

Abstract
Glyoxalase 2 is a β-lactamase fold containing enzyme that appears to be involved with cellular chemical detoxification. Although the cytoplasmic isozyme has been characterized from several organisms, essentially nothing is known about the mitochondrial proteins. As a first step in understanding the structure and function of mitochondrial glyoxalase 2 enzymes, a mitochondrial isozyme (GLX2-5) from Arabidopsis thaliana was cloned, over-expressed, purified, and characterized using metal analyses, EPR and 1H NMR spectroscopies, and X-ray crystallography. The recombinant enzyme was shown to bind 1.04 ± 0.15 equivalents of iron, 1.31 ± 0.05 equivalents of Zn(II), and exhibit kcat and Km values of 129 ± 10 s−1 and 391 ± 48 μM, respectively, when using S-D-lactoylglutathione as the substrate. EPR spectra revealed that recombinant GLX2-5 contains multiple metal centers including a predominant Fe(III)Zn(II) center and an antiferromagnetically-coupled Fe(III)Fe(II) center. Unlike cytosolic glyoxalase 2 from A. thaliana, GLX2-5 does not appear to specifically bind manganese. 1H NMR spectra revealed the presence of at least 8 paramagnetically-shifted resonances that arise from protons in close proximity to a Fe(III)Fe(II) center. Five of these resonances arise from solvent exchangeable protons, and four of these have been assigned to NH protons on metal bound histidines. A 1.74 Å resolution crystal structure of the enzyme revealed that GLX2-5 is a dimeric protein with a metal center that is similar to that of human GLX2. These data demonstrate that mitochondrial glyoxalase 2 can accommodate a number of different metal centers and that the predominant metal center is Fe(III)Zn(II).
The glyoxalase system consists of two enzymes, lactoylglutathione lyase (glyoxalase I, GLX1) and hydroxyacylglutathione hydrolase (glyoxalase II, GLX2) that act coordinately to convert a variety of α-keto aldehydes into hydroxyacids in the presence of glutathione (1). Aromatic and aliphatic α-keto aldehydes react spontaneously with glutathione to form thiohemiacetals, which are converted to S-(2-hydroxyacyl)glutathione derivatives by GLX1. GLX2 hydrolyzes S-(2-hydroxyacyl)glutathione derivatives to regenerate glutathione and produce hydroxyacids. Glyoxalase I, a Zn(II) or Ni(II) metalloprotein, can utilize a number of α-ketoaldehydes (2). However the primary physiological substrate of the enzyme is thought to be methylglyoxal (MG), a cytotoxic and mutagenic compound that is formed primarily as a byproduct of carbohydrate and lipid metabolism (3,4). MG can react with DNA to form modified guanylate residues (5) and inter-strand cross-links (6). It also reacts with proteins to form glycosylamine derivatives of arginine and lysine and hemithioacetals with cysteines (7).
Cells with high glycolytic rates exhibit high rates of methylglyoxal formation and increased levels of GLX1 activity. For instance, increased levels of GLX1 and GLX2 RNA and protein have been detected in tumor cells, including breast carcinoma cells (8). Because selective inhibition of the glyoxalase system could cause cellular toxicity, the glyoxalase enzymes have been investigated as potential anti-tumor and anti-malarial targets in animal systems (1,9). Inhibitors of GLX1 and GLX2 can inhibit the growth of tumor cells in vitro (10,11) and in vivo (12) and have been shown to have anti-proliferative effects on parasitic infections (13). Finally, increased MG levels have been implicated with several complications associated with diabetes mellitus (1,14,15), and changes in glyoxalase enzymes have been linked with neurodegenerative disease (16). Therefore, high levels of MG and the glyoxalase enzymes are associated with several aspects of human disease.
GLXI has been studied extensively in a number of systems with biochemical, computational, and X-ray crystallographic studies providing considerable insight into its kinetic mechanism (reviewed in (17,18)). Insight into the kinetic mechanism of GLX1 has allowed the development of new classes of mechanism-based, competitive inhibitors that can inhibit tumor growth in vitro and in vivo (12,19,20).
Considerably less is known about the structure, reaction mechanism, and physiological role(s) of GLX2. In contrast to GLX1, which is found as a single isozyme, GLX2 exists as multiple isozymes in many organisms, including yeast, plants, and animals. GLX2 activity has been found in both the cytosol and mitochondria (21–23). Furthermore, GLX2 isozymes were found in both the matrix and inter-membrane space of rat liver mitochondria (24,25). In most cases, separate genes encode the cytosolic and mitochondrial isozymes. However, in humans a single gene produces both cytosolic and mitochondrial GLX2 (21). Therefore, the physiological role(s) of GLX2 may be more complex than GLX1.
S-D-lactoylglutathione (SLG) appears to be the preferred substrate for GLX2 from most sources including human, yeast, and plants (26–33). However, GLX2 can hydrolyze many different glutathione thioesters, including S-D-mandeloylglutathione, S-D-acetylglutathione, S-D-acetoacetylglutathione, S-D-formylglutathione, S-D-glycolylglutathione and S-D-lactonylglutathione. Interestingly, glyoxalase II isolated from African trypanosomes prefers thioesters of trypanothione as substrates (34).
GLX2 is one of many protein families characterized to date that contain a metallo-β-lactamase fold. The metallo-β-lactamases typically bind Zn(II) (35,36), while the rubredoxin:oxygen oxidoreductase (ROO) and ZiPD families contain di-Fe and di-Zn centers, respectively (37,38). Arabidopsis cytoplasmic GLX2 (GLX2-2) can bind Zn(II), Fe, and Mn (39–41), while the human enzyme was reported to bind two Zn(II) ions (42). The crystal structure of human cytoplasmic GLX2 showed that one metal binding site consists of 3 conserved histidine residues, a bridging aspartic acid, and a bridging water/hydroxide. The second metal binding site had 2 histidines, 1 bridging Asp, 1 terminally-bound Asp, 1 bridging water/hydroxide, and 1 terminally-bound water (42). These crystallographic data have shown that the structures of the metal binding and active sites of GLX2 are similar to those of metallo-β-lactamase L1 from Stenotrophomonas maltophilia (43). It has recently been suggested that subtle differences in the metal binding ligands of β-lactamase fold containing proteins may be responsible for differences in metal binding properties between the different enzymes (44). This theory is supported by mutational studies on Arabidopsis GLX2-2, which have shown that mutations both in and outside the metal binding ligands can affect the relative amount and identity of metal bound by the enzyme (39). Therefore, metallo-β-lactamase fold containing enzymes can bind several different metals and catalyze a number of different reactions.
The presence of mitochondrial isoforms of GLX2 is intriguing because the GLX1 and its product SLG have only been observed in the cytosol (1). Yet, mitochondrial GLX2 from several sources appears to utilize SLG as its preferred substrate (24,26,28). Thus, the physiological substrate/s and the role(s) of mitochondrial GLX2 are not clear, and further research is required in order to obtain more detailed information on the mitochondrial enzymes.
