

Abstract
Vascular contraction is an important determinant of the peripheral vascular resistance and blood pressure. The mechanisms underlying vascular smooth muscle (VSM) contraction and the pathological changes that occur in hypertension have been the subject of numerous studies and interpretations. Activation of VSM by vasoconstrictor stimuli at the cell surface causes an increase in [Ca2+]i, Ca2+-dependent activation of myosin light chain (MLC) kinase, MLC phosphorylation, actin-myosin interaction and VSM contraction. Additional signaling pathways involving Rho-kinase and protein kinase C (PKC) may increase the myofilament force sensitivity to [Ca2+]i and MLC phosphorylation, and thereby maintain vascular contraction. PKC is a particularly intriguing protein kinase as it comprises a family of Ca2+-dependent and Ca2+-independent isoforms, which have different tissue and subcellular distribution, and undergo differential translocation during cell activation. PKC translocation to the cell surface may trigger a cascade of protein kinases such as mitogen-activated protein kinase (MAPK) and MAPK kinase (MEK) that ultimately interact with the contractile myofilaments and cause VSM contraction. Also, PKC translocation to the nucleus may promote VSM growth and proliferation. Increased PKC expression and activity have been identified in several forms of hypertension. The subcellular location of PKC may determine the state of VSM activity, and may be useful in the diagnosis/prognosis of hypertension. Vascular PKC isoforms may represent specific targets for modulation of VSM hyperactivity, and isoform-specific PKC inhibitors may be useful in treatment of Ca2+ antagonist-resistant forms of hypertension.
Keywords: signal transduction, vascular smooth muscle, calcium, blood pressure
List of abbreviations: AngII, angiotensin II; ATP, adenosine triphosphate; CPI-17, PKC-potentiated phosphatase inhibitor protein-17 kDa; CAM, calmodulin; DAG, diacylglycerol; ET-1, endothelin; IP3, inositol 1,4,5-trisphosphate; MAPK, mitogen-activated protein kinase; MARCKs, myristoylated alanine-rich C-kinase substrate; MEK, MAPK kinase; MLC, myosin light chain; NADP, nicotinamide adenine dinucleotide phosphate; O·2−, superoxide; PDBu, phorbol 12,13-dibutyrate; PIP2, phosphatidylinositol 4,5-bisphosphate; PLD, phospholipase D; PKC, protein kinase C; PMA, phorbol myristate acetate; RACKs, receptors for activated C-kinase; Rho-kinase, Rho-associated kinase; ROS, reactive oxygen species; SHR, spontaneously hypertensive rat; TPA, 12-o-tetradecanoylphorbol-13-acetate; VSM, vascular smooth muscle; WKY, Wistar-Kyoto
Hypertension is a multifactorial disorder that involves pathological changes in the neuronal, renal, and vascular control mechanisms of blood pressure [1]. VSM contraction contributes to the regulation of vascular resistance and blood pressure, and its dysregulation may lead to hypertension. VSM contraction is triggered by an increase in [Ca2+]i due to Ca2+ release from the sarcoplasmic reticulum and Ca2+ entry from the extracellular space through Ca2+ channels. Ca2+ binds calmodulin (CAM) to form a Ca2+-CAM complex, which activates myosin light chain (MLC) kinase, and causes MLC phosphorylation, actin-myosin interaction and VSM contraction (Fig. 1). VSM relaxation is initiated by a decrease in [Ca2+]i due to Ca2+ uptake by the sarcoplasmic reticulum and Ca2+ extrusion by the plasmalemmal Ca2+ pump and Na+-Ca2+ exchanger. The decrease in [Ca2+]i causes dissociation of the Ca2+-CAM complex and the phosphorylated MLC is dephosphorylated by MLC phosphatase [2–4].
Fig. 1.

Mechanisms of VSM contraction. Agonist (A) binds to its receptor (R ), stimulates plasma membrane PLCß, and increases production of IP3 and diacylglycerol (DAG). IP3 stimulates Ca2+ release from the sarcoplasmic reticulum (SR). Agonist also stimulates Ca2+ influx through Ca2+ channels. Ca2+ binds calmodulin (CAM), activates MLC kinase (MLCK), causes MLC phosphorylation, and initiates VSM contraction. DAG activates PKC. PKC phosphorylates CPI-17, which in turn inhibits MLC phosphatase and thereby enhances the myofilament force sensitivity to Ca2+. PKC could phosphorylate calponin (Cap), allowing more actin to bind myosin. PKC may activate a protein kinase cascade involving Raf, MAPK kinase (MEK) and MAPK, leading to phosphorylation of the actin-binding protein caldesmon (CaD). Other pathways of VSM contraction include the RhoA/Rho-kinase pathway, which inhibits MLC phosphatase and further enhances the Ca2+ sensitivity. G, heterotrimeric G-protein; PIP2, phosphatidylinositol 4,5-bisphosphate; PC, phosphatidylcholine; PS, phosphatidylserine; PE, phosphatidylethanolamine; AA, arachidonic acid.
Membrane depolarization in response to electrical stimulation, mechanical stretch or high KCl solution activates voltage-gated Ca2+ channels, increases Ca2+ influx, stimulates MLC phosphorylation, and causes maintained VSM contraction. In contrast, the interaction of a physiological agonist with its receptor results in activation of phospholipase C, hydrolysis of the plasma membrane phosphatidylinositol 4,5-bisphosphate, and increased production of inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) [5,6]. IP3 stimulates Ca2+ release from the sarcoplasmic reticulum and initiates agonist-induced VSM contraction. Agonists also stimulate Ca2+ influx through ligand-gated and store-operated Ca2+ channels, and cause maintained increase in [Ca2+]i, MLC phosphorylation and VSM contraction (Fig. 1). However, Ca2+ channel blockers do not completely inhibit agonist-induced VSM contraction. Although differential dependence on intracellular Ca2+ stores and extracellular Ca2+ for force development could explain the relative insensitivity of agonist-induced contraction to Ca2+ channel blockers in some vessels [2], agonist-induced dissociations between [Ca2+]i and force have been suggested in other vascular preparations. For example, agonist-induced VSM contractions have been observed in Ca2+-free solution and in the absence of increases in [Ca2+]i or MLC phosphorylation. Also, agonist-induced dissociations between [Ca2+]i and MLC phosphorylation have suggested the activation of Ca2+ sensitization pathways involving Rho-kinase and protein kinase C (PKC), which may inhibit MLC phosphatase and thereby enhance VSM contraction [2–4].
Several review articles have examined the biochemical and molecular aspects of VSM contraction and the role of Ca2+ in the regulation of vascular function [2–4]. Because of the small size and diffusible nature of Ca2+, it is feasible to envision its role in transducing the extracellular signal to the contractile myofilaments. Although PKC has been identified for almost 30 years, its role in VSM contraction and control of blood pressure is not as widely perceived as Ca2+ partly because of its relatively large size, its numerous isoforms and substrates, and its differential subcellular distribution during VSM activation. Some important questions are how PKC is identified among other kinases in VSM, and how the PKC signal is transferred from the receptors at the cell surface to the contractile myofilaments in the center of the cell. This review will further examine PKC as a potential modulator of VSM function. We will first provide a brief description of PKC structure, isoforms and protein substrates. The subcellular distribution of PKC isoforms, and the cellular mechanisms that promote PKC translocation during VSM activation will then be discussed. A description of PKC activators and inhibitors will follow. The review will end with an evaluation of the significance of the subcellular location of PKC in determining the state of VSM activity, and its usefulness in the diagnosis/prognosis of VSM hyperactivity disorders such as hypertension. The potential use of isoform-specific PKC inhibitors in treatment of hypertension will also be discussed.
