

Abstract
A comprehensive approach of metabolite balancing, 13C tracer studies, gas chromatography-mass spectrometry, matrix-assisted laser desorption ionization-time of flight mass spectrometry, and isotopomer modeling was applied for comparative metabolic network analysis of a genealogy of five successive generations of lysine-producing Corynebacterium glutamicum. The five strains examined (C. glutamicum ATCC 13032, 13287, 21253, 21526, and 21543) were previously obtained by random mutagenesis and selection. Throughout the genealogy, the lysine yield in batch cultures increased markedly from 1.2 to 24.9% relative to the glucose uptake flux. Strain optimization was accompanied by significant changes in intracellular flux distributions. The relative pentose phosphate pathway (PPP) flux successively increased, clearly corresponding to the product yield. Moreover, the anaplerotic net flux increased almost twofold as a consequence of concerted regulation of C3 carboxylation and C4 decarboxylation fluxes to cover the increased demand for lysine formation; thus, the overall increase was a consequence of concerted regulation of C3 carboxylation and C4 decarboxylation fluxes. The relative flux through isocitrate dehydrogenase dropped from 82.7% in the wild type to 59.9% in the lysine-producing mutants. In contrast to the NADPH demand, which increased from 109 to 172% due to the increasing lysine yield, the overall NADPH supply remained constant between 185 and 196%, resulting in a decrease in the apparent NADPH excess through strain optimization. Extrapolated to industrial lysine producers, the NADPH supply might become a limiting factor. The relative contributions of PPP and the tricarboxylic acid cycle to NADPH generation changed markedly, indicating that C. glutamicum is able to maintain a constant supply of NADPH under completely different flux conditions. Statistical analysis by a Monte Carlo approach revealed high precision for the estimated fluxes, underlining the fact that the observed differences were clearly strain specific.
Amino acid production by Corynebacterium glutamicum is one of the major processes in industrial biotechnology. The world market for amino acids amounts to several billion dollars, among which lysine, with a worldwide production of about 450,000 tons/year, is one of the most important products (17). The progress made in recent years in the area of metabolic engineering allows the optimization of amino acid-producing strains in a targeted and effective way. The targeted optimization of cellular processes requires a detailed understanding and knowledge of intracellular carbon flux distributions and their regulation. Knowledge of metabolic functioning and regulation is often limited, and its increase is still one of the major bottlenecks in metabolic engineering (23). For lysine-producing C. glutamicum, different metabolic network studies by nuclear magnetic resonance (NMR) and labeling analysis of protein hydrolysates have been carried out in continuous culture and have provided detailed insight into its metabolism (6, 7, 14). However these investigations are limited to selected cases due to relatively high experimental effort. Therefore, the need arises for novel approaches in metabolic network analysis that (i) are applicable to industrially relevant batch or fed-batch cultures, (ii) provide high precision of flux estimates, and (iii) allow comparative flux analysis on a broad scale with relatively low experimental effort. In this context, mass spectrometric techniques recently emerged as an alternative and complementary method to NMR in metabolic network analysis (34).
Development of amino acid overproducers is commonly carried out by random mutagenesis and selection (16). The fact that strain optimization is an iterative procedure of mutagenesis and selection results in whole genealogies ranging from wild-type strains over various steps to industrially applied overproducers. Such genealogies build a great reservoir of successful cellular improvement. They are available in either public or industrial strain collections. Due to the lack of effective methods allowing comparative network analysis with relatively low experimental effort, physiological changes introduced in various amino acid-producing mutants of C. glutamicum have remained unknown (17). Analysis of strain improvement genealogies on the level of metabolic fluxes, however, provides a great chance to increase the knowledge of metabolic functioning and regulation and speed up the process of targeted strain improvement. It enables us to identify the detailed consequences of strain selection for properties such as increased production rate, increased product yield, and decreased maintenance demand for the activity of intracellular pathways. By comparing successive generations of a genealogy, one can trace back the history of strain improvement and identify gradual changes linked to gradual optimization, as well as key changes linked to major improvements in production characteristics. Comparison of different producer strains with similar overall behaviors on the level of metabolic fluxes might help to select a certain strain as most promising for further targeted optimization. Combined with more and more powerful sequencing techniques, it might eventually be possible to attribute observed changes on the metabolic level to specific mutations in the key enzymes involved. Since mutations introduced by random mutagenesis are mainly point mutations, they could then easily be applied to targeted strain improvement (13).
In this context, the present paper describes a novel approach of genealogy profiling by comparative metabolic network analysis. Several consecutive generations of a genealogy of lysine-producing Corynebacterium glutamicum previously obtained by random mutagenesis are analyzed in batch cultures by a straightforward and precise approach of metabolite balancing, 13C tracer studies using gas chromatography-mass spectrometry (GC-MS), and matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) MS and isotopomer modeling. By this approach, significant changes of intracellular pathway activities resulting from strain improvement are identified and provide important information on the metabolism of lysine-producing corynebacteria.
MATERIALS AND METHODS
Strains.
In the present work, a genealogy of lysine-producing C. glutamicum strains was studied. The genealogy comprised the five successive generations C. glutamicum ATCC 13032 (wild type), ATCC 13287, ATCC 21253, ATCC 21526, and ATCC 21543, previously obtained by sequential random mutagenesis by UV irradiation or with chemical mutagens, such as nitrosoguanidine, and selection (12). C. glutamicum ATCC 13287 has a requirement for homoserine. C. glutamicum ATCC 21253 has a requirement for homoserine and leucine. The strains C. glutamicum ATCC 21526 and ATCC 21543 have a requirement for homoserine and leucine and are resistant to the lysine analogue S-β-aminoethyl-cysteine. They were selected via their resistance to the growth-inhibiting action of S-β-aminoethyl-cysteine, which was supplied in the selection medium (12). C. glutamicum ATCC 21253 was obtained from the American Type Culture Collection (Manassas, Va.). The other strains were kindly donated by BASF AG (Ludwigshafen, Germany).
Cultivation and tracer experiments.
Tracer experiments were carried out in 25-ml baffled shake flasks containing 5 ml of defined minimal medium with reduced citrate concentration (32) and (i) 100 mM 99% [1-13C]glucose or (ii) a 100 mM 1:1 mixture of naturally labeled and 99% [13C6]glucose, respectively. In cultivations of C. glutamicum ATCC 21526 and ATCC 21543, the medium was additionally amended with 1 mg of pantothenate liter−1, due to the auxotrophic demand identified for these strains. Five hundred microliters of cells grown overnight on LB5G medium (26) and washed twice with minimal medium served as the inoculum. The flasks were incubated on a rotary shaker at 150 rpm and 30°C. The pH was maintained at 6.5 to 7.0 by the addition of 3 N NaOH. Samples taken during the cultivation were analyzed for concentrations of biomass, substrates, and products. Additionally, culture supernatants taken at the end of the lysine production phase were analyzed for labeling patterns of secreted products by GC-MS (lysine, alanine, and valine) and MALDI-TOF MS (trehalose).
Analytics.