In an effort to better understand the specific roles of the different GLX2 isozymes as well as to correlate structural and functional features of this important class of enzymes, we have begun a characterization of the GLX2 isozymes in Arabidopsis thaliana. Five genes have been identified to encode putative GLX2 isozymes in A. thaliana (45). In addition to GLX2-2, three GLX2 isozymes are localized in mitochondria (GLX 2-1, GLX 2-4, and GLX 2-5). The fifth GLX2-like gene, GLX 2-3, encodes a protein that lacks many of the conserved substrate binding residues and may not in fact encode a functional GLX2 enzyme (S. Rhee and C.A. Makaroff, unpublished results). To date detailed structural and kinetic studies have only been conducted on the cytoplasmic isozyme of GLX2 (39–41). A mitochondrial GLX2 isozyme has not been studied in detail from any organism. Therefore, we have over-expressed, purified, and characterized the mitochondrial GLX2-5 isozyme from Arabidopsis using metal analysis, EPR and 1H NMR spectroscopies, and X-ray crystallography.
Biochemical studies indicate that GLX2-5 is a FeZn protein that has a preference for SLG as its substrate. The crystal structure of GLX2-5 reveals that the enzyme has structural features quite similar to that of the human cytoplasmic enzyme and suggested that GLX2-5 is a Fe(III) Zn(II) protein. EPR spectra demonstrate that recombinant GLX2-5 can contain multiple metal centers including an antiferomagnetically-coupled Fe(III)Fe(II) center, but that the predominant form of the enzyme contains an Fe(III)Zn(II) center.
These results represent the first detailed structural characterization of a mitochondrial GLX2 enzyme from any source. They demonstrate for the first time that β-lactamase fold containing proteins can accommodate mixed metal centers and provide new insights into structure function features associated with GLX2 enzymes in particular and β-lactamase fold-containing enzymes in general.
EXPERIMENTAL PROCEDURES
General
PCR reagents, Deep Vent DNA polymerase, restriction enzymes (NdeI and XhoI), and S-D-lactoylglutathione were purchased from Sigma (St. Louis, MO) and New England Biolabs (Beverly, MA). Primers were synthesized by Integrated DNA Technologies (Coralville, IA). All chromatographic steps were carried out on a Pharmacia Biotech Fast Protein Liquid Chromatography (FPLC) system operating at 4 °C. Columns and resins for FPLC were purchased from Pharmacia Biotech. All protein and DNA quantitations were performed on an Agilent 8453 UV-Vis spectrophotometer.
Over-expression and Purification of Arabidopsis GLX2-5
PCR was conducted on a GLX2-5 cDNA (39), with the primers CTCCCATATGCAAATTGAACTGGTGCCTT and CGAGGATCCTCGGTCGACGCTTTTTTTTTTTTTTTTT, which generated (NdeI and XhoI) sites at the 5′ and 3′ ends of the fragment, respectively. For expression in E. coli the leader sequence was removed by placing the N-terminal methionine at amino acid 71 of the predicted protein sequence. This construct yields a protein with the same amino terminus as the cytoplasmic form of the enzyme. The 975 bp GLX2-5 PCR fragment was cloned into pT7-7 using NdeI and XhoI. The resulting plasmid, GLX 2-5/pT7-7, was transformed into DH10B E. coli cells, and the construct was verified by DNA sequencing.
The GLX 2-5/pT7-7 plasmid was transformed into E. coli BL21 (DE3)-Rosetta cells and used for over-expression and protein purification as previously described (31). A 10 mL overnight culture of these cells in LB containing 150 μg/mL ampicillin was used to innoculate 1 L of LB containing 150 μg/mL ampicillin and 8 mL/L glycerol. The cells were allowed to grow at 37 °C with shaking until the cells reached an O.D.600 nm of 0.6–0.8. Protein production was induced by making the culture 0.2 mM in isopropyl thio-β-D-galactoside (IPTG), and the culture was made 250 μM in Fe(NH4)2(SO4)2 and Zn(SO4)2. The cells were allowed to shake at 15 °C for 24 hours. After induction for 24 h, the cells were harvested by centrifugation at 7000 rpm for 8 min and washed three times with cold, sterile ddH2O to remove salts. Harvested cell pellets were stored at −80 °C until further use.
The cell pellet was resuspended in 30 mL of 10 mM MOPS [3-(N-morpholino) propanesulfonic acid] buffer, pH 7.2, containing 0.1 mM PMSF (phenylmethylsulfonyl fluoride). The cells were lysed by passage twice through a French Press at 16000 psi, and the cell debris was removed by centrifugation at 12,500 rpm for 45 min. The cleared supernatant was purified using Fast Protein Liquid Chromatography (FPLC) with a Q-Sepharose column as described previously (31). Enzyme concentrations were determined by measuring the absorbance at 280 nm and using a molar extinction coefficient of 37,800 M−1cm−1. The molar extinction coefficient was determined by PIXE (particle-induced X-ray emission) experiments (46), which were conducted by Dr. W. Meyer-Klaucke at EMBL Outstation, Hamburg, Germany as previously described (38).
Metal analyses
Metal analyses were performed on a Varian-Liberty 150 inductively coupled plasma spectrometer with atomic emission spectroscopy detection (ICP-AES) as described elsewhere (31). The concentration of GLX2-5 was 10 μM in 10 mM MOPS buffer, pH 7.2. A calibration curve with 5 standards and a correlation coefficient of greater than 0.998 was generated using Zn, Mn, Fe, and Cu reference solutions. The following emission wavelengths were chosen to ensure the lowest detection limits possible: Zn, 213.856 nm; Mn, 257.610 nm; Fe, 259.940 nm; and Cu, 324.754 nm. Metal concentrations were obtained and averaged from at least three enzyme preparations.
Steady-state kinetic studies
A series of thiolesters of glutathione were used for the preliminary investigation of substrate preferences of GLX 2-5. The substrates used were S-D-lactoylglutathione (SLG, ɛ240 3,100 M−1cm−1), S-mandeloylglutathione (ɛ263 4,200 M−1 cm−1), S-acetylglutathione (ɛ240 2,980 M−1cm−1), S-acetoacetylglutathione (ɛ240 3,400 M−1cm−1), S-formylglutathione (ɛ240 3,300 M−1cm−1), S-glycolylglutathione (ɛɛ240 3,260 M−1cm−1), and S-lactonylglutathione (ɛɛ240 3,310 M−1cm−1). With the exception of SLG, all substrates were synthesized as described elsewhere (47). Thiolester hydrolysis was monitored at 240 nm (except S-mandeloylglutathione, 263 nm) over 30 s at 25 °C. The concentrations of substrate and enzyme used were 200 μM and 10 μM, respectively.
The steady-state kinetic parameters of the GLX2-5-catalyzed hydrolysis of S-D-lactoylglutathione were determined at 25 °C in 10 mM MOPS, pH 7.2, using an Agilent 5483 Diode Array UV-Vis spectrophotometer. The rate of hydrolysis was monitored by measuring the absorbance at 240 nm for 30 μM to 600 μM S-D-lactoylglutathione over a 30 s reaction period, and the data were analyzed as previously reported (31).
EPR Spectroscopy
Electron paramagnetic resonance (EPR) spectra were recorded on a Bruker ESP-300E spectrometer equipped with an Oxford Instruments ESR-900 helium flow cryostat operating at 4.7 K with 2 mW microwave power at 9.48 GHz, and employing 10 G field modulation at 100 kHz. EPR samples contained 345 μM GLX2-5 in 10 mM MOPS, pH 7.2.