PKC Structure and PKC Isoforms
PKC is a ubiquitous enzyme that was originally described as a Ca2+-activated, phospholipid-dependent protein kinase. Molecular cloning and biochemical analysis have revealed a family of PKC subspecies with closely related structures. The PKC isozymes α, β and γ consist of four conserved (C1–C4) and five variable regions (V1-V5). The C1 region contains cysteine-rich zinc finger-like motifs, is immediately preceded by an autoinhibitory pseudosubstrate sequence, and contains the recognition site for phosphatidylserine, DAG and phorbol ester. The C2 region of some PKC isoforms is rich in acidic residues and binds Ca2+. The C3 and C4 regions form the ATP- and substrate-binding lobes [5,7] (Fig. 2).
Fig. 2.
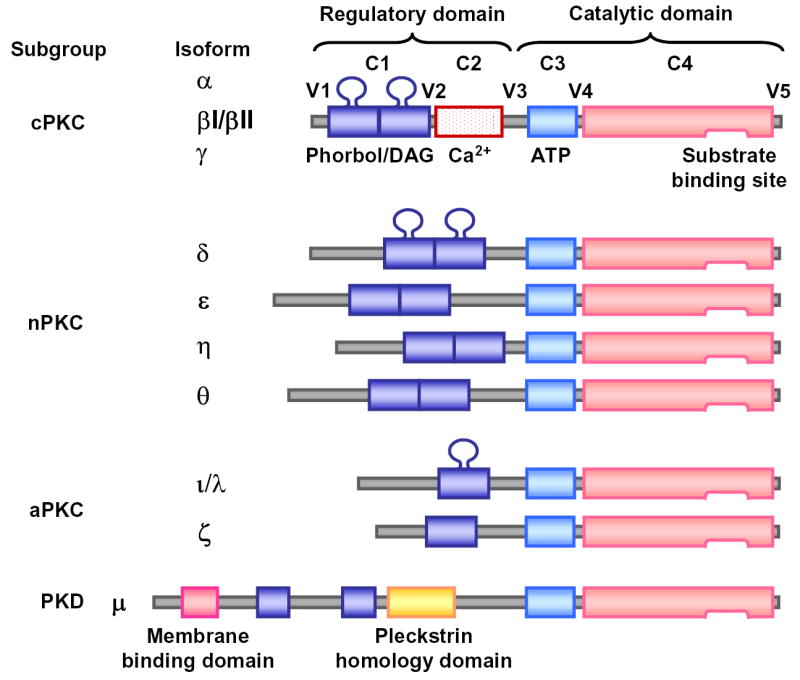
Structure of PKC isoforms. PKC is composed of 4 conserved (C1–C4) and 5 variable (V1–V5) regions. C1 region contains binding sites for DAG, phorbol ester, phosphatidylserine and the PKC antagonist calphostin C. C2 region contains the binding site for Ca2+. C3 and C4 regions contain binding sites for ATP, some PKC antagonists and different PKC substrates. The PKC molecule folds to bring the ATP binding site into proximity with the substrate-binding site. Binding of an endogenous or exogenous pseudosubstrate peptide sequence to the catalytic domain prevents PKC from phosphorylating the true substrate.
PKC isoforms are classified into 3 groups. The conventional PKCs (cPKC) α, βI, βII, and γ have 4 conserved regions (C1–C4) and 5 variable regions (V1–V5). The novel PKCs (nPKC) δ, ɛ, η(L) and θ lack the C2 region and therefore do not require Ca2+ for activation. The atypical PKCs (aPKC) ζ and λ/ι have only one cysteine-rich zinc finger-like motif and are dependent on phosphatidylserine, but not affected by DAG, phorbol esters or Ca2+ (Fig. 2) (Table I).
Table I.
Subcellular Distribution of PKC Isoforms in VSM
| Isoform | M.W. (kDa) | Blood vessel | Resting state | Activated state | Reference |
|---|---|---|---|---|---|
| α-PKC | 74–82 | Bovine aorta | Cytosolic | Membrane | [6] |
| Ferret portal vein | Cytosolic | Surface membrane | [3,15,29] | ||
| Rat aorta | Cytosolic | Nuclear | [6] | ||
| Carotid artery | Cytosolic | Membrane | [6] | ||
| Rat mesenteric artery | Cytosolic/Membrane | Cytosolic/Membrane | [6] | ||
| Coronary artery | Cytosolic | Membrane | [21,22] | ||
| β-PKC | 80–82 | Rat aorta | Cytosolic | Nuclear | [6] |
| Carotid artery | Cytosolic | Membrane | [6] | ||
| γ-PKC | 70–82 | Rat mesenteric artery | Cytosolic | Cytosolic | [6] |
| δ-PKC | 76–82 | Rat aorta | Cytoskeleton
Organelles |
Cytoskeleton
Organelles |
[3,30] |
| Rat mesenteric artery | Membrane | Membrane | [6] | ||
| ɛ-PKC | 90–97 | Ferret aorta | Cytosol | Surface membrane | [3,15] |
| Rat mesenteric artery | Cytosolic/Membrane | Cytosolic/Membrane | [6] | ||
| Coronary artery | Cytosolic | Membrane | [21,22] | ||
| ζ-PKC | 64–82 | Ferret aorta, portal vein | Perinuclear | Intranuclear | [3,15] |
| Rat aorta | Perinuclear | Intranuclear | [3,30] | ||
| Rat mesenteric artery | Cytosolic | Cytosolic | [6] |
PKC Substrates
When PKC is not catalytically active the basic autoinhibitory pseudosubstrate is protected from proteolysis by an acidic patch in the substrate-binding site. As PKC is activated it phosphorylates arginine-rich protein substrates, which neutralize the acidic patch and displace the pseudosubstrate from its binding site in the kinase core [7,8]. The amino acid sequence in the vicinity of the substrate phosphorylation site may assist in PKC substrate recognition. PKC isotypes show specificity in substrate phosphorylation. While α-, β-, and γ-PKC are potent histone kinases, δ-, ɛ-, and η-PKC have a poor capacity to phosphorylate histone IIIS [6].
PKC causes phosphorylation of membrane-bound regulatory proteins in VSM. MARCKS (myristoylated, alanine-rich C kinase substrate), a major PKC substrate, is bound to F-actin and may function as a crossbridge between cytoskeletal actin and the plasma membrane [9]. Also, PKC causes phosphorylation of the inhibitory GTP-binding protein Gi, facilitating the dissociation of the αi subunit from adenylyl cyclase and thereby relieves it from inhibition [6].
PKC also affects plasma membrane channels and pumps. PKC inhibits BKCa channel activity in pulmonary VSM [10]. Also, thromboxane A2-induced inhibition of voltage-gated K+ channels and pulmonary vasoconstriction may involve ζ-PKC [11]. PKC may also phosphorylate and activate plasmalemmal or sarcoplasmic reticulum Ca2+-ATPase, an action that promotes Ca2+ extrusion and may explain the transient nature of the agonist-induced increase in VSM [Ca2+]i. Additionally, the α1 subunit of Na/K-ATPase may serve as a PKC substrate. Furthermore, activated PKC may phosphorylate and activate the Na+/H+ antiport exchanger and thereby increase the cytoplasmic pH [12].
PKC also phosphorylates regulatory proteins in VSM cytoskeleton and contractile myofilaments. PKC phosphorylates vinculin, a cytoskeletal protein localized at adhesion plaques, thus controlling cell shape and adhesion. PKC also phosphorylates CPI-17, which in turn inhibits MLC phosphatase, increases MLC phosphorylation and thereby enhances VSM contraction [13]. The 20 kDa MLC and MLC kinase serve as substrates for PKC, and their phosphorylation could counteract the Ca2+-induced actin-myosin interaction and force development [14]. Also, activation of α-PKC could cause phosphorylation of calponin, an actin-associated regulatory protein, and thereby enhance VSM contraction [6]. A specific link likely exists between each PKC isoform and one or more specific substrates in VSM, and identification of these specific interactions should be further examined in future studies.
