypothesis that post-operative analgesia must be much longer than has been the case in many clinical trials, on the order of several days, in order to effectively prevent peripheral and central sensitization and decrease the incidence of chronic pain syndromes such as phantom limb pain (Katz, 2001; Kelly et al., 2001b). In addition, our results suggest that applying local blockers of spontaneous activity to a limited area of peripheral nerve might have a dramatic impact on the development of neuropathic pain while avoiding the toxicity that occurs with systemic drugs.
Acknowledgments
This work was supported in part by NIH grants DA09444 and DA13471 (L.Y.) and NS39568 and NS045594 (J.Z.).
References
- Araujo MC, Sinnott CJ, Strichartz GR. Multiple phases of relief from experimental mechanical allodynia by systemic lidocaine: responses to early and late infusions. Pain. 2003;103:21–9. doi: 10.1016/s0304-3959(02)00350-0. [DOI] [PubMed] [Google Scholar]
- Barsa J, Batra M, Fink BR, Sumi SM. A comparative in vivo study of local neurotoxicity of lidocaine, bupivacaine, 2-chloroprocaine, and a mixture of 2-chloroprocaine and bupivacaine. Anesth Analg. 1982;61:961–7. [PubMed] [Google Scholar]
- Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Guo HQ, Lee DH, Luo L, Liu C, Kuei C, Velumian AA, Butler MP, Brown SM, Dubin AE. Neuronal hyperpolarization-activated pacemaker channels drive neuropathic pain. J Neurosci. 2003;23:1169–78. doi: 10.1523/JNEUROSCI.23-04-01169.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, King T, Vanderah TW, Ossipov MH, Malan TP, Jr, Lai J, Porreca F. Differential blockade of nerve injury-induced thermal and tactile hypersensitivity by systemically administered brain-penetrating and peripherally restricted local anesthetics. J Pain. 2004;5:281–9. doi: 10.1016/j.jpain.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Covino BG. Recent advances in local anaesthesia. Can Anaesth Soc J. 1986;33:S5–S8. doi: 10.1007/BF03019149. [DOI] [PubMed] [Google Scholar]
- Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–58. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- Eglen RM, Hunter JC, Dray A. Ions in the fire: recent ion-channel research and approaches to pain therapy. Trends Pharmacol Sci. 1999;20:337–42. doi: 10.1016/s0165-6147(99)01372-3. [DOI] [PubMed] [Google Scholar]
- Erichsen HK, Hao JX, Xu XJ, Blackburn-Munro G. A comparison of the antinociceptive effects of voltage-activated Na+ channel blockers in two rat models of neuropathic pain. Eur J Pharmacol. 2003;458:275–82. doi: 10.1016/s0014-2999(02)02792-9. [DOI] [PubMed] [Google Scholar]
- Field MJ, Bramwell S, Hughes J, Singh L. Detection of static and dynamic components of mechanical allodynia in rat models of neuropathic pain: are they signalled by distinct primary sensory neurones? Pain. 1999;83:303–11. doi: 10.1016/s0304-3959(99)00111-6. [DOI] [PubMed] [Google Scholar]
- Gardiner PF, Favron M, Corriveau P. Histochemical and contractile responses of rat medial gastrocnemius to 2 weeks of complete disuse. Can J Physiol Pharmacol. 1992;70:1075–81. doi: 10.1139/y92-149. [DOI] [PubMed] [Google Scholar]
- Gerner P, Mujtaba M, Sinnott CJ, Wang GK. Amitriptyline versus bupivacaine in rat sciatic nerve blockade. Anesthesiology. 2001;94:661–7. doi: 10.1097/00000542-200104000-00021. [DOI] [PubMed] [Google Scholar]
- Goldstein RS, Raber P, Govrin-Lippmann R, Devor M. Contrasting time course of catecholamine accumulation and of spontaneous discharge in experimental neuromas in the rat. Neurosci Lett. 1988;94:58–63. doi: 10.1016/0304-3940(88)90270-4. [DOI] [PubMed] [Google Scholar]