1H NMR Spectroscopy
NMR samples of GLX2-5 contained approximately 10% D2O for locking, and the concentration was 1.8 mM. The samples in D2O were made by performing three or more dilution/concentration cycles in a Centricon-10 to a final concentration of 1.6–1.8 mM. The samples were then loaded into Wilmad 5-mm tubes for NMR. NMR spectra were collected on a Bruker Avance 500 spectrometer operating at 500.13 MHz, 298 K, and a magnetic field of 11.7 T, recycle delay (AQ), 41 ms; sweep width, 400 ppm. Protein chemical shifts were calibrated by assigning the H2O signal the value of 4.70 ppm. A modified presaturation pulse sequence (zgpr) was used to suppress the proton signals originating from water molecules.
X-ray Crystallography
GLX2-5 was prepared and purified as described in the over-expression and purification section. Enzyme purity was ascertained by SDS-PAGE to be >95%, and the concentration was 12 mg/mL (0.4 mM). The sample (2 mL) was drop frozen in liquid nitrogen and shipped on dry ice to the Center for Eukaryotic Structural Genomics, University of Wisconsin, Madison. The coordinates have been deposited on the Protein Databank (accession number: 1XM8).
Molecular Weight Determination
Approximately 5 mg of GLX2-5 was mixed with 7 mg ovalbumin, 10 mg ribonuclease A, and 1 mg Blue Dextran and subjected to chromatography through a Sephadex S200 column in 10 mM MOPS, pH 7.2, containing 0.15 M NaCl. The flow rate was 1 ml/min, and 2.0 ml fractions were collected. Samples containing protein were identified by monitoring A280 and by SDS-PAGE. A sample of GLX2-5 was run as a control.
RESULTS
Over-expression, purification, and characterization of GLX2-5
Sequence comparisons of different GLX2 isozymes in Arabidopsis show that GLX2-5 has a relatively long N-terminal extension that is predicted to target it for localization in the mitochondrion (23). To over-express GLX2-5 in E. coli, this N-terminal leader was removed during subcloning to generate a N-terminus of the protein, MQIELVP, which is similar to that in cytosolic GLX2-2 (23). This modified gene was inserted into pT7-7, to generate the over-expression plasmid GLX2-5/pT7-7. This construct was transformed into E. coli BL21(DE3) Rosetta cells, and small scale growth cultures were used to optimize over-expression conditions for GLX2-5.
During purification using Q-Sepharose chromatography, GLX2-5 eluted from the column at approximately 125 mM NaCl (pH 7.2). SDS-PAGE showed that the purified enzyme was > 95 % pure (data not shown). The eluted protein was initially light blue in color; however, the coloration faded within 15–30 minutes. The procedure described in Experimental Procedures resulted in the highest yield of soluble, over-expressed GLX2-5, and resulted in approximately 50 mg of purified GLX2-5 from a one-liter of culture.
Metal analyses on GLX2-5, over-expressed in the presence of 250 μM Fe(NH4)2(SO4)2 and Zn(SO4)2 in the culture medium, resulted in a purified recombinant enzyme that bound 1.04 ± 0.15 equivalents of iron, 1.31 ± 0.05 equivalents of Zn(II), 0.016 ± 0.02 equivalents of manganese, and <0.001 equivalents of copper. When GLX2-5 was over-expressed in media with no added iron or zinc, the resulting enzyme bound 0.61 ± 0.07 equivalents of iron, 0.58 ± 0.15 equivalents of Zn(II), and no detectable amounts of manganese or copper. Even though this latter sample of GLX2-5 still bound approximately equal amounts of Fe and Zn, the total metal content of the enzyme was roughly only 1.0. Therefore, the over-expression and purification procedure described in Experimental Procedures was used to prepare GLX2-5 for all subsequent studies.
Most glyoxalase II enzymes show a preference for S-D-lactoylglutathione as the substrate, but as described above, SLG has not yet been found in the mitochondrion. Therefore, seven related thioesters of glutathione were used to determine the substrate preference of GLX 2-5. The substrates used were S-lactoylglutathione, S-mandeloylglutathione, S-acetylglutathione, S-acetoacetylglutathione, S-formylglutathione, S-glycolylglutathione, and S-lactonylglutathione. Initial rate assays on these substrates showed that SLG was the preferred substrate. It showed an initial rate of hydrolysis of 2.96 × 10−6 M·s−1. S-acetoacetylglutathione (1.33 × 10−6 M·s−1) and S-glycolylglutathione (0.90 × 10−6 M··s−1), were hydrolyzed at about half the rate of SLG, while S-mandeloylglutathione (0.35 × 10−6 M·s−1), S-acetylglutathione (0.32 × 10−6 M·s−1), and S-lactonylglutathione (0.29 × 10−6 M·s−1) were hydrolyzed at rates about ten-fold lower than SLG. S-formylglutathione was not utilized as a substrate by GLX2-5.
Steady-state kinetic studies were then conducted on recombinant GLX2-5 at 25 °C using SLG as the substrate. The enzyme exhibited a kcat of 129 ± 10 s−1 and a Km of 391 ± 48 μM. These values are on the order of those observed for Arabidopsis GLX2-2 suggesting that SLG is the preferred substrate for GLX2-5. This is consistent with the presence of conserved SLG-binding ligands in the GLX2-5 sequence (Figure 1).
Fig. 1.

Sequence alignment of GLX2-5 from A. thaliana and GLX2 from human.
Spectroscopic studies on GLX2-5
The EPR spectrum of GLX2-5 (Figure 2) is complex containing four distinct components. The two main components of the spectrum are interpreted to result from a protein–bound, magnetically isolated Fe(III) ion and an antiferromagnetically spin-coupled Fe(II)-Fe(III) system, which are described in detail below. In addition, the spectrum contains several minor components. Two of the minor components are likely contaminating species; adventitiously-bound Mn(II) gives rise to a six-line pattern centered at geff. = 2.0 and an isotropic signal at geff. = 4.3 indicative of the presence of mononuclear Fe(III). These two components are commonly seen in the EPR spectra of metalloenzymes, and are present at low levels in the spectrum of GLX2-5. Integration of the spectra of S > ½ species is notoriously unreliable, especially when D is small and multiple Kramers’ doublets are populated, some of which have resonances for which geff. tends to zero. Nevertheless, in the present case the experimental spectrum, Trace A, can be compared with a simulated spectrum, Trace D that does not contain these components. The six-line Mn(II) signal and the geff. = 4.3 isotropic signal are not major components of the spectrum of GLX2-5 and are likely irrelevant to the structure and function of the active site; therefore, they were not studied further.
Fig. 2.

EPR spectrum of GLX2-5. Trace A shows the spectrum of GLX2-5 recorded at 4.7 K. The inset shows an expanded view of the g ~ 1.9 region of the spectrum (B) and a theoretical spectrum (C). The theoretical spectrum was generated using the spin Hamiltonian H = βg1.B.S1 + S1.D1.S1 + βg2.B.S2 + S2.D2.S2 + J.S1.S2, where S1 = 5/2, g1(iso) = 2.0, D1 = 3 cm−1, E1 = 0, S2 = 1, g2(x,y,z) = 2.093, 2.192, 2.322, D2 = 10 cm−1, E = 0, and J = 50 cm−1. This scheme represents a strongly antiferromagnetically coupled Fe(III)-Fe(II) center. Trace D shows a composite simulation of the experimental EPR spectrum. The simulation has two components, one of which is that shown as Trace C, and another, the major component, corresponds to an isolated Fe(III) ion with H = βg.B.S + S.D.S, where g(iso) = 2.0, S = 5/2, D = 0.45 cm−1, E = 0.0855 cm−1 (E/D = 0.19), ΔD = 0.20 cm−1, and ΔE = 0.057 cm−1. An additional component with an isotropic g(eff) = 4.3 is present but was not included in the simulation.