Tissue Distribution of PKC
PKC isoforms are expressed in different proportions in smooth muscle of various vascular beds (Table I). α-PKC is a universal isoform that is expressed in almost all blood vessels tested. γ-PKC is mainly expressed in the neurons and vascular nerve endings. δ-PKC is mainly associated with the vascular cytoskeleton. ζ-PKC is a universal isoform that has been found in many tissues. η/L-PKC has been found in the lung, skin, heart and brain. θ-PKC is mainly expressed in skeletal muscle while ι/λ-PKC is expressed in the ovary and testis [6].
Subcellular Distribution of PKC
The PKC isoforms α, β and γ are mainly localized in the cytosolic fraction of unstimulated cells and undergo translocation to the cell membranes in activated cells (Table I). δ-PKC is located almost exclusively in the particulate fraction of both resting and activated cells. While ζ-PKC is localized in the vicinity of the nucleus of resting and activated mature VSM cells [15], it could play a role in pulmonary vasoconstriction in the perinatal period [16].
Mechanisms of PKC Translocation
An important question is what causes PKC to translocate. Simple diffusion may provide the driving force, while targeting mechanisms allow high-affinity binding when PKC is in the vicinity of its target [15]. Targeting mechanisms may involve one of the following:
A. Conformation-Induced Changes in Hydrophobicity
Binding of Ca2+ or DAG causes conformational change in the PKC molecule that results in exposure of the pseudosubstrate region, increases the hydrophobicity of PKC, and facilitates its binding to membrane lipids [7].
B. Lipid Modification
Lipid modification of proteins changes their subcellular distribution. Myristoylation of MARCKS is required for its binding to actin at the plasma membrane. PKC-mediated phosphorylation of MARCKS causes its displacement from the membrane and interferes with its actin cross-linking. Dephosphorylation of MARCKS causes its re-association with the membrane through its stably attached myristic acid membrane-targeting moiety [17].
The domain architecture of VSM plasma membrane may be regulated. VSM sarcolemma is divided into domains of focal adhesions alternating with caveolae-rich zones, both harboring a subset of membrane-associated proteins. Likewise, sarcolemmal lipids are segregated into domains of cholesterol-rich lipid rafts and glycerophospholipid-rich non-raft regions. The segregation of membrane lipids is critical for preservation of membrane protein architecture and for translocation of proteins to the sarcolemma. In smooth muscle, membrane lipid segregation is supported by annexins that target membrane sites of distinct lipid composition, and each annexin requires different [Ca2+] for its translocation to the sarcolemma, and thus allows a spatially confined, graded response to external stimuli and intracellular PKC [18].
C. Phosphorylation
The change in charge caused by phosphorylation of the protein may affect its affinity for lipid. For example, phosphorylation of MARCKS has an electrostatic effect of equal importance to myristoylation in determining the protein affinity for the membrane. Also, phosphorylation of PKC itself may be required for its activation and translocation, and the phosphorylation sites appear to be located in the catalytic domain of α-, β- and δ-PKC [19].
D. Targeting Sequences
Binding sites for arginine-rich polypeptides have been identified in the PKC molecule distal to the catalytic site and may allow targeting of PKC to specific subcellular locations. Also, receptors for activated C-kinase (RACKS) may allow targeting of PKC to cytoskeletal elements, and a peptide inhibitor derived from the PKC binding proteins annexin I and RACKI may interfere with translocation of β-PKC [20].
Functions of PKC in VSM
PKC plays a pivotal role in cell adjustment to the environment by exerting both positive and negative effects on cellular events. Many physiological functions have been assigned to PKC, including secretion and exocytosis, modulation of ion conductance, gene expression and cell proliferation [5,6]. PKC may also exert negative-feedback control over cell signaling. The negative feedback regulation includes the downregulation of surface receptors and inhibition of agonist-mediated phosphoinositide hydrolysis [5]. Several studies suggest a role for PKC in VSM contraction [3,5,6,21,22]. PKC activation by phorbol esters has been shown to cause significant contraction in isolated vascular preparations [3,6]. Also, PKC inhibitors cause significant inhibition of agonist-induced vascular contraction [21,22]. However, some studies suggest that PKC-mediated phosphorylation of MLC kinase may cause vascular relaxation [14].
PKC Activators
PKC isoforms respond differently to Ca2+, phosphatidylserine, DAG and other phospholipid degradation products. PKC binds Ca2+ in a phospholipid-dependent manner, and Ca2+ may form a “bridge” holding the protein and phospholipid complex together at the membrane [23]. Phosphatidylserine is indispensable for activation of PKC. Phosphatidylinositol and phosphatidic acid activate PKC at high Ca2+ concentrations. DAG activates PKC by reducing its Ca2+ requirement and enhancing its membrane association [5].
PKC activators also include lipids derived from sources other than glycerolipid hydrolysis such as cis-unsaturated free fatty acids and lysophosphatidylcholine, ceramide (a sphingomyelinase product), phosphatidylinositol 3,4,5-trisphosphate and cholesterol sulfate [24]. Phorbol esters such as TPA, PMA and PDBu can substitute for DAG in PKC activation. Phorbol esters stabilize PKC-membrane association by reducing its apparent Km for Ca2+ [6].
Autophosphorylation of PKC may modify its activity or affinity for its substrates. α-, βI- and βII-PKC are synthesized as inactive precursors that require phosphorylation by a putative “PKC kinase” for permissive activation. Also, multiple phosphorylation of α-PKC prevents its down-regulation by phorbol ester. Phosphorylation at the extreme C-terminus of βII-PKC allows the active site to bind ATP and substrate with higher affinity, while phosphorylation of structure determinants in the regulatory region enable higher affinity binding of Ca2+ [25].
PKC Inhibitors
Several PKC inhibitors have been developed (Table II). PKC inhibitors acting in the catalytic domain compete with ATP and therefore may not be specific. PKC Inhibitors acting in the regulatory domain compete at the DAG/phorbol ester or the phosphatidylserine binding site and may be more specific. Extended exposure to phorbol esters can specifically downregulate α-, β-, and γ-PKC, but the tumor promoting properties of phorbol limit its use.
Table II.
Examples of PKC Inhibitors
| Mechanism of Action | Compound | Chemistry | Specificity |
|---|---|---|---|
| Inhibitors in the catalytic domain (Compete with ATP at ATP binding site) | H-7 | 1-(5-isoquinolinesulfonyl)-2-methylpiperazine | Also inhibits cyclic
AMP and cyclic GMP-dependent protein kinases |
| Staurosporine SCH 47112 | Microbial alkaloids, product of Streptomyces | Also inhibits MLC kinase and tyrosine kinase | |
| Chelerythrine | Benzophenanthridine alkaloid | Competitive inhibitor with histone IIIS | |
| Gö6976 | Indocarbazole | Ca2+-dependent isoforms α and βI | |
| GF109203X Ro-318220 | Bisindolylmaleimide derivatives of staurosporine | PKC isozymes α, βI, βII, γ, δ and ɛ | |
| Other: | Aminoacridine
Apigenin Sangivamycin UCN-01, UCN-02 |
||
| Inhibitors in the regulatory domain | Calphostin C (UCN-1028A) | Perylenequinone metabolite isolated from Cladosporium cladosporioides | Binds to the regulatory domain at DAG/phorbol ester binding site |
| Sphingosine | Membrane lipid | Competitive inhibitor with phosphatidylserine | |
| Other: | Adriamycin
Cercosporin Chlorpromazine Dexniguldipine Polymixin B Tamoxifen Trifluoperazine |
The regulatory domain of PKC contains an aminoacid sequence between residues 19 and 36 that resembles the substrate phosphorylation site. Synthetic oligopeptides based on pseudosubstrate sequence are specific PKC inhibitors because they exploit its substrate specificity and do not interfere with ATP binding. The synthetic peptide (19–36) inhibits both PKC autophosphorylation and protein substrate phosphorylation. Replacement of Arg-27 with alanine in the peptide [Ala-27]PKC (19–31) increases the IC50 for inhibition of substrate phosphorylation [8]. Also, a myristoylated peptide based on the substrate motif of α- and β-PKC, myr-ΨPKC, inhibits TPA-Induced PKC activation and phosphorylation of MARCKS [26].