- Govrin-Lippmann R, Devor M. Ongoing activity in severed nerves: source and variation with time. Brain Res. 1978;159:406–10. doi: 10.1016/0006-8993(78)90548-6. [DOI] [PubMed] [Google Scholar]
- Graf BM. The cardiotoxicity of local anesthetics: the place of ropivacaine. Curr Top Med Chem. 2001;1:207–14. doi: 10.2174/1568026013395164. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Jasmin L, Kohan L, Franssen M, Janni G, Goff JR. The cold plate as a test of nociceptive behaviors: description and application to the study of chronic neuropathic and inflammatory pain models. Pain. 1998;75:367–82. doi: 10.1016/s0304-3959(98)00017-7. [DOI] [PubMed] [Google Scholar]
- Ji RR, Woolf CJ. Neuronal plasticity and signal transduction in nociceptive neurons: implications for the initiation and maintenance of pathological pain. Neurobiol Dis. 2001;8:1–10. doi: 10.1006/nbdi.2000.0360. [DOI] [PubMed] [Google Scholar]
- Kalichman MW. Physiologic mechanisms by which local anesthetics may cause injury to nerve and spinal cord. Reg Anesth. 1993;18:448–52. [PubMed] [Google Scholar]
- Katz J. Pre-emptive analgesia: importance of timing. Can J Anaesth. 2001;48:105–14. doi: 10.1007/BF03019721. [DOI] [PubMed] [Google Scholar]
- Kelly DJ, Ahmad M, Brull SJ. Preemptive analgesia I: physiological pathways and pharmacological modalities. Can J Anaesth. 2001a;48:1000–10. doi: 10.1007/BF03016591. [DOI] [PubMed] [Google Scholar]
- Kelly DJ, Ahmad M, Brull SJ. Preemptive analgesia II: recent advances and current trends. Can J Anaesth. 2001b;48:1091–101. doi: 10.1007/BF03020375. [DOI] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–63. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Lai J, Gold MS, Kim CS, Bian D, Ossipov MH, Hunter JC, Porreca F. Inhibition of neuropathic pain by decreased expression of the tetrodotoxin-resistant sodium channel, Na(v)1.8. Pain. 2002;95:143–52. doi: 10.1016/s0304-3959(01)00391-8. [DOI] [PubMed] [Google Scholar]
- Li DF, Bahar M, Cole G, Rosen M. Neurological toxicity of the subarachnoid infusion of bupivacaine, lignocaine or 2-chloroprocaine in the rat. Br J Anaesth. 1985;57:424–9. doi: 10.1093/bja/57.4.424. [DOI] [PubMed] [Google Scholar]
- Lyu YS, Park SK, Chung K, Chung JM. Low dose of tetrodotoxin reduces neuropathic pain behaviors in an animal model. Brain Res. 2000;871:98–103. doi: 10.1016/s0006-8993(00)02451-3. [DOI] [PubMed] [Google Scholar]
- Mao J, Chen LL. Systemic lidocaine for neuropathic pain relief. Pain. 2000;87:7–17. doi: 10.1016/S0304-3959(00)00229-3. [DOI] [PubMed] [Google Scholar]
- Mather LE, Chang DH. Cardiotoxicity with modern local anaesthetics: is there a safer choice? Drugs. 2001;61:333–42. doi: 10.2165/00003495-200161030-00002. [DOI] [PubMed] [Google Scholar]
- McLachlan EM, Janig W, Devor M, Michaelis M. Peripheral nerve injury triggers noradrenergic sprouting within dorsal root ganglia. Nature. 1993;363:543–6. doi: 10.1038/363543a0. [DOI] [PubMed] [Google Scholar]
- Moiniche S, Kehlet H, Dahl JB. A qualitative and quantitative systematic review of preemptive analgesia for postoperative pain relief: the role of timing of analgesia. Anesthesiology. 2002;96:725–41. doi: 10.1097/00000542-200203000-00032. [DOI] [PubMed] [Google Scholar]
- Mosconi T, Kruger L. Fixed-diameter polyethylene cuffs applied to the rat sciatic nerve induce a painful neuropathy: ultrastructural morphometric analysis of axonal alterations. Pain. 1996;64:37–57. doi: 10.1016/0304-3959(95)00077-1. [DOI] [PubMed] [Google Scholar]
- Naguib M, Magboul MM, Samarkandi AH, Attia M. Adverse effects and drug interactions associated with local and regional anaesthesia. Drug Saf. 1998;18:221–50. doi: 10.2165/00002018-199818040-00001. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, Malan TP, Jr, Porreca F. Spinal and supraspinal mechanisms of neuropathic pain. Ann N Y Acad Sci. 2000;909:12–24. doi: 10.1111/j.1749-6632.2000.tb06673.x. [DOI] [PubMed] [Google Scholar]