The major species in the EPR spectrum of GLX2-5 can be confidently assigned to an unusual, protein-bound, magnetically-isolated Fe(III) ion on the basis of computer simulation. The experimental spectrum (Trace A of Figure 2) shows a peak at 750 G (geff. = 9.01), a broad peak at 1410 G, a derivative at 1595 G (geff. = 4.25 G), and a shoulder at 1815 G with broad underlying absorption. Considering the sensitivites of the intensities, resonance positions, and lineshapes of these features to not only the zero-field splitting parameters themselves, but particularly, to the effects of strains in g, D, and E, the theoretical spectrum shown as Trace D of Figure 1 reproduces the features of the experimental spectrum remarkably well. The detailed parameters used for the simulation are given in the legend to Figure 1, but the salient points are (i) an isotropic g = 2.0, (ii) a moderately rhombic E/D, (iii) an unusually low value for D, barely larger than the microwave quantum, and (iv) significant strains in the zero-field splitting parameters. The simulations were extremely sensitive to D, E/D, ΔD, and ΔE; the simulation rapidly becomes unrecognizable as being related to the experimental spectrum upon changing these values, and we estimate maximum errors of ± 15 % for each of these parameters. The main deficiencies of the simulation are the imperfect reproduction of the sharp spike at geff. ~ 4.2 and the underestimation of the intensity of the broad absorption from 1600 – 2200 G. Both of these are likely the result of the overlapping isotropic geff. = 4.3 signal due to adventitious Fe(III), though it is possible that the strains in the zero-field splitting parameters have a more complex distribution than the simple one used in the present work. An additional minor difference between the simulation and experimental spectra is the presence of sharp but very weak features at ~ 3750 G and at ~ 4500 G in the simulation that are not observed experimentally; either strains in g or relaxation effects may be responsible for the absence of these features in the experimental spectrum.
The EPR spectrum of GLX2-5 contained, in addition to the protein-bound Fe(III) that constituted the major species, a rhombic signal with apparent S = ½ and geff. values of 1.93, 1.87, and 1.73 (Figure 2, Trace B). As we are unaware of any mononuclear S = ½ ions that were likely to be present in our preparation that would give rise to these parameters, we were prompted to seek an alternative assignment for this signal. The signal was well-simulated assuming a strongly antiferromagnetically spin-coupled Fe(II)-Fe(III) system and assuming an isotropic gFe(III) = 2.0, a rhombic gFe(II) with g > 2.0, and J ≫ hν. It should be pointed out that there is no unique parameter set for generating this spectrum (or others very similar to it), and the detailed parameters used to obtain Trace D of Figure 2 are given in the figure legend largely for reference. However, they do also serve to illustrate that the otherwise uninterpretable signal at geff. = 1.93, 1.87 and 1.73 can be interpreted in terms of an Fe(II)-Fe(III) mixed valence center using reasonable values for g, individual zero-field splittings, and exchange coupling. Assuming that the signal is indeed due to a di-iron center, and with caveats regarding the quantification of the major S = 5/2 signal in mind, we estimate that the geff. = 1.93, 1.87, 1.73 signal accounts for up to a maximum of 20 % of the total protein-bound iron.
In order to probe further the active site structure of GLX2-5, a 1H NMR spectrum of GLX2-5 was obtained (Figure 3). There were at least 8 paramagnetically-shifted resonances in between 110 and -30 ppm. Previous studies have shown that the T1e of Fe(III) when part of a Fe(III)Zn(II) center is too slow to observe paramagnetically-shifted 1H resonances (48,49). Therefore, the peaks observed in Figure 3 must be due to ligands bound to a Fe(III)Fe(II) center in the sample. Peaks a, c, d and f integrate to 2 protons, while peaks b, g, and h integrate to nearly 1 proton each. Peaks a and d are solvent-exchangeable, and based on their resonance positions and linewidths, these peaks can be assigned to N-H protons on Fe(III)- and Fe(II)-bound histidines, respectively (48,50,51). This result suggests that there are at least 4 histidines bound to the Fe(III)Fe(II) center in this sample. Sequence comparisons of GLX2-5 to human GLX2-2 suggest that there are 5 histidines bound to the metal center in GLX2-5. It is possible that a fifth solvent-exchangeable NH resonance is not observed due to fast exchange with bulk solvent (49,52). The solvent-exchangeable, upfield shifted peak h is very unusual since very few metal binding amino acid ligands are solvent exchangeable and none are known to yield upfield shifted peaks. An upfield shift is suggestive of a π, rather than σ, delocalization mechanism (50). A similarly shifted, solvent exchangeable peak was observed in Fe(III)Fe(II)-containing uteroferrin; however, the assignment of this peak has not been made (50). It has been suggested that this peak arises from a backbone NH in close proximity to the metal center (53). While unambiguous assignments of the remaining peaks are not possible, it is likely that peaks b and c are due to either meta protons (-CH) on Fe(II)-bound histidines or to β-CH2 protons on metal bound histidines (49,50). Ortho protons on metal bound histidines are usually too broad to detect (50). Peaks f and g are probably due to β-CH2 protons on bound Asp/Glu ligands (49,50). Therefore, the NMR spectrum of GLX2-5 confirmed the presence of an Fe(III)Fe(II) center in the enzyme and indicated that the metal binding site is quite similar to that of human GLX2.
Fig. 3.
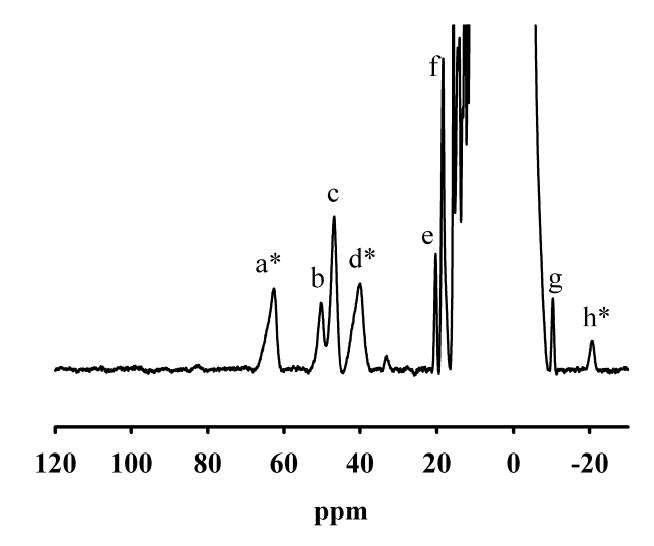
NMR spectrum of 1.6 mM GLX2-5. NMR spectra were collected on a Bruker Avance 500 spectrometer operating at 500.13 MHz, 298 K, and a magnetic field of 11.7 T, recycle delay (AQ), 41 ms; sweep width, 400 ppm. Protein chemical shifts were calibrated by assigning the H2O signal the value of 4.70 ppm. A modified presaturation pulse sequence (zgpr) was used to suppress the proton signals originating from water molecules.