The concentrations of trehalose and different organic acids were measured by high-performance liquid chromatography (Bio-Tek, Neufahrn, Germany) with an Aminex HPX 87-H column (300 by 7.8 mm; Bio-Rad, Hercules, Calif.) and 0.05 N H2SO4 as an isocratic eluent with a flow rate of 0.8 ml min−1 at 45°C. Trehalose was quantified by refractive index detection (model 7515A; ERC, Alteglofsheim, Germany). Organic acids were quantified by UV detection at 210 nm (Bio-Tek). Glucose, pyruvate, and glycerol were analyzed enzymatically (Sigma-Aldrich, Deisenhofen, Germany). Amino acids were separated by high-performance liquid chromatography (Bio-Tek) after precolumn derivatization (AccQ-Tag; Millipore, Milford, Mass.) on a Novapac C18 column (150 by 3.9 mm; Millipore) and were quantified via UV detection at 250 nm (Bio-Tek). Eluents, gradient, flow rate, and column temperature were as specified by the manufacturer (Acc Q-Tag instruction manual; Millipore). Cell density was measured with a spectrophotometer as optical density at 660 nm (OD660) (Pharmacia, Freiburg, Germany) or by gravimetry as cell dry weight. The correlation factors between dry biomass and OD660 in units (grams of dry biomass unit of OD660−1) were determined to be 0.286 (C. glutamicum ATCC 13032), 0.288 (C. glutamicum ATCC 13287), 0.294 (C. glutamicum ATCC 21253), 0.291 (C. glutamicum ATCC 21526), and 0.290 (C. glutamicum ATCC 21543).
GC-MS analysis.
The labeling patterns of extracellular alanine, valine, and lysine were determined by GC-MS after conversion into t-butyl-dimethylsilyl (TBDMS) derivates with dimethyl-t-butyl-silyl-trifluroro-acetamide. For this purpose, 100 μl of cultivation supernatant was lyophilized. The freeze-dried residue was resuspended in 40 μl of dimethylformamide (0.1% pyridine) and 40 μl of N-methyl-t-butyldimethylsilyltrifluoroacetamide (Macherey and Nagel, Easton, Pa.) and incubated at 80°C for 1 h. GC-MS analysis was carried out on a Hewlett-Packard 5890 series II gas chromatograph connected to a Hewlett-Packard 5971 quadrupole mass selective detector (Agilent Technologies, Waldbronn, Germany) with electron impact ionization at 70 eV, and an RTX-5MS column (95% dimethyl-5% diphenylpolysiloxane; 30 m; 320-μm inside diameter; Restek, Bellefonte, Pa.) was used with a column head pressure of 70 kPa and helium as the carrier gas. The column temperature was initially kept at 120°C for 5 min, subsequently increased by 10°C/min up to 270°C, and maintained at that temperature for 4 min. Other temperature settings were 270°C (inlet), 280°C (interface), and 280°C (quadrupole). For analysis, 1 μl of sample was injected. TBDMS-derivatized alanine, valine, and lysine eluted after 7, 12, and 22 min, respectively. All compounds exhibited a high signal intensity for a fragment ion obtained by a mass loss of m-57 from the parent radical due to release of a t-butyl group from the derivatization residue. The fragment ions thus contain the entire carbon skeleton of the corresponding analyte (5). In order to increase the sensitivity, the mass isotopomer fractions m, m + 1, and m + 2 were quantified by selective ion monitoring of the corresponding ion cluster at m/z 260 to 262 (TBDMS-alanine), m/z 288 to 290 (TBDMS-valine), and m/z 431 to 433 (TBDMS-lysine). All measurements were carried out in triplicate.
MALDI-TOF MS analysis.
The mass isotopomer distribution of trehalose in the cultivation supernatant was determined in triplicate by MALDI-TOF MS (Reflex III; Bruker Daltonics, Bremen, Germany) as previously described (32).
Chemicals.
[1-13C]glucose (99%) and [13C6]glucose (99%) were purchased from Euroisotope (Yf-Sur-Lavette, France). Yeast extract and tryptone contained in the LB5G medium were supplied by Difco Laboratories (Detroit, Mich.). All other chemicals were obtained from Sigma-Aldrich and were of analytical grade unless otherwise stated.
Simulations.
All metabolic simulations were carried out on a personal computer using Matlab version 6.1 and Simulink version 3.0 software (Mathworks Inc., Nattick, Mass.).
RESULTS
Network topology.
The topology of the central metabolism of lysine-producing C. glutamicum is displayed in Fig. 1. The metabolic network comprises 48 fluxes, including glycolysis, the pentose phosphate pathway (PPP), the tricarboxylic acid (TCA) cycle, the glyoxylate pathway, pyruvate carboxylation, oxaloacetate decarboxylation, and biosynthesis and secretion of lysine via the two alternative routes of diaminopimelate dehydrogenase and succinylase, respectively. Additionally, biosynthesis and secretion of the by-products glycine, alanine, valine, glutamate, trehalose, acetate, lactate, pyruvate, glycerol, succinate, and α-ketoglutarate are considered (Fig. 1). These compounds were observed in cultivations of the examined strains. A metabolite pool for CO2 was implemented, reflecting its mass isotopomer distribution as a result of the CO2-producing and -consuming reactions in the network. The integration of 13C labeling from CO2 into oxaloacetate-malate in the anaplerotic carboxylation is thus taken into account. The network further contained anabolic fluxes from glucose 6-phosphate, fructose 6-phosphate, ribose 5-phosphate, erythrose 4-phosphate, glyceraldehyde 3-phosphate, 3-phosphoglycerate, the lumped pool of pyruvate-phosphoenolpyruvate, acetyl-coenzyme A (CoA), α-ketoglutarate, oxaloacetate, and diaminopimelate into biomass. Diaminopimelate is considered a separate biomass precursor, because the anabolic fluxes from pyruvate and oxaloacetate into diaminopimelate (cell wall) and lysine (protein) contribute, in addition to the flux of lysine secretion, to the overall flux through the lysine biosynthetic pathway (Fig. 1, ν44). The stoichiometric demand for the different anabolic precursors in relation to the biomass yield was calculated from the biomass composition of C. glutamicum, previously determined by Marx et al. (6). Glucose 6-phosphate isomerase (6, 32), transaldolase and transketolases in the PPP (6), and fumarate hydratase and succinate dehydrogenase in the TCA cycle (7) were previously found to be reversible in C. glutamicum. Accordingly, these reactions are regarded as reversible in the present network. As described previously (32), data in the literature from previous flux measurements for C. glutamicum were taken for the reversibilities of transaldolase and transketolases in the PPP (6).
FIG. 1.