In smooth muscle, α-tocopherol inhibits the expression, activity and phosphorylation of α-PKC. Interestingly, β-tocopherol protects PKC from the inhibitory effects of α-tocopherol [27].
siRNA for specific PKC isoforms are now available and should be useful for studying the role of PKC in various cell function. Also, antisense techniques, transgenic animals and Knock out mice have been useful in studying the effects of PKC downregulation in vivo.
Protein Kinase Cascades during VSM Contraction
The interaction of a PKC isoform with its protein substrate may trigger a cascade of protein kinases that ultimately stimulate VSM contraction. PKC may phosphorylate CPI-17 which in turn inhibits MLC phosphatase, increases MLC phosphorylation and enhances VSM contraction (Fig. 1) [13]. PKC may also phosphorylate the actin binding protein calponin, and thereby reverses its inhibition of actin-activated myosin ATPase, allows more actin to interact with myosin and increases VSM contraction (Fig. 1) [3].
PKC, MAPK, and c-Raf-1 have been implicated in VSM growth. MAPK is a Ser/Thr kinase that is activated by dual phosphorylation at Thr and Tyr residues. MAPK is mainly cytosolic in quiescent VSM cells, but translocates to the nucleus during activation by mitogens. Tyrosine kinase and MAPK activities have been demonstrated in differentiated VSM [15,28]. MAPK transiently translocates to the surface membrane during early activation of VSM, but undergoes redistribution to the cytoskeleton during maintained activation [28]. It appears that during VSM activation DAG causes translocation of cytosolic ɛ-PKC to the surface membrane, where it is fully activated. Activated ɛ-PKC stimulates the translocation of cytosolic MAPK kinase (MEK) and MAPK to the plasmalemma, where they form a surface membrane kinase complex. PKC causes phosphorylation and activation of MEK, which in turn phosphorylates MAPK at both Thr and Tyr residues. Tyrosine phosphorylation targets MAPK to the cytoskeleton, where it phosphorylates caldesmon and reverses its inhibition of MgATPase activity and thus increases actin-myosin interaction and VSM contraction (Fig. 1) [3,28].
PKC in Hypertension
Increased expression/activity of PKC isoforms in VSM could cause excessive vasoconstriction as well as trophic vascular changes leading to increased vascular resistance and hypertension. α-PKC enhances Ca2+-dependent VSM contraction, and its overexpression has been implicated in the pathogenesis of hypertension [29,30]. The Ca2+-independent ɛ-PKC may increase the myofilament sensitivity to [Ca2+]i in VSM during hypertension [3]. δ-PKC is mainly associated with the cytoskeleton and may play a role in vascular remodeling in hypertension [6]. ζ-PKC is localized in the nucleus and may promote the VSM growth associated with hypertension [30]. In the following section we provide specific evidence for a role of PKC in some of the common forms of experimental and human hypertension.
Aortic Constriction Model of Hypertension
PKC activation and translocation have been demonstrated in rat model of pressure overload and left ventricular hypertrophy produced by banding or clipping of the aorta [30]. The increased PKC activity is associated with increased [3H]PDBu binding and PKC concentration in both the cytosolic and membrane fractions [31]. Immunoblot analysis has indicated that the increased PKC activity mainly involves βI-, βII- and ɛ-PKC and is present mainly in the membrane and nuclear-cytoskeletal fractions [31]. Other studies suggest that in VSM of normotensive rats α-PKC is localized in the cytosol, while ζ-PKC is located in the perinuclear area. In VSM of hypertensive rats, α-PKC is hyperactivated and concentrated at the surface membrane while ζ-PKC is localized in the nucleus [30].
Genetic Models of Hypertension
Studies have suggested a role for vascular PKC in the increased blood pressure in spontaneously hypertensive rats (SHR). For instance, the susceptibility of norepinephrine-induced contractions to the PKC inhibitor H-7 is higher in aortas from SHR than WKY. H-7 produces a shift to the right in the dose-response curve for the PKC activator TPA in aortas of SHR, but not WKY [32]. Also, PDBu produces increased contraction and greater reduction in cytosolic PKC activity in SHR aortas than WKY vessels, suggesting functional alterations in PKC during the sustained contraction of VSM in SHR [33].
Other studies have investigated the relation between vascular contraction and PKC activity during the development of hypertension in young (5–6 weeks) SHR. It has been shown that high K+-induced contraction in intact mesenteric arteries and the [Ca2+]i-force relation in α-toxin skinned vessels are not different in SHR and WKY. PDBu augments high K+-evoked contraction and the [Ca2+]i-force relation, and the PKC inhibitors H-7 and calphostin C suppress the responses more in SHR than WKY. These data suggest that the Ca2+ sensitivity of the contractile proteins via PKC is greater in prehypertensive SHR than WKY and may play a role in the development of hypertension [34].
Studies have also investigated possible inborn differences in the proliferation of VSM cells of young (1–2 week) SHR and WKY and the role of PKC before the onset of hypertension. AngII and ET-1 enhance thymidine incorporation into DNA, an indicator of DNA synthesis, in cultured aortic VSM from WKY and SHR. The PKC inhibitor chelerythrine suppresses AngII and ET-1 induced DNA synthesis and VSM growth to a greater extent in cells of SHR than WKY, suggesting an inborn increase in PKC activity in VSM of SHR [35].
To assess the role of PKC in the control of vessel tone in genetic hypertension in vivo, studies have examined the effects of PDBu in the perfused hindlimb of anesthetized SHR and WKY. PDBu infusion into the hindlimb causes prolonged vasoconstriction and elevation of the perfusion pressure that are inhibited by the PKC-inhibitor staurosporine, and the maximal inhibition is greater in SHR than WKY. These data provide evidence for a role of PKC in the control of vascular function and blood pressure in vivo, and further suggest an increase in PKC expression/activity in the SHR model of hypertension [36].
Animal models of salt-sensitive hypertension
The role of vascular PKC in salt-sensitive hypertension is unclear; however, PKC activity may be increased in other tissues in this type of hypertension. In deoxycorticosterone acetate (DOCA)-salt-sensitive hypertensive rats the blood pressure and the heart to body weight ratio are increased. The relative expression of α, γ and ɛ-PKC is increased, whereas δ-PKC is not altered in cardiac extracts of DOCA-salt rats. In cardiac fibroblasts from DOCA-salt rats δ-PKC is increased, suggesting that the hearts of DOCA-salt hypertensive rats are characterized by cell-specific enhanced expression of α, γ, δ or ɛ-PKC [37]. Cardiac PKC may also show significant changes in Dahl salt-sensitive hypertensive rats. Marinobufagenin, an endogenous ligand of α1 Na/K-ATPase, becomes elevated and contributes to hypertension in NaCl-loaded Dahl-Salt sensitive rats [38]. PKC phosphorylates α1 Na/K-ATPase and increases its sensitivity to marinobufagenin [38].
