

Abstract
Sublethal exposure to Escherichia coli endotoxin (lipopolysaccharide, LPS) attenuates the lethal effects of subsequent insults associated with oxidative stress, such as higher LPS dose, septic peritonitis and ischemia. Because administration of the antioxidant ascorbate protects against these same insults and injection of dehydroascorbic acid (DHAA) protects against ischemia, the hypothesis that sublethal LPS increases endogenous ascorbate concentration and recycling (i.e. synthesis from DHAA) was tested. Injection of LPS (5 × 106 EU/kg body weight i.p.) in mice caused a temporary inhibition of food intake, which was significant by 20 h and recovered within 3 days. LPS increased ascorbate concentration in adrenal gland, heart, kidney and liver. LPS had similar effects in wild-type and Slc23a2+/− mice despite the latter’s deficiency in the ascorbate transporter SVCT2. In liver, the ascorbate response to LPS was not accompanied by change in glutathione concentration. LPS decreased gulonolactone oxidase activity, which is rate-limiting for de novo synthesis of ascorbate from glucose, but increased the rate of DHAA reduction to ascorbate. In conclusion, sublethal endotoxin increases ascorbate recycling in liver and ascorbate concentration in liver, adrenal gland, heart and kidney. The enhanced rate of ascorbate production from DHAA may protect these organs against the reactive oxygen species produced by subsequent, potentially lethal challenges.
Keywords: ascorbate, dehydroascorbic acid, lipopolysaccharide, gulonolactone oxidase, transport
INTRODUCTION
Lipopolysaccharide (LPS) is a cell wall constituent of Gram-negative bacteria that is detectable in the plasma of septic patients (1). High plasma levels of LPS are associated with excess risk of morbidity and mortality (2). Paradoxically, injection of LPS at sublethal doses attenuates the lethal effects of subsequent challenges by high-dose LPS, septic peritonitis and ischemia, all of which are insults associated with oxidative stress (3–6). Multiple lines of evidence suggest that the delayed protection conferred by LPS is attributable, in part, to enhanced antioxidant defense. For instance, pretreatment with low-dose LPS decreases the hepatic production of reactive oxygen species elicited by high-dose LPS (7). Intraperitoneal injection of sublethal LPS increases reactive oxygen species in liver within 1 h (8) but subsequently increases the activities of antioxidant enzymes (glutathione peroxidase, superoxide dismutase) and decreases lipid peroxidation in this organ (9). In heart, too, sublethal LPS triggers lipid peroxidation within 1 h but then the oxidative stress declines and antioxidant enzyme activities (catalase, glutathione peroxidase, superoxide dismutase) rise above initial levels within 12–24 h (10). These results are consistent with the hypothesis that LPS injection triggers oxidative stress that is followed by augmentation of antioxidant defenses.
Ascorbate is a low-molecular weight antioxidant that protects against the same insults as does LPS-induced tolerance (11–15). At the cellular level, ascorbate mitigates the reactive oxygen species production triggered by LPS and thereby prevents the induction of nitric oxide synthase and excessive production of nitric oxide that worsens oxidative stress in hepatocytes, endothelial cells and brain astrocytes (14,16–18). Additionally, ascorbate acts through redox-sensitive signaling pathways to induce tolerance in the dendritic cells of the immune system (19). Although most evidence is based on administering exogenous vitamin C, endogenous ascorbate has also been shown to protect rodent hepatocytes from lethal oxidative stress (20).
The ascorbate concentrations in cells may be increased through several physiological mechanisms. Enterocytes absorb ascorbate and its oxidation product, dehydroascorbic acid (DHAA), from ingested food (21). The hepatocytes of most animal species (but not humans) synthesize ascorbate de novo from glucose, through a pathway in which gulonolactone oxidase is the rate-limiting enzyme (22). Ascorbate may be exported from these cells to the extracellular fluid (23) and then taken up by other cells that express the ascorbate transporters SVCT1 and SVCT2 (21). Reduction of DHAA to ascorbate (i.e., ascorbate recycling) also increases intracellular ascorbate concentration and protects against ischemia-reperfusion injury in vivo (24,25). Ascorbate recycling may be stimulated by insults that are associated with oxidative stress, such as smoking (26) and administration of the glutathione synthesis inhibitor buthionine sulfoximine (27). Therefore, the present experiments were designed to test the hypothesis that ascorbate recycling and concentration are increased by LPS.
MATERIALS AND METHODS
Mice
The study protocol was approved by the Institutional Animal Care and Use Committee of the University at Buffalo. Male mice were used from a colony of heterozygous SVCT2 knockout (Slc23a2+/−) mice (28) that was maintained in the specific pathogen-free facility of the University at Buffalo. Slc23a2+/− mice are a valuable model because they survive to adulthood, unlike the homozygous Slc23a2−/− mice that die shortly after birth. Genotyping was performed according to the procedure described previously (29). The mice were allowed free access to water and a commercial diet (PicoLab Mouse Chow 20). The proximate composition of this diet as stated by the manufacturer was 20.5% protein, 9.5% fat and 2.7% crude fiber. It did not contain ascorbic acid and thus negated intestinal absorption of the vitamin. The mice were used for experiments at 11–12 weeks of age. Starting one week before the experiments, the mice were housed individually.