- Radwan IA, Saito S, Goto F. The neurotoxicity of local anesthetics on growing neurons: a comparative study of lidocaine, bupivacaine, mepivacaine, and ropivacaine. Anesth Analg. 2002;94:319–24. doi: 10.1097/00000539-200202000-00016. table. [DOI] [PubMed] [Google Scholar]
- Sakura S, Hashimoto K. Neurotoxicity of subarachnoid bupivacaine. Reg Anesth. 1997;22:198–9. doi: 10.1016/s1098-7339(06)80042-8. [DOI] [PubMed] [Google Scholar]
- Scholz A, Kuboyama N, Hempelmann G, Vogel W. Complex blockade of TTX-resistant Na+ currents by lidocaine and bupivacaine reduce firing frequency in DRG neurons. J Neurophysiol. 1998;79:1746–54. doi: 10.1152/jn.1998.79.4.1746. [DOI] [PubMed] [Google Scholar]
- Seltzer Z, Dubner R, Shir Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain. 1990;43:205–18. doi: 10.1016/0304-3959(90)91074-S. [DOI] [PubMed] [Google Scholar]
- Sheen K, Chung JM. Signs of neuropathic pain depend on signals from injured nerve fibers in a rat model. Brain Res. 1993;610:62–8. doi: 10.1016/0006-8993(93)91217-g. [DOI] [PubMed] [Google Scholar]
- Smith LJ, Shih A, Miletic G, Miletic V. Continual systemic infusion of lidocaine provides analgesia in an animal model of neuropathic pain. Pain. 2002;97:267–73. doi: 10.1016/S0304-3959(02)00028-3. [DOI] [PubMed] [Google Scholar]
- Suter MR, Papaloizos M, Berde CB, Woolf CJ, Gilliard N, Spahn DR, Decosterd I. Development of neuropathic pain in the rat spared nerve injury model is not prevented by a peripheral nerve block. Anesthesiology. 2003;99:1402–8. doi: 10.1097/00000542-200312000-00025. [DOI] [PubMed] [Google Scholar]
- Wall PD, Gutnick M. Ongoing activity in peripheral nerves: the physiology and pharmacology of impulses originating from a neuroma. Exp Neurol. 1974;43:580–93. doi: 10.1016/0014-4886(74)90197-6. [DOI] [PubMed] [Google Scholar]
- Waxman SG, Cummins TR, Dib-Hajj S, Fjell J, Black JA. Sodium channels, excitability of primary sensory neurons, and the molecular basis of pain. Muscle Nerve. 1999;22:1177–87. doi: 10.1002/(sici)1097-4598(199909)22:9<1177::aid-mus3>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Xiao WH, Bennett GJ. Synthetic omega-conopeptides applied to the site of nerve injury suppress neuropathic pains in rats. J Pharmacol Exp Ther. 1995;274:666–72. [PubMed] [Google Scholar]
- Xiao HS, Huang QH, Zhang FX, Bao L, Lu YJ, Guo C, Yang L, Huang WJ, Fu G, Xu SH, Cheng XP, Yan Q, Zhu ZD, Zhang X, Chen Z, Han ZG, Zhang X. Identification of gene expression profile of dorsal root ganglion in the rat peripheral axotomy model of neuropathic pain. Proc Natl Acad Sci USA. 2002;99:8360–5. doi: 10.1073/pnas.122231899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Shimoyama N, Mizuguchi T. Role of the injury discharge in the development of thermal hyperesthesia after sciatic nerve constriction injury in the rat. Anesthesiology. 1993;79:993–1002. doi: 10.1097/00000542-199311000-00018. [DOI] [PubMed] [Google Scholar]
- Yoon YW, Na HS, Chung JM. Contributions of injured and intact afferents to neuropathic pain in an experimental rat model. Pain. 1996;64:27–36. doi: 10.1016/0304-3959(95)00096-8. [DOI] [PubMed] [Google Scholar]
- Zhang JM, Li H, Munir MA. Decreasing sympathetic sprouting in pathologic sensory ganglia: a new mechanism for treating neuropathic pain using lidocaine. Pain. 2004;109:143–9. doi: 10.1016/j.pain.2004.01.033. [DOI] [PubMed] [Google Scholar]
- Zimmermann M. Pathobiology of neuropathic pain. Eur J Pharmacol. 2001;429:23–37. doi: 10.1016/s0014-2999(01)01303-6. [DOI] [PubMed] [Google Scholar]