Renovascular hypertension
PKC may play a role in animal models of renovascular hypertension. Studies have assessed vascular function in aortas from rats with two kidney-one clip (2K-1C) hypertension and age-matched controls. In hypertensive rats, PDBu-induced vascular constriction is enhanced, and superoxide (O.2−) production is increased compared with controls. Vascular dysfunction and O.2− production are normalized by superoxide dismutase and the PKC inhibitor calphostin C. These data suggest that renovascular hypertension in 2K-1C rats is associated with increased vascular O.2− and impaired vasodilator function, possibly due to PKC-mediated activation of NAD(P)H-dependent oxidase [39].
Pulmonary Hypertension
PKC may play a role in the pathogenesis of pulmonary hypertension through specific effects on the pulmonary vessels. For example, insulin-like growth factor I and PKC activation stimulate pulmonary artery VSM cell proliferation. PKC activation also plays a role in pulmonary artery VSM cell proliferation in response to hypoxia. In addition, chronic hypoxia induces exaggerated growth responses in pulmonary artery adventitial fibroblasts via specific PKC isozymes [40].
Human Hypertension
Studies have investigated the role of PKC activation in the increased oxidative stress and growth responses in VSM cells from resistance arteries of patients with essential hypertension. AngII causes an increase in reactive oxygen species (ROS), that is enhanced in VSM from hypertensives compared with normotensive subjects. AngII stimulates phospholipase D (PLD) activity and DNA and protein synthesis to a greater extent in VSM cells from hypertensives than normotensives. The PKC inhibitors chelerythrine and calphostin C partially decrease AngII-elicited signals. These data suggest that in essential hypertension enhanced oxidative stress and augmented growth-promoting actions of AngII are associated with increased activation of PLD- and PKC-dependent pathways and that these processes may contribute to vascular remodeling in hypertension [41].
Hypertension of Pregnancy and Preeclampsia
Normal pregnancy is often associated with decreased blood pressure, increased uterine blood flow and decreased vascular responses to vasoconstrictor agonists [42]. The decreased vascular contraction is associated with decreased PKC activity in the uterine artery of pregnant sheep and the aorta of late pregnant rats [42,43]. Also, the expression and subcellular redistribution of the Ca2+-dependent α-PKC and the Ca2+-independent δ- and ζ-PKC are reduced during late pregnancy in rats [42].
In 5 to 7% of pregnancies, women develop preeclampsia characterized by severe increases in vascular resistance and blood pressure. Because of the difficulty of performing mechanistic studies in pregnant women, animal models of hypertension during pregnancy have been developed. We have recently shown that the blood pressure is increased in pregnant rats treated with the nitric oxide (NO) synthase inhibitor L-NAME. Also, the vascular contraction, PKC activity and the expression and subcellular distribution of α- and δ-PKC are enhanced in L-NAME treated compared with control pregnant rats. These data suggest that increased expression/activity of specific PKC isoforms may be involved in the increased vasoconstriction and vascular resistance during hypertension of pregnancy [42].
PKC may also affect the vascular AngII receptors in preeclampsia. In cultured neonatal rat cardiomyocytes, immunoglobulin from preeclamptic patients enhances AT1 receptor-mediated chronotropic responses, whereas immunoglobulin from controls have no effect. The PKC inhibitor calphostin C prevents the stimulatory effect of immunoglobulin on AT1 receptor. Confocal microscopy of VSM cells show colocalization of purified patient IgG and AT1 receptor antibody. These data suggest that preeclamptic patients develop stimulatory autoantibodies against AT1 receptor, and the effect appears to be PKC-mediated. These novel autoantibodies may participate in the AngII-induced vascular lesions in preeclamptic patients [44].
PKC Inhibitors in Hypertension
Although in vitro studies suggest a role of PKC in VSM contraction particularly in blood vessels of animal models of hypertension, few studies have investigated the in vivo effects of PKC inhibitors. Support for potential benefits of targeting PKC in hypertension came from studies using the antihypertensive compound cicletanine. Cicletanine is effective in salt-sensitive hypertension, in which dysregulation of the sodium pump plays a pathogenic role and marinobufagenin, an endogenous inhibitor of α1 Na/K-ATPase, becomes elevated and contributes to hypertension. Dahl-S rats on 8% NaCl diet exhibit an increase in blood pressure, marinobufagenin excretion, left ventricular mass, and myocardial Na/K-ATPase, βII-PKC and δ-PKC. Cicletanine-treated Dahl-S rats exhibit reduction in blood pressure and left ventricular weight, decreased sensitivity of Na/K-ATPase to marinobufagenin, no increase in βII-PKC, and reduced phorbol diacetate-induced Na/K-ATPase phosphorylation. These data suggest that PKC-induced phosphorylation of cardiac α1 Na/K-ATPase is a likely target for cicletanine in hypertension [38].
The in vivo effects of cicletanine possibly involve an effect on the vasculature. In isolated human mesenteric arteries, marinobufagenin induces sustained vasoconstriction, cicletanine causes relaxation of this contraction, and phorbol diacetate attenuates cicletanine-induced vasorelaxation. In mesenteric artery sarcolemmal membranes, marinobufagenin inhibits Na/K-ATPase activity, cicletanine attenuates Na/K-ATPase inhibition, and phorbol diacetate prevents the cicletanine-induced attenuation of marinobufagenin inhibition of Na/K-ATPase. Cicletanine also causes inhibition of rat brain PKC activity, and the PKC inhibition is not observed in the presence of phorbol diacetate. It appears that PKC phosphorylates α1 Na/K-ATPase and increases its marinobufagenin sensitivity. Cicletanine, via inhibition of PKC, reverses marinobufagenin-induced Na/K-ATPase inhibition and vasoconstriction. PKC is possibly an important factor for cardiotonic steroid-Na/K-ATPase interactions on vascular tone, and may represent a potential target for therapeutic intervention in hypertension [45].
Perspectives
The task of characterizing PKC 30 years ago is now becoming more challenging by the discovery of at least 11 PKC isoforms. Each PKC isoform has a peculiar subcellular distribution, a definite cellular substrate, and a specific cell function. This review highlighted the role of PKC in VSM contraction and the vascular control mechanisms of blood pressure; however, several points need to be clarified and important questions remain to be answered.
In addition to VSM contraction, PKC isoforms may be involved in VSM growth and hypertrophic vascular remodeling in hypertension. For instance, overexpression of α-PKC in A7r5 VSM cells stimulates cell proliferation [46]. Also, ζ-PKC may contribute to aortic VSM growth [15,30]. The increased PKC activity in conjunction with elevation of [Ca2+]i may exert trophic effects on the vasculature and the heart, thereby explaining the narrowing of the lumen in peripheral arteries and the cardiac hypertrophy of long-standing hypertension [12].
Several studies have shown PKC localization to the cell membrane during VSM activation, a property that could be used for the diagnosis/prognosis of VSM hyperactivity associated with hypertension. However, the subcellular redistribution of activated PKC may vary depending on the type and abundance of membrane lipids. For instance, erythrocyte membranes of elderly hypertensive subjects show increased cholesterol/phospholipid ratio and contain higher levels of monounsaturated and lower levels of polyunsaturated fatty acids as compared to normotensive controls. However, the levels of membrane-associated (active/preactive) PKC are not elevated, but rather reduced in elderly hypertensive subjects. These alterations in PKC distribution in elderly subjects are unlikely to be related to the etiopathology of hypertension, but may correspond to adaptive compensatory mechanisms in response to hypertension [47].
PKC inhibitors could be beneficial in modulation of VSM function in hypertension particularly when used in combination with other modes of treatment. PKC inhibitors could potentiate the vascular effects of Ca2+ channel blockers. Also, targeting Ca2+-independent PKC isoforms could be effective in Ca2+ antagonist-resistant forms of hypertension. The effects of PKC inhibitors on vascular function and blood pressure could also be potentiated by inhibitors of Rho-kinase and MAPK-dependent pathways. This is supported by reports that activation of RhoA/Rho-kinase and subsequent inhibition of MLC phosphatase contribute to VSM contraction and the enhanced vascular tone in hypertension [48,49].