Experimental protocol
Mice were subjected to three studies. The first determined if intraperitoneal injection of lipopolysaccharide (LPS from E. coli 0127:B8, catalog number L3880, 106 EU/mg, Sigma-Aldrich), at doses of 0.2 × 106 and 5 × 106 EU/kg body weight, altered food intake and survival. Wild-type mice received either dose at 1 h before the 12-h dark period. Food intake and survival were monitored at 20–24 h intervals, from 24 h before to 68 h after injection.
The second study examined the effects of LPS and partial SVCT2-deficiency on ascorbate, glutathione and gulonolactone oxidase at 20 h post-injection. Wild-type and SVCT2-deficient (Slc23a2+/−) mice received an intraperitoneal injection of either LPS (5 × 106 EU/kg) or vehicle (sterile phosphate-buffered saline, 5 ml/kg) at 1 h before the dark period. The mice were killed by cervical dislocation at 20 h post-injection. They were immediately decapitated and blood was drained from the body into heparinized vials for 20 s, then the blood was centrifuged to separate the plasma. Liver, heart, spleen, brain, adrenal gland, kidney and hind limb skeletal muscle were also collected, so that the study included organs that express predominantly SVCT1 or SVCT2 (28–31). The heart was blotted to remove blood from its chambers. Some liver samples were used immediately for determination of gulonolactone oxidase activity. Liver samples for RNA analysis were placed in RNAlater (Ambion). The plasma and remaining tissue samples were frozen in liquid nitrogen and then transferred to a −80 °C freezer for storage until analysis for ascorbate and glutathione.
The third study determined the time course of changes in organ ascorbate concentration and the effect of LPS on hepatic ascorbate recycling rate. Wild-type mice were randomly assigned to receive injection of LPS (5 × 106 EU/kg) or vehicle (sterile phosphate-buffered saline, 5 ml/kg) at 1 h before the dark period. The mice were killed by cervical dislocation at 1, 3, 6 or 20 h post-injection and liver, kidney and spleen were collected. From each liver collected at 20 h post-injection, a portion was used for determination of ascorbate recycling rate. The remaining samples were frozen in liquid nitrogen and then transferred to a −80 °C freezer for storage until analysis.
Biochemical analysis
Ascorbate was extracted from plasma and tissues and its concentration was measured by HPLC with electrochemical detection, according to a procedure we described previously (11). Hepatic ascorbate content was calculated as the product of the ascorbate concentration times the organ weight. The concentrations of total glutathione (i.e. reduced glutathione and glutathione disulfide combined) and glutathione disulfide were measured by enzyme-linked spectrophotometric assay (32,33) in livers that had been stored at −80 °C for up to 2 weeks. Preliminary studies showed that glutathione concentration and redox state were maintained under these storage conditions. The concentration of reduced glutathione was calculated as the difference between the levels of total glutathione and glutathione disulfide.
Gulonolactone oxidase mRNA expression was determined by realtime RT-PCR and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA expression, as described previously (29). Gulonolactone oxidase activity was measured by incubating liver homogenate in phosphate-buffered saline containing L-gulonolactone (5 mmol/L) or vehicle for 30 min at 37 °C (29). The reaction was terminated by rapid freezing and the ascorbate was extracted and assayed by HPLC with electrochemical detection. For each liver, the activity was calculated as the difference between the rates of ascorbate production by the incubate that received L-gulonolactone and the one that did not.
Ascorbate recycling rate was measured by incubating liver homogenate with DHAA (1 mmol/L) in phosphate-buffered saline for 30 min at 37 °C and then assaying the ascorbate concentration by HPLC. DHAA was dissolved immediately before incubation and its contamination by ascorbate was found to be 0.1%. To calculate the recycling rate, the amounts of ascorbate measured in the DHAA solution and in homogenates incubated without DHAA were subtracted from the amount of ascorbate measured in homogenates incubated with DHAA. The rate for each liver was normalized to the concentration of total protein that was measured by a modified Lowry method.
Statistical analysis
Data are presented as means ± SEM and n indicates the number of mice. The homogeneity of variance in the untransformed or cubic transformed data was evaluated by Levene’s test. If homogeneity was observed in the variance then the differences between means were evaluated using 2-way ANOVA, 1-way ANOVA with repeated measures and the Tukey-Kramer multiple comparison test, or the two-tailed t test for pooled samples. If heterogeneity was observed in the variance then the differences between means were evaluated using the nonparametric Mann-Whitney U test. P < 0.05 was considered significant.
RESULTS
LPS at a dose of 5 × 106 EU/kg led to a temporary inhibition of food intake, which was significant at 20 h and recovered at 68 h post-injection, whereas food intake remained constant after the lower dose of LPS (0.2 × 106 EU/kg) (Fig. 1). Neither dose of LPS affected survival, which was 100% at 68 h post-injection.
FIGURE 1.