Metabolic network of C. glutamicum, including transport fluxes, fluxes between intermediary metabolite pools, and anabolic fluxes. The following key flux parameters for the production of lysine are highlighted: the flux-partitioning ratios between the PPP and glycolysis (φPPP), between anaplerosis and the TCA cycle (φPC), between dehydrogenase and the succinylase branch in lysine biosynthesis (φDH), and between glyoxylate and the TCA cycle (φICL) and the reversibility of glucose 6-phosphate isomerase (ζPGI), of bidirectional fluxes between C4 metabolites of the TCA cycle and C3 metabolites of glycolysis (ζPC/PEPCK), and of bidirectional fluxes between the pools of oxaloacetate and succinate (ζTCA). DH, diaminopimelate dehydrogenase branch in lysine biosynthesis; SC, succinylase branch in lysine biosynthesis; DAP, diaminopimelic acid; PGI, phosphoglucose isomerase; PC, pyruvate carboxylase; PEPCK, phosphoenolpyruvate carboxylase; ICL, isocitrate lyase; BM, biomass; ΔPDC, Δ1-piperidine-2,6-dicarboxylate. The subscript ex indicates extracellular pools of substrates and products.
Model implementation.
The metabolic network was implemented in Matlab, including a graphical representation of the network in the Simulink toolbox of Matlab and an interactive routine in Matlab for flux estimation, as previously described (31, 32). Additionally, the flux software comprises a novel tool for statistical analysis of obtained metabolic flux distributions using the statistics toolbox of Matlab and a direct link to Excel (Microsoft, Redmond, Wash.), which allows the effective transfer of measurement data from Excel into the flux estimation software and of flux distributions or statistics obtained into Excel for further processing. The metabolic model in Simulink calculates the distribution of 13C labeling in the network for a given set of fluxes and a given labeling of the input tracer substrate using the isotopomer-mapping matrix approach of Schmidt et al. (18). Thus, the corresponding positional isotopomer labels of all metabolites of the network are calculated in vector form by a relaxation method applying the ordinary differential equation algorithm Ode15s. The positional isotopomer distribution vectors calculated by the model are then transformed into mass isotopomer distribution vectors by appropriate transformation matrices and corrected for natural isotopes as described below. The model has proven to be very robust even for highly reversible reactions.
Correction for natural isotopes.
The presence of naturally occurring 13C in the carbon skeleton of all metabolites of the network is considered by appropriate definition of the substrate isotopomer distribution used as input to the simulation model. In the case of derivatized compounds, e.g., those used for GC-MS analysis, naturally occurring 13C in added derivatization residues also has to be taken into account. Moreover, naturally occurring isotopes of oxygen, nitrogen, hydrogen, and silicon in all of the MS analytes have to be considered. To this end, mass isotopomer distribution vectors obtained from the simulations are multiplied with suitable correction matrices (27). The data on natural isotope abundance are taken from Rosman and Taylor (15). By this approach, simulated mass isotopomer distributions are corrected for the presence of natural isotopes and can thus be directly compared with experimental mass isotopomer distributions in parameter estimation.
Parameter estimation.
For parameter estimation, the isotopomer model calculating the 13C label distribution in the network was coupled with an iterative optimization algorithm, in which the Matlab function fmincon for multidimensional constrained nonlinear minimization was applied as previously described (32). In each optimization step, metabolite balancing was automatically performed for all branch points of the network. The algorithm involved two parallel metabolic networks for calculation of the 13C label distribution in order to consider the labeling data from the two parallel tracer experiments for each strain with (i) [1-13C]glucose and (ii) a mixture of naturally labeled and 99% [13C6]glucose in the same parameter optimization. Following previous procedures for metabolic flux analysis from MS data (32), the different mass isotopomer labelings from the MS analysis of the two parallel experiments were considered as experimental data in the parameter optimization. As described in the appendix, the network was overdetermined. A least-squares approach was therefore possible. A weighted sum of least squares (SLS) was used as the error criterion (equation 1).
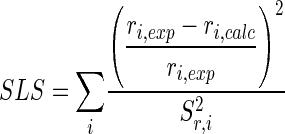 |
(1) |
The differences between experimental (rexp) and calculated (rcalc) mass isotopomer ratios were normalized, and the resulting relative experimental errors of the corresponding MS measurements (sr,i) were used for weighting. By this approach, data with relatively small errors contributed to a greater extent, whereas data with a relatively high uncertainty had a minor influence on the overall optimization result. Since the nonlinear structure of isotopomer models potentially leads to local minima (30), multiple parameter initialization was used to investigate whether an obtained flux distribution represented a global optimum. The lower and upper boundaries of free flux-partitioning ratios (φA) were 0 (for 0% flux into branch A) and 1 (for 100% flux into branch A). For flux reversibilities (ζ), the boundaries were set at 0 (irreversible) and 25 (highly reversible). Thus, a backflux can reach the 25-fold value compared to the net flux of the reversible reaction under consideration.
Statistical evaluation.
Statistical analysis aiming at the quantification of accuracy and confidence for the intracellular flux distributions obtained from the corresponding data sets for the different strains was carried out using a Monte Carlo approach. Monte Carlo approaches can provide precise information on the error distribution of flux parameters (10, 19). For the present work, statistical analysis is of great importance in order to identify whether differences observed among intracellular flux distributions for the examined mutants can really be attributed to strain-specific differences. For each strain, the statistical analysis was carried out by multiple parameter estimation runs, in which the experimental data, comprising measured mass isotopomer ratios and measured fluxes, and fixed flux parameters were varied statistically. The statistical variation was done such that random errors were added to the data sets, assuming a normal distribution of measurement errors around previously obtained mean values. The normally distributed random errors were generated using the statistics toolbox of Matlab. The errors considered were the errors of the triplicate measurements of the MS analysis and the deviation observed between the two parallel incubations for each strain. By this approach, the statistical analysis yields information on accuracy and confidence directly related to the corresponding tracer experiments performed. For the reversibilities of the PPP enzymes, standard deviations of ±20% were assumed. Subsequently, 250 independent parameter estimations were carried out for each strain, yielding 250 flux distributions with a corresponding mean value and a standard deviation for each intracellular flux parameter, from which 90% confidence limits for the single parameters were calculated according to the method of Wiechert et al. (30).
Characteristics of growth and product formation.