In addition to VSM, changes in PKC activity in the endothelium could contribute to the regulation of vascular function and blood pressure. PKC has been implicated in the endothelial dysfunction in blood vessels of SHR and DOCA hypertensive rats [50,51]. NO plays a major role in the regulation of blood pressure, and an effect of PKC on NO production/activity could cause hypertension. For instance, renovascular hypertension in 2K-1C rats is associated with impaired vasodilation and increased vascular O.2− likely secondary to a PKC-mediated activation of membrane-associated NAD(P)H-dependent oxidase [38,52].
PKC may also contribute to the neuronal control mechanisms of blood pressure. For example, the expression and redistribution of PKC isozymes is increased in brain tissue of SHR [53]. Additionally, PKC may affect the renin-angiotensin-aldosterone system and the renal control mechanism of the blood pressure. AngII infusion in rats causes hypertension and endothelial dysfunction and increases O.2− production in vascular tissue. Some of the effects of AngII on VSM are mediated by ET-1, a known activator of PKC. AngII stimulates ET-1 production and thereby vascular PKC activity to a greater extent in blood vessels of SHR compared with normotensive control rats [54]. Studies have examined whether angiotensin converting enzyme inhibitors such as enalapril modulates vascular PKC activity. It has been shown that cytosolic PKC activity is higher in aortic media of SHR than in WKY or enalapril-treated SHR, and the changes in PKC activity are closely associated with the blood pressure. The membrane PKC activity is detected in samples of SHR, but not in WKY or enal-SHR. Also, the levels of α-PKC enzyme and mRNA are higher in SHR than in WKY or enal-SHR. These data suggest that the beneficial effects of angiotensin converting enzyme inhibitors in hypertension may involve changes in vascular PKC activity and the levels of α-PKC enzyme and mRNA in VSM [55]. PKC could also affect the Na+/Ca2+ exchange mechanism in the renal arterioles leading to defective renal vasodilation in salt-sensitive hypertension [56].
Targeting PKC isoforms could be useful in other disorders/complications associated with hypertension such as diabetes, obesity, insulin resistance and the metabolic syndrome. For example, glucose increases endothelial cell permeability in association with activation of α-PKC. Glucose also alters Na+/H+ antiport activity and gene expression in VSM cells via activation of PKC. Additionally, an antisense complementary to the mRNA initiation codon regions for α- and β-PKC promotes their downregulation and inhibits insulin-induced glucose uptake. Furthermore, inhibitors of β-PKC ameliorate the vascular dysfunction in diabetic rats and attenuate the progression of experimental diabetic nephropathy in hypertension [57,58]. Another major complication associated with hypertension is left ventricular overload and cardiac hypertrophy. Alterations in PKC expression and autophosphorylation have been demonstrated during the progression of pressure overload-induced left ventricular hypertrophy [59]. Also, tissue levels of AngII increase during progression or ventricular hypertrophy and heart failure in hypertensive rats, and may have differential effects on β- and ɛ-PKC [60].
Upregulation of PKC appears to play a pathogenic role in multiple morbidities such as hypertension, atherogenesis, insulin resistance, and cancer promotion - the “PKC syndrome'” [61]. Thus, to evaluate therapeutic efficacy of PKC inhibitors in human, studies in animal models of hypertension that have other comorbidities such as hypercholesterolemia, diabetes, and old age should be carried out. Also, treatment of a particular PKC-mediated pathological condition would require selective inhibition of the expression/activity of the specific PKC isoform involved. An important area of research has been to develop knock-out mice and transgenic animals that lack certain PKC isoforms. Other areas of research have focused on developing pharmacological tools that inhibit specific PKC subspecies. The first generation of PKC antagonists are not selective and none has fulfilled the specificity criteria in the laboratory. The new PKC antagonists appear to be more selective; however, rigorous experiments are needed before these compounds can be used safely in human.
Acknowledgments
This work was supported by grants from National Heart, Lung, and Blood Institute (HL-65998, HL-70659). RA Khalil is an Established Investigator of the American Heart Association.
References
- 1.Cain AE, Khalil RA. Pathophysiology of essential hypertension: role of the pump, the vessel, and the kidney. Semin Nephrol. 2002;22(1):3–16. [PubMed] [Google Scholar]
- 2.Khalil RA, van Breemen C. Mechanisms of calcium mobilization and homeostasis in vascular smooth muscle and their relevance to hypertension. In: Laragh JH, Brenner BM, editors. Hypertension: Pathophysiology, Diagnosis and Management. New York: Raven Press, 1995. p. 523:40.
- 3.Horowitz A, Menice CB, Laporte R, Morgan KG. Mechanisms of smooth muscle contraction. Physiol Rev. 1996;76(4):967–1003. doi: 10.1152/physrev.1996.76.4.967. [DOI] [PubMed] [Google Scholar]
- 4.Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83(4):1325–58. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- 5.Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of PKC. Science. 1992;258:607–14. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- 6.Kanashiro CA, Khalil RA. Signal transduction by protein kinase C in mammalian cells. Clin Exp Pharmacol Physiol. 1998;25(12):974–85. doi: 10.1111/j.1440-1681.1998.tb02170.x. [DOI] [PubMed] [Google Scholar]
- 7.Newton AC. Protein kinase C: structure, function, and regulation. J Biol Chem. 1995;270:28495–8. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- 8.House C, Kemp BE. Protein kinase C contains a pseudosubstrate prototope in its regulatory domain. Science. 1987;238:1726–8. doi: 10.1126/science.3686012. [DOI] [PubMed] [Google Scholar]
- 9.Hartwig JH, Thelen M, Rosen A, Janmey PA, Nairn AC, Aderem A. MARCKS is an actin filament crosslinking protein regulated by protein kinase C and calcium-calmodulin. Nature. 1992;356:618–22. doi: 10.1038/356618a0. [DOI] [PubMed] [Google Scholar]
- 10.Barman SA, Zhu S, White RE. Protein kinase C inhibits BKCa channel activity in pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2004;286:L149–55. doi: 10.1152/ajplung.00207.2003. [DOI] [PubMed] [Google Scholar]
- 11.Cogolludo A, Moreno L, Bosca L, Tamargo J, Perez-Vizcaino F. Thromboxane A2-induced inhibition of voltage-gated K+ channels and pulmonary vasoconstriction: role of protein kinase Czeta. Circ Res. 2003;93(7):656–63. doi: 10.1161/01.RES.0000095245.97945.FE. [DOI] [PubMed] [Google Scholar]
- 12.Aviv A. Cytosolic Ca2+, Na+/H+ antiport, protein kinase C trio in essential hypertension. Am J Hypertens. 1994;7(2):205–12. doi: 10.1093/ajh/7.2.205. [DOI] [PubMed] [Google Scholar]
- 13.Woodsome TP, Eto M, Everett A, Brautigan DL, Kitazawa T. Expression of CPI-17 and myosin phosphatase correlates with Ca2+ sensitivity of protein kinase C-induced contraction in rabbit smooth muscle. J Physiol. 2001;535(Pt 2):553–64. doi: 10.1111/j.1469-7793.2001.t01-1-00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inagaki M, Yokokura H, Itoh T, Kanmura Y, Kuriyama H, Hidaka H. Purified rabbit brain protein kinase C relaxes skinned vascular smooth muscle and phosphorylates myosin light chain. Arch Biochem Biophys. 1987;254(1):136–41. doi: 10.1016/0003-9861(87)90089-0. [DOI] [PubMed] [Google Scholar]
- 15.Khalil RA, Morgan KG. Enzyme translocations during smooth muscle activation. In: Barany M, editor. Biochemistry of Smooth Muscle Contraction. New York: Academic Press, 1996. p. 307–18.