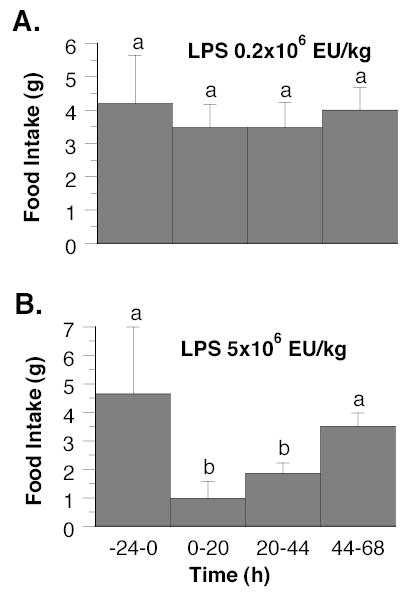
Food intake in wild-type mice from 24 h before to 68 h after intraperitoneal injection at time zero of LPS 0.2 × 106 EU/kg (Panel A) and 5 × 106 EU/kg (Panel B). Values are means ± SEM, n = 4–7. Differences between means were evaluated using either 1-way ANOVA with repeated measures and the Tukey-Kramer multiple comparisons test (Panel A) or the Mann-Whitney U test (Panel B). Within each panel the means without a common letter differ, P < 0.5.
The ascorbate concentrations of plasma and organs that express predominantly the SVCT1 isoform of ascorbate transporter (kidney, liver) are presented in Table 1. Partial SVCT2-deficiency (Slc23a2+/− genotype) did not change the ascorbate concentration in plasma or kidney. Although partial SVCT2-deficiency increased hepatic ascorbate concentration (Table 1), the livers of SVCT2-deficient and wild-type mice did not differ significantly in ascorbate content (2.20 ± 0.18 umol and 1.97 ± 0.14 umol, respectively), organ weight (885 ± 23 mg and 900 ± 27 mg), gulonolactone oxidase:GAPDH mRNA expression ratio (0.722 ± 0.206 and 0.716 ± 0.239) or gulonolactone oxidase activity (70.8 ± 8.8 nmol ascorbate/mg protein and 65.4 ± 3.5 nmol ascorbate/mg protein). The ascorbate concentrations in organs that express predominantly the SVCT2 isoform of ascorbate transporter (adrenal gland, brain, heart, skeletal muscle, spleen) are presented in Table 2. SVCT2-deficiency was associated with decreased ascorbate concentrations in adrenal gland, brain, heart and skeletal muscle.
TABLE 1.
Ascorbate concentrations in plasma and predominantly SVCT1-expressing organs of mice at 20 h after intraperitoneal injection of LPS (5 × 106 EU/kg body weight) or vehicle (5 ml phosphate-buffered saline/kg)1
| Slc23a2 genotype | Treatment | Plasma (umol/L) | Kidney (nmol/mg) | Liver (nmol/mg) |
|---|---|---|---|---|
| +/+ | Vehicle | 97 ± 10 | 0.75 ± 0.03 | 1.86 ± 0.07 |
| +/+ | LPS | 87 ± 6 | 0.75 ± 0.06 | 2.49 ± 0.13 |
| +/− | Vehicle | 77 ± 8 | 0.68 ± 0.05 | 2.02 ± 0.09 |
| +/− | LPS | 85 ± 6 | 0.75 ± 0.05 | 2.92 ± 0.20 |
| P-value2 | ||||
| Genotype | 0.2 | 0.5 | 0.03 | |
| LPS | 0.9 | 0.5 | 0.0001 | |
Values are means ± SEM, n = 5–7.
Differences between means were evaluated by 2-way ANOVA. There were no genotype × LPS interactions.
TABLE 2.
Ascorbate concentrations in predominantly SVCT2-expressing organs of mice at 20 h after intraperitoneal injection of LPS (5 × 106 EU/kg) or vehicle (5 ml phosphate-buffered saline/kg)1
| Slc23a2 genotype | Treatment | Adrenal gland (nmol/mg) | Brain (nmol/mg) | Heart (nmol/mg) | Skeletal muscle (nmol/mg) | Spleen (nmol/mg) |
|---|---|---|---|---|---|---|
| +/+ | Vehicle | 10.36 ± 0.59 | 3.55 ± 0.30 | 0.37 ± 0.04 | 0.32 ± 0.02 | 3.13 ± 0.29 |
| +/+ | LPS | 12.11 ± 0.69 | 3.65 ± 0.26 | 0.49 ± 0.02 | 0.35 ± 0.03 | 2.62 ± 0.29 |
| +/− | Vehicle | 6.87 ± 0.20 | 2.78 ± 0.06 | 0.21 ± 0.01 | 0.22 ± 0.01 | 2.42 ± 0.18 |
| +/− | LPS | 9.12 ± 0.49 | 2.56 ± 0.08 | 0.37 ± 0.04 | 0.24 ± 0.01 | 2.05 ± 0.36 |
| P-value2 | ||||||
| Genotype | 0.0001 | 0.01 | 0.0003 | 0.0001 | 0.3 | |
| LPS | 0.004 | 0.8 | 0.0005 | 0.2 | 0.7 | |
Values are means ± SEM, n = 5–7.