The cultivation of all of the strains examined showed a typical two-phase profile with an initial phase of exponential growth from about hours 0 to 7 and a phase of lysine production from hours 7 to 25. This is exemplified for C. glutamicum ATCC 13032, ATCC 23287, and ATCC 21543 (Fig. 2). Drastic differences were observed in the formation of the main product, lysine, and biomass. While the wild-type strain, C. glutamicum ATCC 13032, produced only 1 mM lysine in 25 h, much higher lysine concentrations of 15 and 16 mM were observed for C. glutamicum ATCC 13287 and ATCC 21543, respectively (Fig. 2). The beginning of lysine secretion coincided with the depletion of threonine, known as an inhibitor of the aspartokinase in the lysine biosynthetic pathway, from the medium (Fig. 2). The wild-type strain, C. glutamicum ATCC 13032, continued to grow until the glucose was completely consumed (Fig. 2A). In contrast, all other strains, which are hsd-negative mutants and therefore cannot grow in the absence of threonine, showed a distinct reduction of growth and substrate uptake during the phase of lysine production, as shown for C. glutamicum ATCC 13287 and ATCC 21543 (Fig. 2B and C). The observed doubling of the biomasses of C. glutamicum ATCC 13287 and ATCC 21543 after the depletion of threonine can be explained by the utilization of endogenous threonine (26). The main lysine formation activity of C. glutamicum ATCC 13287 was observed from hours 7 to 15 (Fig. 2B). From hours 15 to 25, lysine formation was weak for this strain. In contrast, C. glutamicum ATCC 21543 showed a rather linear increase in lysine concentration during the whole production phase. Cultivation of C. glutamicum ATCC 21253 and 21526 revealed a typical two-phase profile similar to that of C. glutamicum ATCC 21543 (data not shown). Accordingly, lysine production was characterized by reduced substrate uptake and an almost linear increase of the lysine concentration. The measured yields of all strains during the lysine production phase from hours 7 to 25, which was of central interest in the present work, are given in Table 1. The data shown are mean values from two parallel incubations, (i) on [1-13C]glucose and (ii) on a mixture of naturally labeled and [13C6]glucose, respectively. As shown, the strains differed markedly in the stoichiometry of growth and product formation. The biomass yield of the parent strain, C. glutamicum ATCC 13032, was 60.7 mg (dry mass) of cells mmol of glucose−1 and thus was significantly higher than the values of the succeeding generations. This is mainly due to the auxotrophy for homoserine introduced in the latter strains, causing a growth reduction with the depletion of threonine and methionine in the medium. The biomass yields among the different mutants were rather similar, with values between 39.4 and 43.3 mg (dry mass) of cells mmol of glucose−1. The most remarkable difference between the parent strain, C. glutamicum ATCC 13032, and the succeeding mutants of the genealogy is the achieved increase in lysine yield from 11.8 mmol mol of glucose−1 to values between 173.1 (for C. glutamicum ATCC 13287) and 249.2 (for C. glutamicum ATCC 21543) mmol mol of glucose−1 related to strain optimization (Table 1). Thus, the product yield increased by >20-fold through five cycles of random mutagenesis and selection. However, the increase in yield was not equally distributed over the single cycles but mainly showed two major steps, after the first and fourth mutagenesis selection cycles, which were accompanied by a 15-fold and a further 1.5-fold increase in yield, respectively. In addition to lysine, different by-products, such as other amino acids, organic acids, and the disaccharide trehalose, were formed (Table 1). The by-products stemmed from different parts of the central metabolism, such as glucose 6-phosphate (trehalose), the upper glycolysis (glycine and glycerol), pyruvate (alanine, valine, lactate, and pyruvate), acetyl-CoA (acetate), and the TCA cycle (glutamate, α-ketoglutarate, and succinate). In comparison to the wild-type strain, the formation of by-products was decreased significantly in all succeeding generations of lysine-producing mutants. This is reflected in the amounts of carbon secreted in the form of by-products during the phase of lysine production. Whereas C. glutamicum ATCC 13032 secreted 62.3 mol carbon of by-product during the lysine production phase, only 35.4 to 46.3 mol carbon of by-product was produced by the other strains. This was mainly due to a marked decrease in compounds originating from pyruvate and from the TCA cycle, while the formation of by-products from glyceraldehyde stayed relatively constant in all strains. Interestingly, the mutants with increased lysine production revealed >2-fold-higher trehalose secretion than C. glutamicum ATCC 13032. Furthermore, in regard to the stoichiometry, the measured yields in the parallel flasks for each strain showed excellent agreement, as depicted by the deviation between the two parallel flasks (Table 1). It can be concluded that the cells were provided with comparable conditions in parallel flasks and thus revealed comparable characteristics of growth, substrate uptake, and product formation. Therefore, the consideration of combined labeling data from the two parallel experiments for the calculation of the intracellular flux distribution is justified for each strain. The provision of identical conditions in the parallel flasks was aimed at in the experimental setup, where the only difference between two parallel flasks for a strain was the addition of the stock solution of the corresponding tracer substrate.
FIG. 2.

Cultivation profiles of C. glutamicum ATCC 13032 (A), ATCC 13287 (B), and ATCC 21543 (C) in batch culture on defined medium with 100 mM glucose as a carbon source. The concentrations of glucose (glc), biomass (CDM), lysine (lys), and threonine (thr) are shown. The data are mean values of two parallel cultivations.
TABLE 1.
Stoichiometry of successive generations of a genealogy of lysine-producing C. glutamicum strains during lysine production phase
| Product | Yield for C. glutamicuma:
|
||||
|---|---|---|---|---|---|
| ATCC 13032 | ATCC 13287 | ATCC 21253 | ATCC 21526 | ATCC 21543 | |
| Biomass | 60.65 ± 1.94 | 43.26 ± 1.10 | 39.40 ± 1.00 | 42.84 ± 1.05 | 41.35 ± 0.10 |
| Lysine | 11.84 ± 0.09 | 173.13 ± 5.75 | 199.85 ± 6.64 | 183.67 ± 6.86 | 249.15 ± 6.20 |
| Valine | 50.26 ± 0.70 | 6.71 ± 0.33 | 23.07 ± 1.12 | 19.96 ± 0.49 | 19.90 ± 0.68 |
| Alanine | 41.01 ± 2.09 | 7.49 ± 0.17 | 12.55 ± 0.29 | 16.72 ± 0.66 | 10.57 ± 0.67 |
| Glycine | 7.86 ± 0.34 | 29.67 ± 1.25 | 31.45 ± 1.33 | 4.94 ± 0.21 | 48.37 ± 3.98 |
| Glutamate | 0.32 ± 0.15 | 0.24 ± 0.02 | 0.23 ± 0.02 | 0.00 ± 0.00 | 0.22 ± 0.04 |
| Glycerol | 47.55 ± 2.34 | 31.23 ± 0.28 | 29.75 ± 0.27 | 56.91 ± 3.70 | 31.51 ± 2.39 |
| Trehalose | 5.04 ± 0.18 | 11.57 ± 1.03 | 14.71 ± 1.31 | 14.35 ± 1.13 | 12.48 ± 1.05 |
| α-Ketoglutarate | 7.50 ± 0.68 | 3.12 ± 0.33 | 13.42 ± 1.40 | 5.41 ± 0.13 | 6.60 ± 0.64 |
| Succinate | 25.15 ± 3.60 | 5.44 ± 0.43 | 35.64 ± 2.81 | 0.33 ± 0.00 | 16.56 ± 1.06 |
| Acetate | 24.66 ± 0.21 | 10.59 ± 0.03 | 14.57 ± 0.04 | 24.02 ± 0.89 | 6.39 ± 0.85 |
| Lactate | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 35.34 ± 4.87 | 1.21 ± 0.18 |
| Pyruvate | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 | 19.29 ± 0.03 | 0.00 ± 0.00 |
Experimental data are given as mean values of two parallel incubations on (i) [1-13C]glucose and (ii) a 1:1 mixture of naturally labeled and [13C6]glucose with corresponding deviations between the two incubations. All yields are given in millimoles of product moles of glucose−1 except the yield for biomass, which is given in milligrams of dry biomass millimoles of glucose−1.