- 16.Cogolludo A, Moreno L, Lodi F, Tamargo J, Perez-Vizcaino F. Postnatal maturational shift from PKCzeta and voltage-gated K+ channels to RhoA/Rho kinase in pulmonary vasoconstriction. Cardiovasc Res. 2005;66(1):84–93. doi: 10.1016/j.cardiores.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 17.Thelen M, Rosen A, Nairn AC, Aderem A. Regulation by phosphorylation of reversible association of a myristoylated protein kinase C substrate with the plasma membrane. Nature. 1991;351(6324):320–2. doi: 10.1038/351320a0. [DOI] [PubMed] [Google Scholar]
- 18.Draeger A, Wray S, Babiychuk EB. Domain architecture of the smooth-muscle plasma membrane: regulation by annexins. Biochem J. 2005;387(Pt 2):309–14. doi: 10.1042/BJ20041363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cazaubon SM, Parker PJ. Identification of the phosphorylated region responsible for the permissive activation of protein kinase C. J Biol Chem. 1993;268(23):17559–63. [PubMed] [Google Scholar]
- 20.Ron D, Mochly-Rosen D. Agonists and antagonists of protein kinase C function, derived from its binding proteins. J Biol Chem. 1994;269(34):21395–8. [PubMed] [Google Scholar]
- 21.Dallas A, Khalil RA. Ca2+ antagonist-insensitive coronary smooth muscle contraction involves activation of ɛ-protein kinase C-dependent pathway. Am J Physiol Cell Physiol. 2003;285(6):C1454–63. doi: 10.1152/ajpcell.00066.2003. [DOI] [PubMed] [Google Scholar]
- 22.McNair LL, Salamanca DA, Khalil RA. Endothelin-1 promotes Ca2+ antagonist-insensitive coronary smooth muscle contraction via activation of ɛ-protein kinase C. Hypertension. 2004;43(4):897–904. doi: 10.1161/01.HYP.0000118520.92686.3b. [DOI] [PubMed] [Google Scholar]
- 23.Bazzi MD, Nelseusten GL. Protein kinase C interaction with calcium: a phospholipid-dependent process. Biochemistry. 1990;29:7624–30. doi: 10.1021/bi00485a012. [DOI] [PubMed] [Google Scholar]
- 24.Nishizuka Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 1995;9:484–96. [PubMed] [Google Scholar]
- 25.Edwards AS, Newton AC. Phosphorylation at conserved carboxyl-terminal hydrophobic motif regulates the catalytic and regulatory domains of protein kinase C. J Biol Chem. 1997;272:18382–90. doi: 10.1074/jbc.272.29.18382. [DOI] [PubMed] [Google Scholar]
- 26.Eicholtz T, de Bont DB, Widt J, Liskamp RMJ, Ploegh HL. A myristoylated pseudo substrate peptide, a novel protein kinase C inhibitor. J Biol Chem. 1993;268:1982–6. [PubMed] [Google Scholar]
- 27.Clement S, Tasinato A, Boscoboinik D, Azzi A. The effect of α-tocoferol on the synthesis, phosphorylation and activity of protein kinase C in smooth muscle cells after phorbol 12-myristate 13-acetate. Eur J Biochem. 1997;246:745–9. doi: 10.1111/j.1432-1033.1997.t01-2-00745.x. [DOI] [PubMed] [Google Scholar]
- 28.Khalil RA, Menice CB, Wang CL, Morgan KG. Phosphotyrosine-dependent targeting of mitogen-activated protein kinase in differentiated contractile vascular cells. Circ Res. 1995;76(6):1101–8. doi: 10.1161/01.res.76.6.1101. [DOI] [PubMed] [Google Scholar]
- 29.Khalil RA, Lajoie C, Morgan KG. In situ determination of [Ca2+]i threshold for translocation of the α-protein kinase C isoform. Am J Physiol. 1994;266:C1544–51. doi: 10.1152/ajpcell.1994.266.6.C1544. [DOI] [PubMed] [Google Scholar]
- 30.Liou YM, Morgan KG. Redistribution of protein kinase C isoforms in association with vascular hypertrophy of rat aorta. Am J Physiol. 1994;267:C980–9. doi: 10.1152/ajpcell.1994.267.4.C980. [DOI] [PubMed] [Google Scholar]
- 31.Gu X, Bishop SP. Increased protein kinase C and isozyme redistribution in pressure-overload cardiac hypertrophy in the rat. Circ Res. 1994;75(5):926–31. doi: 10.1161/01.res.75.5.926. [DOI] [PubMed] [Google Scholar]
- 32.Shibata R, Morita S, Nagai K, Miyata S, Iwasaki T. Effects of H-7 (protein kinase inhibitor) and phorbol ester on aortic strips from spontaneously hypertensive rats. Eur J Pharmacol. 1990;175(3):261–71. doi: 10.1016/0014-2999(90)90563-l. [DOI] [PubMed] [Google Scholar]
- 33.Bazan E, Campbell AK, Rapoport RM. Protein kinase C activity in blood vessels from normotensive and spontaneously hypertensive rats. Eur J Pharmacol. 1992;227(3):343–8. doi: 10.1016/0922-4106(92)90014-m. [DOI] [PubMed] [Google Scholar]
- 34.Sasajima H, Shima H, Toyoda Y, Kimura K, Yoshikawa A, Hano T, Nishio I. Increased Ca2+ sensitivity of contractile elements via protein kinase C in alpha-toxin permeabilized SMA from young spontaneously hypertensive rats. Cardiovasc Res. 1997;36(1):86–91. doi: 10.1016/s0008-6363(97)00131-4. [DOI] [PubMed] [Google Scholar]
- 35.Rosen B, Barg J, Zimlichman R. The effects of angiotensin II, endothelin-1, and protein kinase C inhibitor on DNA synthesis and intracellular calcium mobilization in vascular smooth muscle cells from young normotensive and spontaneously hypertensive rats. Am J Hypertens. 1999;12(12 Pt 1–2):1243–51. doi: 10.1016/s0895-7061(99)00158-2. [DOI] [PubMed] [Google Scholar]
- 36.Bilder GE, Kasiewski CJ, Perrone MH. Phorbol-12,13-dibutyrate-induced vasoconstriction in vivo: characterization of response in genetic hypertension. J Pharmacol Exp Ther. 1990;252(2):526–30. [PubMed] [Google Scholar]
- 37.Fareh J, Touyz RM, Schiffrin EL, Thibault G. Altered cardiac endothelin receptors and protein kinase C in deoxycorticosterone-salt hypertensive rats. J Mol Cell Cardiol. 2000;32(4):665–76. doi: 10.1006/jmcc.2000.1110. [DOI] [PubMed] [Google Scholar]
- 38.Fedorova OV, Talan MI, Agalakova NI, Droy-Lefaix MT, Lakatta EG, Bagrov AY. Myocardial PKC beta2 and the sensitivity of Na/K-ATPase to marinobufagenin are reduced by cicletanine in Dahl hypertension. Hypertension. 2003;41(3):505–11. doi: 10.1161/01.HYP.0000053446.43894.9F. [DOI] [PubMed] [Google Scholar]
- 39.Heitzer T, Wenzel U, Hink U, Krollner D, Skatchkov M, Stahl RA, MacHarzina R, Brasen JH, Meinertz T, Munzel T. Increased NAD(P)H oxidase-mediated superoxide production in renovascular hypertension: evidence for an involvement of protein kinase C. Kidney Int. 1999;55(1):252–60. doi: 10.1046/j.1523-1755.1999.00229.x. [DOI] [PubMed] [Google Scholar]