Differences between means were evaluated by 2-way ANOVA (adrenal gland, heart, skeletal muscle, spleen) or the Mann-Whitney U test (brain). There were no genotype × LPS interactions detected by ANOVA.
The effects of LPS (5 × 106 EU/kg) on ascorbate levels were examined at 20 h post-injection in Slc23a2+/− and wild-type mice. No significant interactions were found between genotype and LPS treatment. LPS did not change plasma or renal ascorbate concentration at this time point (Table 1), but it increased hepatic ascorbate concentration by 38% (Table 1) and hepatic ascorbate content by 45% (2.44 ± 0.15 umol for LPS and 1.68 ± 0.06 umol for vehicle, n = 12, P = 0.0001). LPS also increased ascorbate concentrations in adrenal gland and heart by 26% and 42%, respectively, but did not change the concentrations in brain, skeletal muscle or spleen at 20 h (Table 2).
Glutathione was measured in liver and spleen of Slc23a2+/− and wild-type mice at 20 h post- injection, to compare this low-molecular weight antioxidant to ascorbate. Neither partial SVCT2- deficiency nor LPS treatment altered the concentrations of total or reduced glutathione in these organs (Table 3). Thus the effects of SVCT2-deficiency and LPS were different for glutathione than for ascorbate.
TABLE 3.
Total glutathione and reduced glutathione concentrations in liver and spleen of mice at 20 h after intraperitoneal injection of LPS (5 × 106 EU/kg) or vehicle (5 ml phosphate-buffered saline/kg)1
| Slc23a2 genotype | Treatment | Total glutathione in liver (nmol/mg) | Reduced glutathione in liver (nmol/mg) | Total glutathione in spleen (nmol/mg) | Reduced glutathione in spleen (nmol/mg) |
|---|---|---|---|---|---|
| +/+ | Vehicle | 7.38 ± 0.33 | 6.98 ± 0.34 | 2.79 ± 0.13 | 2.49 ± 0.16 |
| +/+ | LPS | 7.61 ± 0.49 | 7.12 ± 0.49 | 2.64 ± 0.23 | 2.46 ± 0.24 |
| +/− | Vehicle | 8.07 ± 0.62 | 7.70 ± 0.56 | 3.09 ± 0.11 | 2.76 ± 0.09 |
| +/− | LPS | 7.07 ± 0.55 | 6.74 ± 0.52 | 2.93 ± 0.11 | 2.60 ± 0.11 |
| P-value2 | |||||
| Genotype | 0.9 | 0.7 | 0.2 | 0.1 | |
| LPS | 0.4 | 0.4 | 0.5 | 0.3 | |
Values are means ± SEM, n = 5–7.
Differences between means were evaluated by 2-way ANOVA. There were no genotype × LPS interactions.
Time-course experiments with wild-type mice showed that LPS (5 × 106 EU/kg) elicited a transient rise in the ascorbate concentration in kidney that was significant at 3 and 6 h (increases of 20% and 25%, respectively) but dissipated by 20 h post-injection (Fig. 2). In these experiments, LPS also elevated ascorbate concentration in liver at 3 and 6 h by, respectively, 40% and 43% (P = 0.0001 at both times), whereas it caused a 17% increase at 20 h that did not achieve significance (P = 0.05, Fig. 2). In spleen, LPS decreased ascorbate concentration transiently at 3 and 6 h post-injection by 22% (P = 0.0001) and 21% (P = 0.0002), respectively (Fig. 2).
FIGURE 2.
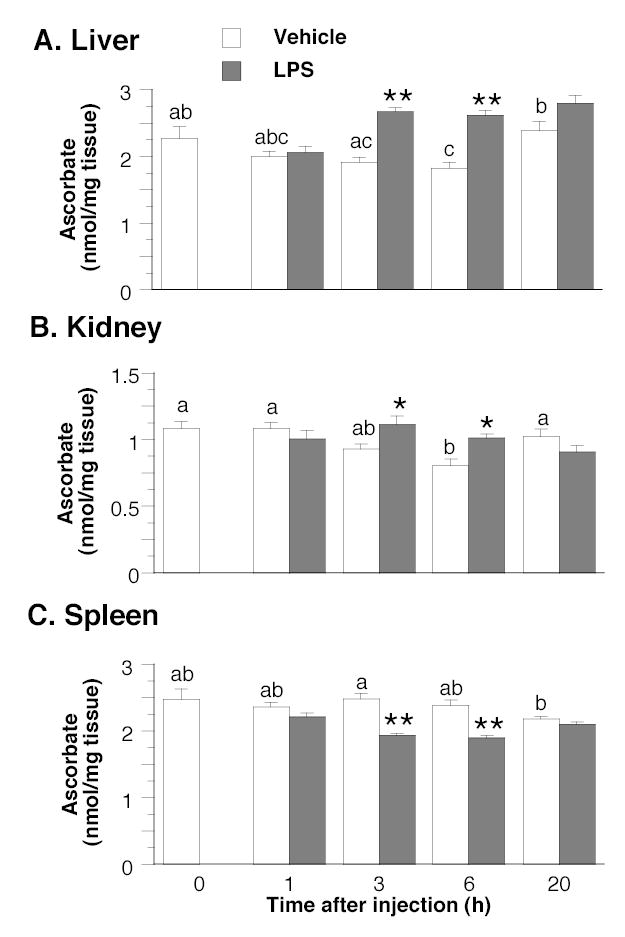
Time course of ascorbate concentrations in liver (Panel A), kidney (Panel B) and spleen (Panel C) of wild-type mice after intraperitoneal injection of LPS (5 × 106 EU/kg) or vehicle (5 ml phosphate-buffered saline/kg). Values are means ± SEM, n = 6. In each panel, the effect of time was evaluated in the vehicle control group by 1-way ANOVA and the Tukey-Kramer multiple comparisons test. Means without a common letter differ, P < 0.5. To assess the effects of LPS, differences between means at each time point were evaluated using the two-tailed t test for pooled samples. Asterixes indicate the times at which significant differences occurred between the LPS and vehicle control groups; *P < 0.05, **P < 0.001.