13C labeling analysis by MS.
To elucidate intracellular flux distributions in the different strains, mass spectrometric analysis of the secreted products alanine, valine, lysine, and trehalose was carried out. The labeling patterns of alanine, valine, and lysine were analyzed in selective ion-monitoring mode by GC-MS after conversion into the corresponding TBDMS derivates, whereas labeling analysis of trehalose was carried out by MALDI-TOF MS. The results for the tracer experiments with (i) [1-13C]glucose and (ii) a mixture of naturally labeled and [13C6]glucose are given in Tables 2 and 3. A manual inspection of the mass isotopomer ratios obtained allows interesting conclusions about the metabolic profiles of the strain genealogy examined. For example, the mass isotopomer fractions of alanine clearly differ in the wild-type strain, C. glutamicum ATCC 13032, and C. glutamicum ATCC 21543 grown on [1-13C]glucose (Table 2). Compared to C. glutamicum ATCC 13032, the singly and doubly labeled fractions of alanine were clearly decreased in C. glutamicum ATCC 21543. Based on the fact that, from the entry point of glucose into the metabolism to the secretion point of alanine, the 13C label of the applied tracer substrate [1-13C]glucose is specifically lost in the decarboxylation reaction of the PPP, an increase of flux through the PPP results in a decrease of 13C-labeled mass isotopomer fractions (34). The labeling patterns of alanine thus shed light on the increased fluxes through the PPP in lysine-producing C. glutamicum ATCC 21543 compared to the wild type. This is confirmed by the corresponding labeling patterns of valine and lysine from the tracer experiment with [1-13C]glucose. In comparison, the data for the other strains range between those for the wild-type strain and C. glutamicum ATCC 21543. For the observed lysine yields (Table 1), this shows that the flux through the PPP might be coupled to the flux through the lysine biosynthetic pathways. Measurement of the trehalose labeling from the tracer experiment with [1-13C]glucose as the tracer substrate revealed the clear presence of the m + 1 fraction of single-labeled trehalose (Table 1). For all strains, the m + 1 fraction amounted to ∼30% of the fraction of double-labeled trehalose. As described previously, m + 1 mass isotopomers of trehalose are exclusively formed by the reversible action of glucose 6-phosphate isomerase (33). Thus, glucose 6-phosphate isomerase seemed to be highly reversible in all of the strains examined. As shown by the small deviations resulting from the triplicate measurements performed, the GC-MS analysis of TBDMS-derivatized alanine, valine, and lysine allowed a rather precise estimation of the mass isotopomer ratios. In comparison, slightly higher deviations resulted for the labeling analysis of trehalose by MALDI-TOF MS.
TABLE 2.
13C mass isotopomer ratios of secreted products alanine, valine, lysine, and trehalose in successive generations of a genealogy of lysine-producing C. glutamicum strains cultivated on [1-13C]glucose
| Analyte | Mass isotopomer ratio | Source | Value for C. glutamicuma:
|
||||
|---|---|---|---|---|---|---|---|
| ATCC 13032 | ATCC 13287 | ATCC 21253 | ATCC 21526 | ATCC 21543 | |||
| Alanine | m + 1/m | Exp | 0.75 ± 0.02 | 0.73 ± 0.02 | 0.70 ± 0.02 | 0.70 ± 0.02 | 0.67 ± 0.02 |
| Calc | 0.74 | 0.69 | 0.72 | 0.68 | 0.64 | ||
| m + 2/m | Exp | 0.24 ± 0.01 | 0.22 ± 0.01 | 0.20 ± 0.01 | 0.20 ± 0.00 | 0.19 ± 0.01 | |
| Calc | 0.23 | 0.21 | 0.22 | 0.21 | 0.20 | ||
| m + 2/m + 1 | Exp | 0.32 ± 0.00 | 0.30 ± 0.01 | 0.28 ± 0.00 | 0.29 ± 0.01 | 0.29 ± 0.01 | |
| Calc | 0.31 | 0.31 | 0.31 | 0.31 | 0.31 | ||
| Valine | m + 1/m | Exp | 1.24 ± 0.01 | 1.16 ± 0.01 | 1.19 ± 0.00 | 1.18 ± 0.04 | 1.06 ± 0.04 |
| Calc | 1.24 | 1.14 | 1.19 | 1.12 | 1.04 | ||
| m + 2/m | Exp | 0.62 ± 0.01 | 0.56 ± 0.01 | 0.57 ± 0.01 | 0.57 ± 0.02 | 0.48 ± 0.02 | |
| Calc | 0.61 | 0.53 | 0.57 | 0.52 | 0.46 | ||
| m + 2/m + 1 | Exp | 0.50 ± 0.01 | 0.48 ± 0.01 | 0.48 ± 0.01 | 0.48 ± 0.01 | 0.45 ± 0.01 | |
| Calc | 0.49 | 0.47 | 0.48 | 0.46 | 0.44 | ||
| Lysine | m + 1/m | Exp | 1.70 ± 0.03 | 1.57 ± 0.02 | 1.57 ± 0.04 | 1.55 ± 0.01 | 1.46 ± 0.01 |
| Calc | 1.72 | 1.59 | 1.63 | 1.56 | 1.46 | ||
| m + 2/m | Exp | 1.24 ± 0.04 | 1.12 ± 0.01 | 1.11 ± 0.03 | 1.08 ± 0.04 | 0.98 ± 0.02 | |
| Calc | 1.31 | 1.14 | 1.19 | 1.10 | 0.98 | ||
| m + 2/m + 1 | Exp | 0.73 ± 0.02 | 0.72 ± 0.01 | 0.71 ± 0.01 | 0.70 ± 0.03 | 0.67 ± 0.01 | |
| Calc | 0.76 | 0.71 | 0.73 | 0.70 | 0.67 | ||
| Trehalose | m + 2/m + 1 | Exp | 2.64 ± 0.13 | 2.39 ± 0.12 | 2.22 ± 0.11 | 2.36 ± 0.12 | 2.23 ± 0.11 |
| Calc | 2.72 | 2.47 | 2.30 | 2.37 | 2.32 | ||
Experimental data with standard deviation (Exp) obtained by GC-MS (for TBDMS-derivatized alanine, valine, and lysine) or MALDI-TOF MS (for trehalose) and values predicted by the solution of the mathematical model corresponding to the optimized set of fluxes (Calc).
TABLE 3.