- 40.Das M, Dempsey EC, Bouchey D, Reyland ME, Stenmark KR. Chronic hypoxia induces exaggerated growth responses in pulmonary artery adventitial fibroblasts: potential contribution of specific protein kinase c isozymes. Am J Respir Cell Mol Biol. 2000;22(1):15–25. doi: 10.1165/ajrcmb.22.1.3536. [DOI] [PubMed] [Google Scholar]
- 41.Touyz RM, Schiffrin EL. Increased generation of superoxide by angiotensin II in smooth muscle cells from resistance arteries of hypertensive patients: role of phospholipase D-dependent NAD(P)H oxidase-sensitive pathways. J Hypertens. 2001;19(7):1245–54. doi: 10.1097/00004872-200107000-00009. [DOI] [PubMed] [Google Scholar]
- 42.Khalil RA, Granger JP. Vascular mechanisms of increased arterial pressure in preeclampsia: lessons from animal models. Am J Physiol Regul Integr Comp Physiol. 2002;283(1):R29–45. doi: 10.1152/ajpregu.00762.2001. [DOI] [PubMed] [Google Scholar]
- 43.Magness RR, Rosenfeld CR, Carr BR. Protein kinase C in uterine and systemic arteries during ovarian cycle and pregnancy. Am J Physiol. 1991;260(3 Pt 1):E464–70. doi: 10.1152/ajpendo.1991.260.3.E464. [DOI] [PubMed] [Google Scholar]
- 44.Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jupner A, Baur E, Nissen E, Vetter K, Neichel D, Dudenhausen JW, Haller H, Luft FC. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest. 1999;103(7):945–52. doi: 10.1172/JCI4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bagrov AY, Dmitrieva RI, Dorofeeva NA, Fedorova OV, Lopatin DA, Lakatta EG, Droy-Lefaix MT. Cicletanine reverses vasoconstriction induced by the endogenous sodium pump ligand, marinobufagenin, via a protein kinase C dependent mechanism. J Hypertens. 2000;18(2):209–15. doi: 10.1097/00004872-200018020-00012. [DOI] [PubMed] [Google Scholar]
- 46.Wang S, Desai D, Wright G, Niles RM, Wright GL. Effects of protein kinase C alpha overexpression on A7r5 smooth muscle cell proliferation and differentiation. Exp Cell Res. 1997;236(1):117–26. doi: 10.1006/excr.1997.3714. [DOI] [PubMed] [Google Scholar]
- 47.Escriba PV, Sanchez-Dominguez JM, Alemany R, Perona JS, Ruiz-Gutierrez V. Alteration of lipids, G proteins, and PKC in cell membranes of elderly hypertensives. Hypertension. 2003;41(1):176–82. doi: 10.1161/01.hyp.0000047647.72162.a8. [DOI] [PubMed] [Google Scholar]
- 48.Seko T, Ito M, Kureishi Y, Okamoto R, Moriki N, Onishi K, Isaka N, Hartshorne DJ, Nakano T. Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circ Res. 2003;92(4):411–8. doi: 10.1161/01.RES.0000059987.90200.44. [DOI] [PubMed] [Google Scholar]
- 49.Lee DL, Webb RC, Jin L. Hypertension and RhoA/Rho-kinase signaling in the vasculature: highlights from the recent literature. Hypertension. 2004;44(6):796–9. doi: 10.1161/01.HYP.0000148303.98066.ab. [DOI] [PubMed] [Google Scholar]
- 50.Fatehi-Hassanabad Z, Fatehi M, Shahidi MI. Endothelial dysfunction in aortic rings and mesenteric beds isolated from deoxycorticosterone acetate hypertensive rats: possible involvement of protein kinase C. Eur J Pharmacol. 2004;494(2–3):199–204. doi: 10.1016/j.ejphar.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 51.Soloviev AI, Parshikov AV, Stefanov AV. Evidence for the involvement of protein kinase C in depression of endothelium-dependent vascular responses in spontaneously hypertensive rats. J Vasc Res. 1998;35(5):325–31. doi: 10.1159/000025602. [DOI] [PubMed] [Google Scholar]
- 52.Ungvari Z, Csiszar A, Huang A, Kaminski PM, Wolin MS, Koller A. High pressure induces superoxide production in isolated arteries via protein kinase C-dependent activation of NAD(P)H oxidase. Circulation. 2003;108(10):1253–8. doi: 10.1161/01.CIR.0000079165.84309.4D. [DOI] [PubMed] [Google Scholar]
- 53.Hughes-Darden CA, Wachira SJ, Denaro FJ, Taylor CV, Brunson KJ, Ochillo R, Robinson TJ. Expression and distribution of protein kinase C isozymes in brain tissue of spontaneous hypertensive rats. Cell Mol Biol (Noisy-le-grand) 2001;47(6):1077–88. [PubMed] [Google Scholar]
- 54.Schiffrin EL. Endothelin: potential role in hypertension and vascular hypertrophy. Hypertension. 1995;25(6):1135–43. doi: 10.1161/01.hyp.25.6.1135. [DOI] [PubMed] [Google Scholar]
- 55.Kanayama Y, Negoro N, Okamura M, Konishi Y, Nishimura M, Umetani N, Inoue T, Takeda T. Modulation of protein kinase C in aorta of spontaneously hypertensive rats with enalapril treatment. Osaka City Med J. 1994;40(2):83–97. [PubMed] [Google Scholar]
- 56.Bell PD, Mashburn N, Unlap MT. Renal sodium/calcium exchange; a vasodilator that is defective in salt-sensitive hypertension. Acta Physiol Scand. 2000;168(1):209–14. doi: 10.1046/j.1365-201x.2000.00671.x. [DOI] [PubMed] [Google Scholar]
- 57.Ishii H, Jirousek MR, Daisuke K, Takagi C, Xia P, Clermont A, Bursell SE, Kern TS, Ballas LM, Health WF, Stramm LE, Feener EP, King GL. Amelioration of vascular dysfunction in diabetes rats by an oral PKC β inhibitor. Science. 1996;272:728–31. doi: 10.1126/science.272.5262.728. [DOI] [PubMed] [Google Scholar]
- 58.Kelly DJ, Zhang Y, Hepper C, Gow RM, Jaworski K, Kemp BE, Wilkinson-Berka JL, Gilbert RE. Protein kinase C beta inhibition attenuates the progression of experimental diabetic nephropathy in the presence of continued hypertension. Diabetes. 2003;52(2):512–8. doi: 10.2337/diabetes.52.2.512. [DOI] [PubMed] [Google Scholar]
- 59.Bayer AL, Heidkamp MC, Patel N, Porter M, Engman S, Samarel AM. Alterations in protein kinase C isoenzyme expression and autophosphorylation during the progression of pressure overload-induced left ventricular hypertrophy. Mol Cell Biochem. 2003;242:145–52. [PubMed] [Google Scholar]
- 60.Inagaki K, Iwanaga Y, Sarai N, Onozawa Y, Takenaka H, Mochly-Rosen D, Kihara Y. Tissue angiotensin II during progression or ventricular hypertrophy to heart failure in hypertensive rats; differential effects on PKC epsilon and PKC beta. J Mol Cell Cardiol. 2002;34(10):1377–85. doi: 10.1006/jmcc.2002.2089. [DOI] [PubMed] [Google Scholar]
- 61.McCarty MF. Up-regulation of intracellular signalling pathways may play a central pathogenic role in hypertension, atherogenesis, insulin resistance, and cancer promotion--the 'PKC syndrome'. Med Hypotheses. 1996;46(3):191–221. doi: 10.1016/s0306-9877(96)90243-1. [DOI] [PubMed] [Google Scholar]