The effects of LPS on gulonolactone oxidase activity and ascorbate recycling in the livers of wild-type mice were determined at 20 h post-injection. LPS decreased gulonolactone oxidase activity to 51 ± 3 nmol ascorbate/(mg protein 30 min), which was significantly lower than the vehicle-injected control value of 69 ± 5 nmol ascorbate/(mg protein 30 min) (n = 6, P = 0.03). In contrast, LPS stimulated ascorbate recycling, since the rates were 107 ± 4 nmol ascorbate/(mg protein 30 min) in the LPS-injected group and 90 ± 5 nmol ascorbate/(mg protein 30 min) in the vehicle-injected group (n = 6, P = 0.02).
DISCUSSION
The present model found that the ascorbate concentrations in adrenal gland, heart, kidney and liver were increased by LPS (5 × 106 EU/kg i.p.). This dose was sublethal, as indicated by the reversibility of its initial inhibition of food intake and the 100% survival in mice at 68 h post-injection. It is unlikely that the drop in food intake caused the rise in ascorbate concentrations, since previous studies showed that food restriction decreased ascorbate concentrations in adrenal gland and liver (34,35). We did not find evidence that the rise in ascorbate concentration above control was a rebound or overshoot phenomenon triggered by depletion of ascorbate, because time-course experiments showed that ascorbate levels in liver and kidney were unchanged at 1 h and elevated at 3 h post-LPS compared to vehicle control. Although the ascorbate concentration in spleen did fall after sublethal LPS exposure, this change was short-lived and did not lead to an overshoot. Higher doses of LPS depleted ascorbate from the heart and lung of guinea pigs but this may have been a consequence of lethal endotoxemia (36,37).
A stimulatory effect of partial SVCT2-deficiency on hepatic ascorbate concentration was observed in the present experiments but not in an earlier study (28). However, the present and earlier results (28) both supported an important role for SVCT2 in organs that express predominantly this transporter isoform, by showing that ascorbate concentrations in adrenal gland, brain, heart and skeletal muscle were lower for Slc23a2+/− than for wild-type mice. LPS may act independently of SVCT2 to raise ascorbate concentration in adrenal gland, heart, kidney and liver, because no interaction between LPS and SVCT2-deficiency was observed. Even in organs for which SVCT2 is the only known transporter mediating direct uptake of ascorbate, namely the adrenal gland (30) and heart (31), SVCT2-deficiency did not alter the response to LPS.
Ascorbate recycling may be an important pathway for accumulating ascorbate in the adult brain, heart and liver, because exogenous DHAA was more effective than exogenous ascorbic acid for rapidly conferring protection against ischemia-reperfusion injury in these organs (24,25,38). However, the brain differed from heart and liver in its endogenous ascorbate response to systemic LPS. The dose of LPS administered in the present experiments did not alter cerebral ascorbate concentration, possibly because of the blood-brain barrier. Indeed, when low doses of LPS are injected by the intraperitoneal route into normal rodents, the endotoxin may not cross the blood-brain barrier (39).
The liver is an important organ for ascorbate synthesis and storage that contains approximately one-third of the body’s ascorbate content (40). Hepatocytes synthesize ascorbate de novo from glucose (22). However, it is unlikely that de novo synthesis accounted for the elevated ascorbate concentration and content in the livers of LPS-injected mice because we found that gulonolactone oxidase activity was diminished at 20 h post-injection. Although the time course of this change in the hepatic enzyme was not determined, it may have resulted from the decreased food intake induced by LPS because a previous study found that food restriction lowered gulonolactone oxidase activity (34).