13C mass isotopomer ratios of secreted products alanine, valine, and lysine in successive generations of a genealogy of lysine-producing C. glutamicum strains cultivated on a 1:1 mixture of naturally labeled and [13C6]glucose
| Analyte | Mass isotopomer ratio | Source | Value for C. glutamicuma:
|
||||
|---|---|---|---|---|---|---|---|
| ATCC 13032 | ATCC 13287 | ATCC 21253 | ATCC 21526 | ATCC 21543 | |||
| Alanine | m + 1/m | Exp | 0.43 ± 0.02 | 0.43 ± 0.01 | 0.41 ± 0.00 | 0.41 ± 0.01 | 0.40 ± 0.02 |
| Calc | 0.41 | 0.40 | 0.40 | 0.41 | 0.40 | ||
| m + 2/m | Exp | 0.35 ± 0.01 | 0.35 ± 0.01 | 0.35 ± 0.01 | 0.34 ± 0.01 | 0.33 ± 0.01 | |
| Calc | 0.33 | 0.32 | 0.32 | 0.33 | 0.32 | ||
| m + 2/m + 1 | Exp | 0.81 ± 0.01 | 0.81 ± 0.03 | 0.86 ± 0.02 | 0.86 ± 0.03 | 0.84 ± 0.02 | |
| Calc | 0.80 | 0.80 | 0.80 | 0.81 | 0.80 | ||
| Valine | m + 1/m | Exp | 0.59 ± 0.01 | 0.55 ± 0.01 | 0.57 ± 0.01 | 0.56 ± 0.02 | 0.55 ± 0.01 |
| Calc | 0.60 | 0.59 | 0.58 | 0.59 | 0.57 | ||
| m + 2/m | Exp | 1.29 ± 0.01 | 1.29 ± 0.03 | 1.24 ± 0.01 | 1.27 ± 0.04 | 1.33 ± 0.02 | |
| Calc | 1.28 | 1.27 | 1.27 | 1.32 | 1.27 | ||
| m + 2/m + 1 | Exp | 2.20 ± 0.02 | 2.34 ± 0.04 | 2.18 ± 0.05 | 2.24 ± 0.05 | 2.41 ± 0.04 | |
| Calc | 2.15 | 2.17 | 2.18 | 2.23 | 2.24 | ||
| Lysine | m + 1/m | Exp | 1.61 ± 0.05 | 1.58 ± 0.01 | 1.46 ± 0.02 | 1.50 ± 0.03 | 1.43 ± 0.02 |
| Calc | 1.55 | 1.58 | 1.48 | 1.52 | 1.42 | ||
| m + 2/m | Exp | 2.44 ± 0.04 | 2.41 ± 0.01 | 2.25 ± 0.04 | 2.32 ± 0.03 | 2.14 ± 0.06 | |
| Calc | 2.38 | 2.41 | 2.24 | 2.30 | 2.13 | ||
| m + 2/m + 1 | Exp | 1.51 ± 0.02 | 1.53 ± 0.00 | 1.54 ± 0.04 | 1.54 ± 0.03 | 1.50 ± 0.02 | |
| Calc | 1.53 | 1.53 | 1.51 | 1.51 | 1.50 | ||
Experimental data with standard deviation (Exp) obtained after TBDMS derivatization by GC-MS and values predicted by the solution of the mathematical model corresponding to the optimized set of fluxes (Calc).
Estimation of intracellular flux distributions.
A central issue of the present work was the comparison of the lysine-producing C. glutamicum genealogy on the level of intracellular fluxes through the central metabolism. For this purpose, the experimental data obtained were used to calculate metabolic flux distributions for each strain by applying the flux estimation software implemented in Matlab as described above. The parameter estimation was carried out by minimizing the deviation between experimental and calculated mass isotopomer ratios. The approach utilized metabolite balancing during each step of the optimization, including (i) stoichiometric data on product secretion (Table 1) and (ii) stoichiometric data on the anabolic demand for biomass precursors (Table 4). The set of intracellular fluxes that gave the minimum deviation between experimental and simulated labeling patterns was taken as the best estimate for the intracellular flux distribution. For all strains examined, identical flux distributions were obtained with multiple initialization values for the flux parameters, suggesting that global minima were identified in all cases. Obviously, excellent agreement between experimentally determined and calculated mass isotopomer ratios was achieved for all five strains examined (Tables 2 and 3). The sum of the squares of the weighted relative deviations between experimental and calculated mass isotopomer ratios (equation 1) were 0.020 (for C. glutamicum ATCC 13032), 0.029 (for C. glutamicum ATCC 13287), 0.040 (for C. glutamicum ATCC 21253), 0.027 (for C. glutamicum ATCC 21526), and 0.022 (for C. glutamicum ATCC 21543). The intracellular flux distributions obtained for all strains are shown in Fig. 3. For all strains, the glucose uptake flux during lysine production was set to 100%, and the other fluxes in the network are given as relative molar fluxes normalized to the glucose uptake flux.
TABLE 4.
Anabolic demand for intracellular metabolites in successive generations of a genealogy of lysine-producing C. glutamicum strains during lysine production phase
| Metabolite | Precursor demand (mmol mol of glucose−1)a
|
||||
|---|---|---|---|---|---|
| ATCC 13032 | ATCC 13287 | ATCC 21253 | ATCC 21526 | ATCC 21543 | |
| Glucose 6-phosphate | 12.43 ± 0.40 | 8.87 ± 0.23 | 8.08 ± 0.10 | 8.78 ± 0.10 | 8.48 ± 0.02 |
| Fructose 6-phosphate | 4.31 ± 0.14 | 3.07 ± 0.08 | 2.80 ± 0.04 | 3.04 ± 0.07 | 2.94 ± 0.01 |
| Pentose 5-phosphate | 53.31 ± 1.71 | 38.03 ± 0.97 | 34.63 ± 0.44 | 37.66 ± 0.92 | 36.35 ± 0.09 |
| Erythrose 4-phosphate | 16.25 ± 0.52 | 11.59 ± 0.30 | 10.56 ± 0.13 | 11.48 ± 0.28 | 11.08 ± 0.03 |
| Glyceraldehyde 3-phosphate | 7.82 ± 0.25 | 5.58 ± 0.14 | 5.08 ± 0.06 | 5.53 ± 0.14 | 5.33 ± 0.01 |
| 3-Phosphoglycerate | 78.42 ± 2.51 | 55.94 ± 1.42 | 50.94 ± 0.65 | 55.40 ± 1.35 | 53.47 ± 0.13 |
| Pyruvate/phosphoenolpyruvate | 120.88 ± 3.88 | 86.22 ± 2.19 | 78.52 ± 0.99 | 85.39 ± 2.09 | 82.42 ± 0.20 |
| α-Ketoglutarate | 103.71 ± 3.33 | 73.98 ± 1.88 | 67.37 ± 0.85 | 73.26 ± 1.79 | 70.71 ± 0.17 |
| Oxaloacetate | 54.83 ± 1.76 | 39.11 ± 1.00 | 35.62 ± 0.45 | 38.73 ± 0.95 | 37.38 ± 0.09 |
| Acetyl-CoA | 151.63 ± 4.86 | 108.16 ± 2.75 | 98.50 ± 1.25 | 107.11 ± 2.62 | 103.38 ± 0.25 |
| Diaminopimelateb | 8.86 ± 0.28 | 6.32 ± 0.16 | 5.75 ± 0.07 | 6.26 ± 0.15 | 6.04 ± 0.01 |
| Lysineb | 12.25 ± 0.39 | 8.74 ± 0.22 | 7.96 ± 0.10 | 8.65 ± 0.21 | 8.35 ± 0.02 |
Experimental data are given as mean values of two parallel incubations on (i) [1-13C]glucose and (ii) a 1:1 mixture of naturally labeled and [13C6]glucose with deviations between the two incubations. The estimation of precursor demands was based on the experimental biomass yield obtained for each strain (Table 1) and the biomass composition previously measured for C. glutamicum (6).