Crystallographic Studies on GLX2-5
The X-ray crystal structure of GLX2-5 at pH 4.5 was solved to 1.74 Å resolution and is shown in Figure 4 (Table 1). The enzyme crystallized as a dimer with monomers of 254 amino acids, and each monomer has an αββα motif, which is found in all enzymes containing a metallo-β-lactamase fold (44). The interface between the two monomers contains Lys10, His56, Tyr57, Lys140, Phe142, Glu143, Lys177, Lys203, Asp253, and Phe254 from both subunits. The dimer is held together by a number of electrostatic interactions such as Lys10 forming contacts with Asp11 and Glu170, Asp253 forming a contact with His56, and Phe254 hydrogen bonding to Tyr57. One molecule of acetate was found in each subunit and was located near (distances < 3.0 Å) Arg248, Lys251, Lys140, and Ser137. One molecule of PEG was found in subunit A and was shown to be positioned near Lys202. The C-terminus of each monomer lies in the dimer interface, which is solvent accessible, while the N-termini are solvent-exposable.
Fig. 4.

Ribbon structure of GLX2-5 from Arabidopsis thaliana. The coordinates have been deposited on the Protein Databank (accession number: 1XM8). Figure was rendered using Raswin Molecular Graphics, Windows version 2.7.2.1.1.
Table 1.
Comparisons of the active sites of human GLX2-2 and A. thaliana GLX2-5a
| Metalb | Enzyme | Residue | Atom | Distance (Å) | Bond angle (°)c | Second Sphere ligands |
|---|---|---|---|---|---|---|
| M1 | GLX2-5 | His54 | Nɛ2 | 2.1 | 119 | Thr53 |
| GLX2-2 | His54 | Nɛ2 | 2.3 | 104 | ||
| M1 | GLX2-5 | His56 | Nδ1 | 2.1 | 104 | |
| GLX2-2 | His56 | Nδ1 | 2.3 | 95 | Glu146 via water | |
| M1 | GLX2-5 | His112 | Nɛ2 | 2.1 | 129 | Lys140 carbonyl |
| GLX2-2 | His110 | Nɛ2 | 2.3 | 161 | ||
| M1 | GLX2-5 | Bridging | 2.0 | |||
| GLX2-2 | H2O/OH− | 2.1 | ||||
| M1 | GLX2-5 | None | ||||
| GLX2-2 | Asp134 | Oδ2 | 2.2 | 78 | Asp134 carbonyl | |
| M2 | GLX2-5 | Asp58 | Oδ2 | 2.0 | 90 | |
| GLX2-2 | Asp58 | Oδ2 | 2.3 | 87 | ||
| M2 | GLX2-5 | His59 | Nɛ2 | 1.9 | 107 | Asp29 |
| GLX2-2 | His59 | Nɛ2 | 2.2 | 101 | ||
| M2 | GLX2-5 | Asp131 | Oδ2 | 2.0 | 81 | |
| GLX2-2 | Asp134 | Oδ2 | 2.2 | 78 | ||
| M2 | GLX2-5 | His169 | Nɛ2 | 1.9 | 141 | Asp11 |
| GLX2-2 | His173 | Nɛ2 | 2.1 | 151 | ||
| M2 | GLX2-5 | Bridging | 2.1 | |||
| GLX2-2 | H2O/OH− | 2.1 |
Structures for human GLX2-2 (Protein Data Bank number: 1QH3) and A. thaliana GLX2-5 (Protein Data Bank number: 1XM8) were analyzed with Raswin v. 2.7.2.1.
M1 is Zn(II) for both enzymes; M2 is Zn(II) for human GLX2-2 and Fe for A. thaliana GLX2-5.
Bond angle is defined as the bridging H2O/OH− - metal – ligating atom of ligand.
Each monomer binds two heavy atoms, and the metal ions are bound near the surfaces of the monomers in between the β sheets of the αββα motif. Since EPR studies have predicted that a Fe(III)Zn(II) center is the predominant one in recombinant GLX2-5, this center is shown in Figures 4 and 5. However, we cannot completely rule out the possibility of a dinuclear Fe or Zn center. The metal centers face one another with the metal ion in site 1 of each subunit being 17.8 Å apart (Figure 4). The metal ions in each subunit are separated by 3.3(4) Å (Figure 5). The metal ion in site 1 is coordinated tetrahedrally by His54, His56, His112, and a bridging water/hydroxide (Table 1). The metal ion in site 2 is coordinated in a trigonal bipyramid geometry by Asp58, His59, Asp131, His169, and a bridging water/hydroxide, with Asp131 and Asp58 in the apical positions of the trigonal bipyramid. One oxygen (Oδ1) of Asp58 appears to be in position (2.78 Å from bridging hydroxide oxygen) to form a hydrogen bond to the bridging water/hydroxide. A similar role was predicted for Asp120 in metallo-β-lactamase L1 (2.83 Å from Oδ1 to hydroxide oxygen) (43).
Fig. 5.
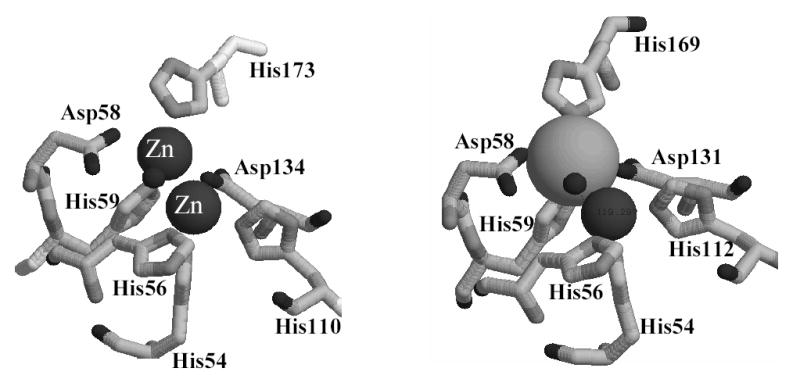
Active site of GLX2-5 from Arabidopsis thaliana. The coordinates have been deposited on the Protein Databank (accession number: 1XM8). Figure was rendered using Raswin Molecular Graphics, Windows version 2.7.2.1.1.
The crystal structure of human GLX2 complexed with glutathione and S-hydroxybromophenylcarbamoyl glutathione (SHBPCG) showed that Arg249, Lys252, Lys143, Tyr175, and Tyr145 are within 13 Å of the metal ion in site 1 and interact with glutathione and SHBPCG. By aligning the amino acid sequences of human GLX2 and GLX2-5 (Figure 1), it is clear that all of the glutathione and inhibitor binding residues of human GLX2 are conserved in GLX2-5 except Tyr145, which is replaced by a Phe in GLX2-5. As in human GLX2, all of residues (Phe142, Tyr171, Arg248, Lys140, and Lys251) are within 13 Å of the metal ion in site 1 of GLX2-5, and we predict that these ligands are involved in substrate/inhibitor binding.
One common structural feature of enzymes that have a metallo-β-lactamase fold is an extensive H-bonding network around the metal binding ligands (Table 1). In particular, the ligands that coordinate the metal ion in site 2 typically form hydrogen bonds to two Asp residues. In GLX2-5, His169 (Nδ1) forms a H-bond with Oδ1 of Asp11 (2.86 Å), and Asp11 (Oδ2) forms a hydrogen bond with the side-chain of Lys10. The Oδ2 oxygen of Asp11 is also positioned to hydrogen bond to a solvent molecule, which H-bonds to a second solvent molecule that hydrogen bonds to Asp11 (Oδ2) in molecule B of the structure. The Nδ1 nitrogen of His59 forms a hydrogen bond to the Oδ2 of Asp29 (2.819 Å), while the Oδ1 oxygen of Asp29 forms a hydrogen bond to the backbone amine of Thr53. The metal binding ligands in site 2 also form hydrogen bonds with amino acids near the active site: Nδ1 of His54 hydrogen bonds to the Oγ1 of Thr53 and the carbonyl oxygen of Lys140 (which corresponds to Lys143 in human GLX2) forms a hydrogen bond with the Nδ1 of His112. Therefore, the hydrogen bonding network is also conserved in GLX2-5.