Ascorbate recycling is an alterative pathway by which the liver produces ascorbate for local use and export (23). LPS increased the activity of the facilitative glucose transporter isoform mediating DHAA uptake (55 kDa isoform of GLUT1) in hepatocytes (41). Once inside the cells, DHAA can be reduced to ascorbate by glutathione directly or by enzymes that transfer reducing equivalents from glutathione and NAD(P)H. It is not likely that LPS accelerated the reduction of DHAA through increases in the supply of glutathione because we observed no change in the concentrations of total or reduced glutathione in liver at 20 h post-injection. It is possible that LPS altered glutathione levels at earlier time points that were not examined. However, the inference that DHAA reduction rate can increase independently of glutathione is consistent with previous reports that accelerated rates of ascorbate recycling in erythrocytes and skeletal muscle cells were induced by stressors that did not raise intracellular glutathione levels (26,27). In contrast, thioredoxin, thioredoxin reductase and protein disulfide isomerase are important catalysts of intracellular DHAA reduction (21,42,43) and their expression levels were elevated by sublethal doses of LPS (42–44). Taken together, the increases in GLUT1 (41), DHAA-reducing enzymes (44–46) and rate of DHAA reduction (present study) are consistent with the hypothesis that LPS stimulates ascorbate recycling.
In conclusion, sublethal endotoxin increases ascorbate recycling in liver and ascorbate concentration in liver, kidney, adrenal gland and heart. The enhanced rate of ascorbate production from DHAA may protect these organs against the reactive oxygen species produced by subsequent, potentially lethal challenges.
Acknowledgments
The excellent technical assistance of Ewa Jaworski, Marlene MacLean, James Boyer and Lana Burl is gratefully acknowledged.
Footnotes
Supported by grants from NIH (R01KD54728 to SMK) and the University at Buffalo Foundation (to JXW).
References
- 1.Venet C, Zeni F, Viallon A, Ross A, Pain P, Gery P, Page D, Vermesch R, Bertrand M, et al. Endotoxaemia in patients with severe sepsis or septic shock. Intensive Care Med. 2000;26:538–44. doi: 10.1007/s001340051201. [DOI] [PubMed] [Google Scholar]
- 2.Opal SM. The clinical relevance of endotoxin in human sepsis: a critical analysis. J Endotoxin Res. 2002;8:473–6. doi: 10.1179/096805102125001109. [DOI] [PubMed] [Google Scholar]
- 3.Barroso-Aranda J, Schmid-Schonbein GW, Zweifach BW, Mathison JC. Polymorphonuclear neutrophil contribution to induced tolerance to bacterial lipopolysaccharide. Circ Res. 1991;69:1196–206. doi: 10.1161/01.res.69.5.1196. [DOI] [PubMed] [Google Scholar]
- 4.Cavaillon JM, Adrie C, Fitting C, Adib-Conquy M. Endotoxin tolerance: is there a clinical relevance? J Endotoxin Res. 2003;9:101–7. doi: 10.1179/096805103125001487. [DOI] [PubMed] [Google Scholar]
- 5.Ebisawa Y, Kono T, Yoneda M, Asama T, Chisato N, Sugawara M, Ishikawa K, Iwamoto J, Ayabe T, et al. Direct evidence that induced nitric oxide production in hepatocytes prevents liver damage during lipopolysaccharide tolerance in rats. J Surg Res. 2004;118:183–9. doi: 10.1016/S0022-4804(03)00348-2. [DOI] [PubMed] [Google Scholar]
- 6.Echtenacher B, Mannel DN. Requirement of TNF and TNF receptor type 2 for LPS-induced protection from lethal septic peritonitis. J Endotoxin Res. 2002;8:365–9. doi: 10.1179/096805102125000696. [DOI] [PubMed] [Google Scholar]
- 7.Bautista AP, Spitzer JJ. Acute endotoxin tolerance downregulates superoxide anion release by the perfused liver and isolated hepatic nonparenchymal cells. Hepatology. 1995;21:855–62. [PubMed] [Google Scholar]
- 8.Brackett DJ, Lai EK, Lerner MR, Wilson MF, McCay PB. Spin trapping of free radicals produced in vivo in heart and liver during endotoxemia. Free Radic Res Commun. 1989;7:315–24. doi: 10.3109/10715768909087957. [DOI] [PubMed] [Google Scholar]
- 9.Ben-Shaul V, Sofer Y, Bergman M, Zurovsky Y, Grossman S. Lipopolysaccharide-induced oxidative stress in the liver: comparison between rat and rabbit. Shock. 1999;12:288–93. doi: 10.1097/00024382-199910000-00007. [DOI] [PubMed] [Google Scholar]
- 10.Maulik N, Watanabe M, Engelman D, Engelman RM, Kagan VE, Kisin E, Tyurin V, Cordis GA, Das DK. Myocardial adaptation to ischemia by oxidative stress induced by endotoxin. Am J Physiol. 1995;269:C907–16. doi: 10.1152/ajpcell.1995.269.4.C907. [DOI] [PubMed] [Google Scholar]