Diaminopimelate and lysine are regarded as separate anabolic precursors. This is due to the fact that anabolic fluxes from pyruvate and oxaloacetate into diaminopimelate (cell wall) and lysine (protein) contribute, in addition to the flux of lysine secretion, to the overall flux through the lysine biosynthetic pathway.
FIG.3.
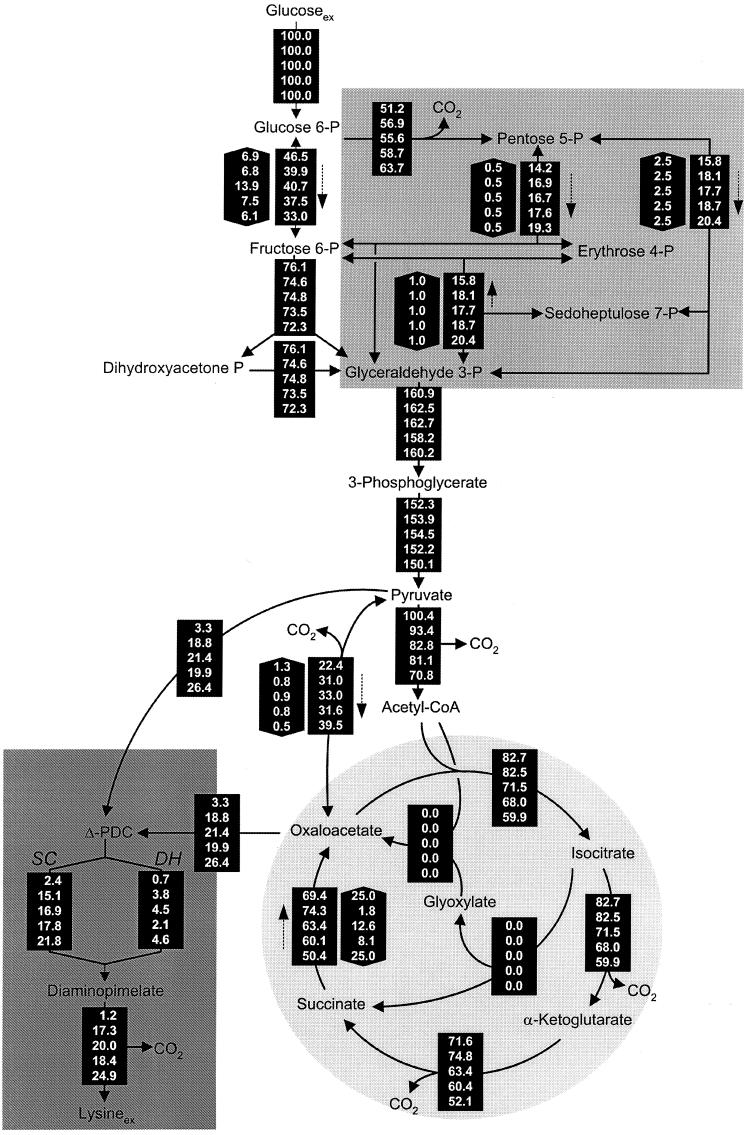
In vivo carbon flux distributions in the central metabolisms of C. glutamicum ATCC 13032, ATCC 13287, ATCC 21253, ATCC 21526, and ATCC 21543 (displayed in that order from top to bottom for each reaction) during the phase of lysine production in batch culture estimated from the best fit to the experimental results using a comprehensive approach of combined metabolite balancing and 13C tracer experiments with labeling measurement of the secreted products lysine, alanine, valine, and trehalose by GC-MS and MALDI-TOF MS, respectively. Net fluxes are given in square boxes; the numbers in hexagonal boxes represent flux reversibilities, and for reversible reactions, the direction of the net flux is indicated by a dashed arrow. All fluxes are expressed as molar percentages of the mean specific glucose uptake rate during lysine production (1.08 mmol g−1 h−1 for ATCC 13032, 1.13 mmol g−1 h−1 for ATCC 13287, 1.13 mmol g−1 h−1 for ATCC 21253, 1.05 mmol g−1 h−1 for ATCC 21526, and 1.19 mmol g−1 h−1 for ATCC 21543).
DISCUSSION
Fluxes at the G6P node into glycolysis and PPP.
As shown in Fig. 3, the wild-type strain, C. glutamicum ATCC 13032, exhibited a relative flux of 51.2% into the PPP, which is equal to a flux-partitioning ratio between PPP and glycolysis (φPPP) of 0.52. In comparison, all lysine-producing mutants exhibited an increased relative flux into the PPP (Fig. 3). Accordingly, φPPP was significantly higher, showing values of 0.59 (for C. glutamicum ATCC 13287), 0.58 (for C. glutamicum ATCC 21253), 0.61 (for C. glutamicum ATCC 21526), and 0.66 (for C. glutamicum ATCC 21543). In all five strains, the carbon flux into the PPP was much higher than the flux required for the anabolic demands of the PPP intermediates ribose 5-phosphate and erythrose 4-phosphate (Table 4). Thus, the major portion of carbon was redirected into glycolysis at the levels of fructose 6-phosphate and glyceraldehyde 3-phosphate. Major carbon fluxes through the PPP were previously quantified for lysine-producing C. glutamicum using NMR spectroscopy (6, 7) and MALDI-TOF MS (32). Interestingly, a strong correlation between the flux through the PPP and the flux through the lysine biosynthetic pathway for product formation and anabolic demand resulted (Fig. 4A). An increased flux into lysine biosynthesis was directly coupled to an enhanced flux through the PPP. The increased NADPH demand in the lysine-producing mutants thus seemed to be compensated for by an increased NADPH supply in the PPP. The overall contribution of the PPP to the NADPH balance in the different strains is discussed in a description of the functioning and regulation of the NADPH metabolism below. As indicated by the small confidence intervals of the fluxes into PPP and glycolysis in all strains (Table 5), an extremely precise determination of the flux partitioning at this node was possible by the chosen comprehensive approach of MS and metabolite balancing. The different flux-partitioning ratios in PPP and glycolysis observed for the different C. glutamicum strains of the genealogy can therefore be clearly described as strain specific. Glucose 6-phosphate isomerase, catalyzing the interconversion of glucose 6-phosphate and fructose 6-phosphate, was found to be highly reversible in all of the strains examined. For the wild-type strain, C. glutamicum ATCC 13032, the anabolic demand for precursors in the PPP and the upper glycolysis was equal to 7.5% of the total glucose entering the cell, while the corresponding demand in the lysine-producing mutants C. glutamicum ATCC 13287, ATCC 21253, ATCC 21526, and ATCC 21543 was only between 5.0 and 5.5% (Table 4). Additionally, these strains, which all exhibited significantly reduced growth, showed a significantly higher trehalose secretion of up to 2.9% of the glucose uptake flux. In contrast, only 1.0% of the glucose was converted into trehalose in C. glutamicum ATCC 13032. Therefore, the decreased amount of carbon, withdrawn from upper glycolysis and the PPP for growth in the lysine-producing mutants, was accompanied by an increased amount of carbon withdrawn from these metabolic reactions in the form of by-products. As a consequence, all five strains revealed very similar fluxes through the reactions of the lower glycolysis between 2-phosphoglycerate and pyruvate. The fluxes through glyceraldehyde 3-phosphate were 160.9 (for C. glutamicum ATCC 13032), 162.5 (for C. glutamicum ATCC 13287), 162.7 (for C. glutamicum ATCC 21253), 158.2 (for C. glutamicum ATCC 21526), and 160.2% (for C. glutamicum ATCC 21543). In summary, the flux through the lower glycolysis showed comparable values in all strains. It cannot be concluded from the given data that the increase of trehalose formation is a direct consequence of the decreased anabolic demands of glucose 6-phosphate, fructose 6-phosphate, and glyceraldehyde 3-phosphate (in the upper glycolysis) and of pentose 5-phosphate and erythrose 4-phosphate (in the PPP). However, it seems possible—assuming a constant capacity of the lower glycolytic reaction chain—that strains of C. glutamicum may secrete excess carbon as by-products such as trehalose under conditions of reduced growth.