GLX2-5 exists as a monomer in solution
In contrast to the situation in the crystal structure, GLX2-5 was found to exist as a monomer when a sample of recombinant protein was subjected to gel filtration chromatography (Figure 6). GLX2-5 was found to elute between ovalbumin (44 kDa) and ribonuclease A (13 kDa) during gel filtration chromatography. The molecular weight of the recombinant GLX2-5 monomer is 28.2 kDa, while a dimer would be expected to run as a 56 kDa protein. It is possible that weak interactions between the GLX2-5 monomers result in some dimerization of the enzyme in the cell that are disrupted by the ionic strength of the buffer during gel flitration. However, it is more likely that the dimers observed in the crystal structure of both GLX2-5 and human GLX2 are likely the result of the conditions used for crystallization.
Fig. 6.

Elution profile from gel filtration column. Approximately 5 mg of GLX2-5, 7 mg Ovalbumin, 10 mg Ribonuclease A, and 1 mg Blue Dextran were separated on a Sephadex S200 column. Samples containing protein were identified by monitoring A280 and by SDS-PAGE.
DISCUSSION
Mitochondrial GLX2-5 from A. thaliana was successfully over-expressed and purified with single step chromatography. The resulting protein was shown to bind significant amounts of both Fe and Zn(II), but unlike Arabidopsis GLX2-2 (40,41), very little Mn was bound to the enzyme. The complex EPR spectra of GLX2-5 are very reminiscent of those of Arabidopsis GLX2-2 (40,41), which contain overlapping signals that have been assigned to Fe(III)Zn(II), antiferromagnetically-coupled Fe(III)Fe(II), and ferromagnetically-coupled Mn(II)Mn(II) centers. However, there are no signals that could be assigned to a ferromagnetically-coupled Mn(II)Mn(II) center in the spectrum of GLX2-5, and the relatively weak, multi-line signal at g ~ 2 is probably due to adventitiously bound, mononuclear Mn(II). The EPR spectrum of GLX2-5 does suggest the presence of a magnetically-uncoupled Fe(III) center that, given the metal analyses and precedents in the literature (54–56), can be assigned to a Fe(III)Zn(II) center. This center is the predominant one in GLX2-5 accounting for nearly 70% of the EPR-active Fe(III) in the spectrum. There is also a significant amount of an antiferromagnetically-coupled Fe(III)Fe(II) center, that accounts for nearly 30% of the Fe in the sample. Since there are roughly equal amounts of Fe and Zn in GLX2-5, there must also be a significant amount of dinuclear Zn(II) containing enzyme in this sample. Therefore, like recombinant GLX2-2 (40,41), recombinant mitochondrial GLX2-5 contains a mixture of metal centers.
The 1H NMR spectrum of GLX2-5 arises from protons in close proximity to the Fe(III)Fe(II) center in the sample. The crystal structure of GLX2-5 (Figure 5) shows that there are 5 bound histidines to the metal center; however, the NMR spectra only revealed 4 solvent-exchangeable NH resonances. It was speculated above that one of the histidine NH groups might be in rapid exchange with bulk solvent and therefore not observed in the NMR spectrum. The crystal structure (Figure 5) shows that the NH group on His56 is pointed directly into the media, and this positioning provides an excellent explanation of why the fifth solvent-exchangeable NH resonance is not observed. His56 is also the only histidine that is bound via the δN; therefore, there would only be one meta CH proton expected in the NMR spectrum. Previous model studies by Que revealed that meta protons on imidzoles/histidines bound to Fe(III) exhibit resonance lines from 70–90 ppm, while those bound to Fe(II) exhibit resonances from 30–60 ppm (50). Since there are no resonances in the 70–90 ppm region (Figure 3) that could be assigned to a meta proton, this result indicates that His56 is coordinated to an Fe(II). Since it is most likely that Zn(II) will replace Fe(II) in the Fe(III)Zn(II) analog of GLX2-5, this result suggests that the metal bound in the site containing His56 in Figure 5 is Zn(II).
To further probe the structure of GLX2-5, the enzyme was sent to the Center for Eukaryotic Structural Genomics for crystal structure determination. Even though our spectroscopic results indicate that the GLX2-5 sample is heterogeneous in terms of metal centers, this sample yielded suitable crystals for structure determination. Although very similar in many regards, the crystal structures of human GLX2 and GLX2-5 show some significant structural differences between the two enzymes. Both enzymes crystallized as dimers; however, the dimer interface in human GLX2 is made up entirely of α-helices in the C-termini of the monomers. The metal centers in human GLX2 face each other but are separated by >36 Å. Conversely, the dimer interface in GLX2-5 is extensive with multiple interactions between amino acids in each monomer and a Zn(II)—Zn(II) distance of 18 Å (Figure 4).
Perhaps more interesting are our results concerning the metal centers in the two enzymes, which also show several differences. The crystal structure of human GLX2 showed 2 Zn(II) ions separated by 3.4–3.5 Å, and each Zn(II) was coordinated by 6 ligands in an octahedral geometry (42). The metal ions in GLX2-5 are coordinated by the same amino acid ligands that coordinate the Zn(II) ions in human GLX2. However, there is no evidence for additional solvent molecules coordinated to the metal ions in GLX2-5, and the metal ions have trigonal bipyramidal and tetrahedral geometries. The Zn(II) ions in human GLX2 are bridged by a hydroxide and by Oδ2 of Asp134 (42), while there is no evidence of a bridging Asp (Asp131) in GLX2-5. Despite missing the bridging Asp, the M-M bond distance is 0.1 Å shorter in GLX2-5 than in the human enzyme (42). In addition, the metal-ligand distances are shorter in GLX2-5 than in human GLX2-2 (Table 1). There are also large differences in the bond angles (bridging hydroxide/water – metal- atom in ligand) in the active sites of the two enzymes (Table 1).
Despite the differences in metal ions and their geometry and in some first sphere ligands in human GLX2 and GLX2-5, the hydrogen bonding network around the metal centers is remarkably conserved. Asp11 and Asp29 form H-bonds to the metal binding histidines in the Zn2 site of human GLX2 (42), and these same residues form hydrogen bonds with metal binding ligands of site 2 in GLX2-5. The different metal contents of the two enzymes cannot be directly attributed to different metal ligands, but may be related to different 2nd sphere interactions. Two of the second sphere ligands in human GLX2 (Thr53 and Lys143 (Lys140 in GLX2-5)) form interactions with His residues in the metal ions of site 1 in GLX2-5. However, two key second sphere ligand interactions observed in the structure of human GLX2 are missing in GLX2-5. Glu146 (incorrectly labeled Asp146 in Table 1 of reference (42)) was shown to interact with His56, via a water molecule, in the structure of human GLX2. There is no evidence of a water molecule in the structure of GLX2-5 between His56 and Glu143, and these two residues are 5.2 Å apart and probably do not interact. The structure of human GLX2 also suggested an interaction of the carbonyl of Asp134 with the side chain of Asp134 (42); this interaction is not observed in GLX2-5. At this time it is not clear if these differences in second sphere influence the metal binding properties of the enzyme, or if they result from the different geometries of the metal centers in GLX2-5.