- 11.Armour J, Tyml K, Lidington D, Wilson JX. Ascorbate prevents microvascular dysfunction in the skeletal muscle of the septic rat. J Appl Physiol. 2001;90:795–803. doi: 10.1152/jappl.2001.90.3.795. [DOI] [PubMed] [Google Scholar]
- 12.Kanter M, Coskun O, Armutcu F, Uz YH, Kizilay G. Protective effects of vitamin C, alone or in combination with vitamin A, on endotoxin-induced oxidative renal tissue damage in rats. Tohoku J Exp Med. 2005;206:155–2. doi: 10.1620/tjem.206.155. [DOI] [PubMed] [Google Scholar]
- 13.Nathens AB, Neff MJ, Jurkovich GJ, Klotz P, Farver K, Ruzinski JT, Radella F, Garcia I, Maier RV. Randomized, prospective trial of antioxidant supplementation in critically ill surgical patients. Ann Surg. 2002;236:814–22. doi: 10.1097/00000658-200212000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu F, Tyml K, Wilson JX. Ascorbate inhibits iNOS expression in endotoxin- and IFN gamma-stimulated rat skeletal muscle endothelial cells. FEBS Lett. 2002;520:122–6. doi: 10.1016/s0014-5793(02)02804-1. [DOI] [PubMed] [Google Scholar]
- 15.Wu F, Wilson JX, Tyml K. Ascorbate protects against impaired arteriolar constriction in sepsis by inhibiting inducible nitric oxide synthase expression. Free Radic Biol Med. 2004;37:1282–9. doi: 10.1016/j.freeradbiomed.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 16.De la Fuente M, Victor VM. Ascorbic acid and N-acetylcysteine improve in vitro the function of lymphocytes from mice with endotoxin-induced oxidative stress. Free Radic Res. 2001;35:73–84. doi: 10.1080/10715760100300611. [DOI] [PubMed] [Google Scholar]
- 17.Kim JY, Lee SM. Effect of ascorbic acid on hepatic vasoregulatory gene expression during polymicrobial sepsis. Life Sci. 2004;75:2015–26. doi: 10.1016/j.lfs.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 18.Korcok J, Wu F, Tyml K, Hammond RR, Wilson JX. Sepsis inhibits reduction of dehydroascorbic acid and accumulation of ascorbate in astroglial cultures: intracellular ascorbate depletion increases nitric oxide synthase induction and glutamate uptake inhibition. J Neurochem. 2002;81:185–93. doi: 10.1046/j.1471-4159.2002.00814.x. [DOI] [PubMed] [Google Scholar]
- 19.Tan PH, Sagoo P, Chan C, Yates JB, Campbell J, Beutelspacher SC, Foxwell BM, Lombardi G, George AJ. Inhibition of NF-{kappa}B and oxidative pathways in human dendritic cells by antioxidative vitamins generates regulatory T cells. J Immunol. 2005;174:7633–44. doi: 10.4049/jimmunol.174.12.7633. [DOI] [PubMed] [Google Scholar]
- 20.Chan TS, Shangari N, Wilson JX, Chan H, Butterworth RF, O’Brien PJ. The biosynthesis of ascorbate protects isolated rat hepatocytes from cumene hydroperoxide-mediated oxidative stress. Free Radic Biol Med. 2005;38:867–73. doi: 10.1016/j.freeradbiomed.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 21.Wilson JX. Regulation of vitamin C transport. Annu Rev Nutr. 2005;25:105–25. doi: 10.1146/annurev.nutr.25.050304.092647. [DOI] [PubMed] [Google Scholar]
- 22.Banhegyi G, Braun L, Csala M, Puskas F, Mandl J. Ascorbate metabolism and its regulation in animals. Free Radic Biol Med. 1997;23:793–803. doi: 10.1016/s0891-5849(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 23.Upston JM, Karjalainen A, Bygrave FL, Stocker R. Efflux of hepatic ascorbate: a potential contributor to the maintenance of plasma vitamin C. Biochem J. 1999;342:49–56. [PMC free article] [PubMed] [Google Scholar]
- 24.De Tata V, Brizzi S, Saviozzi M, Lazzarotti A, Fierabracci V, Malvaldi G, Casini A. Protective role of dehydroascorbate in rat liver ischemia-reperfusion injury. J Surg Res. 2005;123:215–21. doi: 10.1016/j.jss.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 25.Guaiquil VH, Golde DW, Beckles DL, Mascareno EJ, Siddiqui MA. Vitamin C inhibits hypoxia-induced damage and apoptotic signaling pathways in cardiomyocytes and ischemic hearts. Free Radic Biol Med. 2004;37:1419–29. doi: 10.1016/j.freeradbiomed.2004.06.041. [DOI] [PubMed] [Google Scholar]
- 26.Lykkesfeldt J, Viscovich M, Poulsen HE. Ascorbic acid recycling in human erythrocytes is induced by smoking in vivo. Free Radic Biol Med. 2003;35:1439–47. doi: 10.1016/j.freeradbiomed.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 27.Savini I, Catani MV, Duranti G, Ceci R, Sabatini S, Avigliano L. Vitamin C homeostasis in skeletal muscle cells. Free Radic Biol Med. 2005;38:898–907. doi: 10.1016/j.freeradbiomed.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 28.Sotiriou S, Gispert S, Cheng J, Wang Y, Chen A, Hoogstraten-Miller S, Miller GF, Kwon O, Levine M, et al. Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nat Med. 2002;8:514–7. doi: 10.1038/0502-514. [DOI] [PubMed] [Google Scholar]
- 29.Kuo SM, MacLean ME, McCormick K, Wilson JX. Gender and sodium-ascorbate transporter isoforms determine ascorbate concentrations in mice. J Nutr. 2004;134:2216–21. doi: 10.1093/jn/134.9.2216. [DOI] [PubMed] [Google Scholar]
- 30.Takanaga H, Mackenzie B, Hediger MA. Sodium-dependent ascorbic acid transporter family SLC23. Pflugers Arch. 2004;447:677–82. doi: 10.1007/s00424-003-1104-1. [DOI] [PubMed] [Google Scholar]
- 31.Wilson JX, MacLean ME, Jaworski EJ, McCormick K, Kuo SM. The contributions of sodium-dependent vitamin C transporters SVCT1 and SVCT2 to tissue concentrations of ascorbate. FASEB J. 2004;18:A694. [Google Scholar]
- 32.Griffith OW. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal Biochem. 1980;106:207–12. doi: 10.1016/0003-2697(80)90139-6. [DOI] [PubMed] [Google Scholar]
- 33.Paradkar PN, Blum PS, Berhow MA, Baumann H, Kuo SM. Dietary isoflavones suppress endotoxin-induced inflammatory reaction in liver and intestine. Cancer Lett. 2004;215:21–8. doi: 10.1016/j.canlet.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 34.Stubbs DW, Griffin JF. Effects of fasting on gulonolactone hydrolase, gulonate NADP oxidoreductase, and hepatic ascorbate in male and female rats. Proc Soc Exp Biol Med. 1973;144:195–8. doi: 10.3181/00379727-144-37555. [DOI] [PubMed] [Google Scholar]
- 35.Davies JE, Hughes RE. A note on the effect of food restriction on tissue ascorbic acid in guinea-pigs. Br J Nutr. 1977;38:299–300. doi: 10.1079/bjn19770090. [DOI] [PubMed] [Google Scholar]
- 36.Benito E, Bosch MA. Impaired phosphatidylcholine biosynthesis and ascorbic acid depletion in lung during lipopolysaccharide-induced endotoxaemia in guinea pigs. Mol Cell Biochem. 1997;175:117–23. doi: 10.1023/a:1006883628365. [DOI] [PubMed] [Google Scholar]
- 37.Rojas C, Cadenas S, Herrero A, Mendez J, Barja G. Endotoxin depletes ascorbate in the guinea pig heart. Protective effects of vitamins C and E against oxidative stress. Life Sci. 1996;59:649–57. doi: 10.1016/0024-3205(96)00346-3. [DOI] [PubMed] [Google Scholar]
- 38.Huang J, Agus DB, Winfree CJ, Kiss S, Mack WJ, McTaggart RA, Choudhri TF, Kim LJ, Mocco J, et al. Dehydroascorbic acid, a blood-brain barrier transportable form of vitamin C, mediates potent cerebroprotection in experimental stroke. Proc Natl Acad Sci USA. 2001;98:11720–4. doi: 10.1073/pnas.171325998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh AK, Jiang Y. How does peripheral lipopolysaccharide induce gene expression in the brain of rats? Toxicology. 2004;201:197–207. doi: 10.1016/j.tox.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 40.Toutain PL, Bechu D, Hidiroglou M. Ascorbic acid disposition kinetics in the plasma and tissues of calves. Am J Physiol. 1997;273:R1585–97. doi: 10.1152/ajpregu.1997.273.5.R1585. [DOI] [PubMed] [Google Scholar]
- 41.Battelino T, Goto M, Krzisnik C, Zeller WP. Tissue glucose transport and glucose transporters in suckling rats with endotoxic shock. Shock. 1996;6:259–62. doi: 10.1097/00024382-199610000-00006. [DOI] [PubMed] [Google Scholar]
- 42.Li X, Qu ZC, May JM. GSH is required to recycle ascorbic acid in cultured liver cell lines. Antioxid Redox Signal. 2001;3:1089–97. doi: 10.1089/152308601317203594. [DOI] [PubMed] [Google Scholar]
- 43.May JM, Mendiratta S, Hill KE, Burk RF. Reduction of dehydroascorbate to ascorbate by the selenoenzyme thioredoxin reductase. J Biol Chem. 1997;272:2607–10. doi: 10.1074/jbc.272.36.22607. [DOI] [PubMed] [Google Scholar]
- 44.Jung HI, Lim HW, Kim BC, Park EH, Lim CJ. Differential thioredoxin reductase activity from human normal hepatic and hepatoma cell lines. Yonsei Med J. 2004;45:263–72. doi: 10.3349/ymj.2004.45.2.263. [DOI] [PubMed] [Google Scholar]
- 45.Paver JL, Freedman RB, Parkhouse RM. Induction of expression of protein disulphide-isomerase during lymphocyte maturation stimulated by bacterial lipopolysaccharide. FEBS Lett. 1989;242:357–62. doi: 10.1016/0014-5793(89)80501-0. [DOI] [PubMed] [Google Scholar]
- 46.Sano H, Sata T, Nanri H, Ikeda M, Shigematsu A. Thioredoxin is associated with endotoxin tolerance in mice. Crit Care Med. 2002;30:190–4. doi: 10.1097/00003246-200201000-00027. [DOI] [PubMed] [Google Scholar]