Results presented in this study represent the first detailed structural characterization of a mitochondrial glyoxalase 2 isozyme from any source. They demonstrated that some mitochondrial glyoxalase enzymes are very similar to the cytoplasmic enzymes and have a preference for SLG. Furthermore, these results indicate that while the Arabidopsis GLX2 isozymes can utilize a number of different metal centers, it appears that a Fe(III)Zn(II) center is the predominant form of GLX2-5. While further studies are required on other GLX2 and β-lactamase-fold containing enzymes to better understand factors that control metal specificity in these enzymes, our results suggest that subtle alterations in second sphere ligands may directly metal preference in GLX2 enzymes. Differences in second sphere ligands may explain why human GLX2 appears to have a preference for a dinuclear Zn site, while the majority of recombinant Arabidopsis GLX2-5 contains a Fe(III)Zn(II) site.
Table 2.
Crystallographic and refinement statisticsa
| PDB ID | 1XM8 |
| Space group | P1 21 1 |
| Resolution range (Å) | 33.71-1.74 |
| Unique reflections | 51,949 |
| Rmerge (%)b | 7.9 (54.5) |
| <I/σ(I)> (%)b | 21.2 (4.3) |
| Completeness (%)b | 95.4 (97.4) |
| R/Rfree (%) | 17.9/28.4 |
| Root mean square deviation bond length (Å) | 0.020 |
| Root mean square deviation bond angle (degree) | 1.7 |
| Number of water molecules | 547 |
| Method to solve structure | SAD |
Additional details about crystallographic and refinement statistics can be found on the RCSB website (Protein Data Bank ID number: 1XM8).
Values for the highest resolution shell are in parentheses.
Footnotes
The authors would like to thank Wolfram Meyer–Klaucke from the EMBL Outstation Hamburg for performing the PIXE experiments, Damodaran Krishnan for assistance in obtaining the NMR spectra, and G. E. Wesenberg, D. W. Smith, G. N. Phillips Jr., E. Bitto, C. A. Bingman, S. T. M. Allard at the Center For Eukaryotic Structural Genomics at the University of Wisconsin for determining the crystal structure of GLX2-5. Funds to purchase the 500 MHz NMR were provided by the Hayes Investment Fund (Ohio Board of Regents). The authors would also like to thank NIH (GM40075 to M.W.C., AI056231 and EB001980 to B.B.) and NSF (MCB-0041105 to C.A.M.) for funding this work.
Abbreviations: FPLC, Fast Performance liquid chromatography; ICP-AES, Inductively coupled plasma with atomic emission spectroscopy; IPTG, isopropylthio-β-D-galactoside; LB, Luria-Bertani; MG, methylglyoxal; MOPS, 3-(N-morpholino)propanesulfonic acid; PIXE, particle induced X-ray emission; PMSF, phenylmethylsulfonyl fluoride; SLG, S-D-lactoylglutathione.
References
- 1.Thornalley P. Mol Aspects Med. 1993;14:287–371. doi: 10.1016/0098-2997(93)90002-u. [DOI] [PubMed] [Google Scholar]
- 2.Davidson G, Clugston SL, Honek JF, Maroney MJ. Inorg Chem. 2000;39:2962–2963. doi: 10.1021/ic0001208. [DOI] [PubMed] [Google Scholar]
- 3.Thornalley PJ. Chem Biol Inter. 1998;111–112:137–151. doi: 10.1016/s0009-2797(97)00157-9. [DOI] [PubMed] [Google Scholar]
- 4.Thornalley P. Crit Rev Oncol Hematol. 1995;20:99–128. doi: 10.1016/1040-8428(94)00149-n. [DOI] [PubMed] [Google Scholar]
- 5.Papoulis A, Al-Abed Y, Bucala R. Biochemistry. 1995;34:648–655. doi: 10.1021/bi00002a032. [DOI] [PubMed] [Google Scholar]
- 6.Rahman A, Shahabuddin A, Hadi S. J Biochem Toxicol. 1990;5:161–166. doi: 10.1002/jbt.2570050305. [DOI] [PubMed] [Google Scholar]
- 7.Lo T, Westwood M, McLellan A, Selwood T, Thornalley P. J Biol Chem. 1994;269:32299–32305. [PubMed] [Google Scholar]
- 8.Rulli A, Carli L, Romani R, Baroni T, Giovannini E, Rosi G, Talesa V. Breast Cancer Res Treat. 2001;66:67–72. doi: 10.1023/a:1010632919129. [DOI] [PubMed] [Google Scholar]
- 9.Thornalley PJ. Biochem Soc Trans. 1993;21:531–534. doi: 10.1042/bst0210531. [DOI] [PubMed] [Google Scholar]
- 10.Norton SJ, Elia AC, Chyan MK, Gillis G, Frenzel C, Principato GB. Biochem Soc Trans. 1993;21:545–548. doi: 10.1042/bst0210545. [DOI] [PubMed] [Google Scholar]
- 11.Allen RE, Lo TWC, Thornalley PJ. Biochem Soc Trans. 1993;21:535–539. doi: 10.1042/bst0210535. [DOI] [PubMed] [Google Scholar]
- 12.Creighton DJ, Zheng ZB, Holewinski RJ, Hamilton DS, Eiseman JL. Biochem Soc Trans. 2003;31:1378–1382. doi: 10.1042/bst0311378. [DOI] [PubMed] [Google Scholar]
- 13.Thornalley PJ, Strath M, Wilson RJH. Biochem Pharmacol. 1994;47:418–420. doi: 10.1016/0006-2952(94)90035-3. [DOI] [PubMed] [Google Scholar]
- 14.McLellan AC, Thornalley PJ, Benn J, Sonksen PH. Clin Sci. 1994;87:21–29. doi: 10.1042/cs0870021. [DOI] [PubMed] [Google Scholar]
- 15.Ratliff DM, Vander Jagt DJ, Eaton RP, Vander Jagt DL. J Clin Endocrinol Metabol. 1996;81:488–492. doi: 10.1210/jcem.81.2.8636255. [DOI] [PubMed] [Google Scholar]
- 16.Chen F, Wollmer MA, Hoerndli F, Munch G, Kuhla B, Rogaev EI, Tsolaki M, Papassotiropoulos A, Gotz J. Proc Natl Acad Sci. 2004;101:7687–7692. doi: 10.1073/pnas.0402338101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Creighton DJ, Hamilton DS. Arch Biochem Biophys. 2001;387:1–10. doi: 10.1006/abbi.2000.2253. [DOI] [PubMed] [Google Scholar]
- 18.Richter U, Krauss M. J Am Chem Soc. 2001;123:6973–6982. doi: 10.1021/ja0105966. [DOI] [PubMed] [Google Scholar]
- 19.Sharkey EM, O’Neill HB, Kavarana MJ, Wang HB, Creighton DJ, Sentz DL, Eiseman JL. Cancer Chemother Pharmacol. 2000;46:156–166. doi: 10.1007/s002800000130. [DOI] [PubMed] [Google Scholar]
- 20.Kavarana MJ, Kovaleva EG, Creighton DJ, Wollman MB, Eiseman JL. J Med Chem. 1999;42:221–228. doi: 10.1021/jm9708036. [DOI] [PubMed] [Google Scholar]
- 21.Cordell PA, Futers TS, Grant PJ, Pease RJ. J Biol Chem. 2004;279:28653–28661. doi: 10.1074/jbc.M403470200. [DOI] [PubMed] [Google Scholar]
- 22.Bito A, Haider M, Hadler I, Breitenbach M. J Biol Chem. 1997;272:21509–21519. doi: 10.1074/jbc.272.34.21509. [DOI] [PubMed] [Google Scholar]
- 23.Maiti MK, Krishnasamy S, Owen HA, Makaroff CA. Plant Mol Biol. 1997;35:471–481. doi: 10.1023/a:1005891123344. [DOI] [PubMed] [Google Scholar]
- 24.Talesa V, Uotila L, Koivusalo M, Principato G, Giovannini E, Rosi G. Biochim Biophys Acta. 1989;993:7–11. doi: 10.1016/0304-4165(89)90135-9. [DOI] [PubMed] [Google Scholar]
- 25.Talesa V, Uotila L, Koivusalo M, Principato G, Giovannini E, Rosi G. Biochim Biophys Acta. 1988;955:103–110. doi: 10.1016/0167-4838(88)90183-5. [DOI] [PubMed] [Google Scholar]
- 26.Bito A, Haider M, Briza P, Strasser P, Breitenbach M. Protein Express Purif. 1999;17:456–464. doi: 10.1006/prep.1999.1151. [DOI] [PubMed] [Google Scholar]
- 27.Norton SJ, Talesa V, Yuan WJ, Principato GB. Biochem Internat. 1990;22:411–418. [PubMed] [Google Scholar]
- 28.Talesa V, Rosi G, Contenti S, Mangiabene C, Lupattelli M, Norton SJ, Giovannini E, Principato GB. Biochem Internat. 1990;22:1115–1120. [PubMed] [Google Scholar]
- 29.Talesa V, Principato GB, Norton SJ, Contenti S, Mangiabene C, Rosi G. Biochem Internat. 1990;20:53–58. [PubMed] [Google Scholar]
- 30.Talesa V, Rosi G, Bistoni F, Marconi P, Norton SJ, Principato GB. Biochem Internat. 1990;21:397–403. [PubMed] [Google Scholar]
- 31.Crowder MW, Maiti MK, Banovic L, Makaroff CA. FEBS Lett. 1997;418:351–354. doi: 10.1016/s0014-5793(97)01416-6. [DOI] [PubMed] [Google Scholar]
- 32.Ridderstrom M, Mannervik B. Biochem J. 1997;322:449–454. doi: 10.1042/bj3220449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ridderstrom M, Saccucci F, Hellman U, Bergman T, Principato G, Mannervik B. J Biol Chem. 1996;271:319–323. doi: 10.1074/jbc.271.1.319. [DOI] [PubMed] [Google Scholar]
- 34.Irsch T, Krauth-Siegel RL. J Biol Chem. 2004;279:22209–22217. doi: 10.1074/jbc.M401240200. [DOI] [PubMed] [Google Scholar]
- 35.Crowder MW, Walsh TR. Research Signpost. 1999;3:105–132. [Google Scholar]
- 36.Cricco JA, Vila AJ. Curr Pharma Design. 1999;5:915–927. [PubMed] [Google Scholar]
- 37.Frazao C, Silva G, Gomes CM, Matias P, Coelho R, Sieker L, Macedo S, Liu MY, Oliveira S, Teixeira M, Xavier AV, Rodrigues-Pousada C, Carrondo MA, Le Gall J. Nature Struct Biol. 2000;7:1041–1045. doi: 10.1038/80961. [DOI] [PubMed] [Google Scholar]
- 38.Vogel A, Schilling O, Niecke M, Bettmer J, Meyer-Klaucke W. J Biol Chem. 2002;277:29078–29085. doi: 10.1074/jbc.M112047200. [DOI] [PubMed] [Google Scholar]
- 39.Zang TM, Hollman DA, Crawford PA, Crowder MW, Makaroff CA. J Biol Chem. 2001;276:4788–4795. doi: 10.1074/jbc.M005090200. [DOI] [PubMed] [Google Scholar]
- 40.Schilling O, Wenzel N, Naylor M, Vogel A, Crowder M, Makaroff C, Meyer-Klaucke W. Biochemistry. 2003;42:11777–11786. doi: 10.1021/bi034672o. [DOI] [PubMed] [Google Scholar]
- 41.Wenzel NF, Carenbauer AL, Pfiester MP, Schilling O, Meyer-Klaucke W, Makaroff CA, Crowder MW. J Biol Inorg Chem. 2004;9:429–438. doi: 10.1007/s00775-004-0535-2. [DOI] [PubMed] [Google Scholar]
- 42.Cameron AD, Ridderstrom M, Olin B, Mannervik B. Structure. 1999;7:1067–1078. doi: 10.1016/s0969-2126(99)80174-9. [DOI] [PubMed] [Google Scholar]
- 43.Ullah JH, Walsh TR, Taylor IA, Emery DC, Verma CS, Gamblin SJ, Spencer J. J Mol Biol. 1998;284:125–136. doi: 10.1006/jmbi.1998.2148. [DOI] [PubMed] [Google Scholar]
- 44.Melino SCc, Dragani B, Aceto A, Petruzzelli R. Trends Biochem Sci. 1998;23:381–382. doi: 10.1016/s0968-0004(98)01264-x. [DOI] [PubMed] [Google Scholar]
- 45.Maiti MK, Krishnasamy S, Owen HA, Makaroff CA. Plant Mol Biol. 1997;35:471–481. doi: 10.1023/a:1005891123344. [DOI] [PubMed] [Google Scholar]
- 46.Johansson, S. A. E., and Campbell, J. L. (1988) PIXE: a novel technique for elemental analysis, Wiley, New York
- 47.Uotila L. Methods Enzymol. 1981;77:424–430. doi: 10.1016/s0076-6879(81)77045-9. [DOI] [PubMed] [Google Scholar]
- 48.Borovik AS, Papaefthymiou V, Taylor LF, Anderson OP, Que L. J Am Chem Soc. 1989;111:6183–6195. [Google Scholar]
- 49.Battistuzzi G, Dietrich M, Locke R, Witzel H. Biochem J. 1997;323:593–596. doi: 10.1042/bj3230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lauffer RB, Antanaitis BC, Aisen P, Que L. J Biol Chem. 1983;258:14213–14218. [PubMed] [Google Scholar]
- 51.Wang Z, Ming LJ, Que L, Vincent JB, Crowder MW, Averill BA. Biochemistry. 1992;31:5263–5268. doi: 10.1021/bi00138a004. [DOI] [PubMed] [Google Scholar]
- 52.Bertini I, Turano P, Vila AJ. Chem Rev. 1993;93:2833–2932. [Google Scholar]
- 53.Scarrow RC, Pyrz JW, Que L. J Am Chem Soc. 1990;112:657–665. [Google Scholar]
- 54.Beck JL, de Jersey J, Zerner B, Hendrich MP, Debrunner PG. J Am Chem Soc. 1988;110:3317–3318. [Google Scholar]
- 55.Merkx M, Averill BA. Biochemistry. 1998;37:11223–11231. doi: 10.1021/bi980389r. [DOI] [PubMed] [Google Scholar]
- 56.Yu L, Haddy A, Rusnak F. J Am Chem Soc. 1995;117:10147–10148. [Google Scholar]