FIG. 4.

Impact of strain optimization on metabolic key flux parameters in the central metabolisms of five successive generations of lysine-producing C. glutamicum. (A) Flux through the PPP related to flux through the lysine pathway. (B) Flux-partitioning ratio at the pyruvate (PYR) node (φPC) related to the demand for oxaloacetate (OAA) for anabolism and lysine synthesis. (C) Flux reversibility at the pyruvate node TCA cycle (ζPC/PEPCK) related to flux through the lysine pathway. (D) Flux through isocitrate dehydrogenase related to flux through the lysine pathway. (E) Flux reversibility in the TCA cycle (ζTCA) related to flux through fumarate hydratase. (F) Flux-partitioning ratio at the oxaloacetate node (φOAA) related to flux through the lysine pathway.
TABLE 5.
Statistical evaluation of comparative metabolic flux analysis of a genealogy of lysine-producing C. glutamicum strains by 13C tracer studies with MS and metabolite balancing
| Flux parameter | Value for C. glutamicuma:
|
||||
|---|---|---|---|---|---|
| ATCC 13032 | ATCC 13287 | ATCC 21253 | ATCC 21526 | ATCC 21543 | |
| Fluxes | |||||
| Glucose 6-phosphate dehydrogenase (ν3) | [49.6 52.9] | [55.3 58.7] | [53.4 57.5] | [56.6 61.0] | [61.6 65.8] |
| Glucose 6-phosphate isomerase (ν5 − ν6) | [44.8 48.2] | [38.1 41.5] | [38.8 42.8] | [35.2 39.7] | [30.9 35.1] |
| Aldolase | [75.5 76.8] | [73.9 75.2] | [74.1 75.6] | [72.6 74.3] | [71.6 73.1] |
| Pyruvate kinase (ν20) | [151.0 153.6] | [152.8 154.9] | [153.0 155.9] | [150.8 153.6] | [148.8 151.4] |
| Pyruvate dehydrogenase (ν28) | [97.0 104.8] | [90.8 96.0] | [81.1 84.7] | [78.2 84.4] | [68.4 73.4] |
| Pyruvate carboxylase (ν30) | [45.6 55.7] | [52.7 62.1] | [58.1 65.6] | [54.1 64.8] | [55.4 64.5] |
| Phosphoenolpyruvate carboxykinase (ν31) | [25.7 31.8] | [22.3 30.7] | [25.1 32.6] | [22.9 33.1] | [22.9 33.1] |
| Isocitrate dehydrogenase (ν33) | [77.0 87.2] | [78.4 84.5] | [69.6 73.6] | [64.3 71.7] | [57.2 62.4] |
| Isocitrate lyase (ν34) | [0.0 3.6] | [0.0 0.4] | [0.0 0.0] | [0.0 0.9] | [0.0 0.9] |
| Lysine synthetic pathway (ν44) | [3.2 3.4] | [17.8 19.8] | [21.0 21.7] | [18.6 21.0] | [25.4 27.4] |
| Succinylase branch of lysine synthesis (ν45) | [1.1 1.5] | [14.2 15.7] | [14.9 19.4] | [13.0 18.7] | [19.0 18.7] |
| Dehydrogenase branch of lysine synthesis (ν46) | [0.2 1.1] | [3.3 4.4] | [1.9 6.5] | [1.1 6.9] | [1.0 7.5] |
| Flux partitioning ratios | |||||
| ΦPPP [ν3/(ν3 + ν5 − ν6)] | [0.51 0.54] | [0.57 0.61] | [0.56 0.60] | [0.59 0.63] | [0.64 0.68] |
| ΦPC [(ν30 − ν31)/(ν30 − ν31 + ν28)] | [0.15 0.20] | [0.24 0.26] | [0.28 0.29] | [0.26 0.29] | [0.34 0.37] |
| ΦDH (ν46/(ν45 + ν46) | [0.07 0.33] | [0.18 0.23] | [0.09 0.30] | [0.06 0.34] | [0.04 0.28] |
| Flux reversibilities | |||||
| ζPGI[(ν6/(ν5 − ν6)] | [3.5 11.7] | [4.2 9.7] | [5.7 23.7] | [3.4 11.8] | [4.1 8.6] |
| ζPC/PEPCK [(ν31/(ν30 − ν31)] | [1.1 1.6] | [0.7 1.0] | [0.8 1.0] | [0.7 1.0] | [0.4 0.6] |
| ζTCA [(ν42/ν41 − ν42)] | [10.2 25.0] | [0.7 3.1] | [0.3 25.0] | [0.0 18.8] | [7.1 25.0] |
Mean values and 90% confidence intervals of flux parameters obtained by a Monte Carlo approach, including 250 independent parameter estimation runs with varied experimental data for each strain. The flux numbers according to Fig. 1 are given for the examined reactions.
Fluxes at the pyruvate node: connection among glycolysis, the TCA cycle, and lysine formation.
Pyruvate is one of the key metabolites of the intermediary metabolism of C. glutamicum. It is the connection among glycolysis, the TCA cycle, anaplerosis, lysine synthesis, and the pathways of different by-products (Fig. 1). The flux distributions obtained reveal drastic changes of the fluxes around the pyruvate node through strain optimization (Fig. 3). The fraction of carbon entering the lysine biosynthetic pathway from the pyruvate node exhibited a marked increase in the lysine-producing mutants. Thus, the lysine pathway comprises the anabolic flux toward diaminopimelate and proteinogenic lysine plus the flux of secreted lysine. Whereas a relative flux of 3.3% into the lysine pathway resulted for the wild-type strain, C. glutamicum ATCC 13032, an eightfold-higher value of 26.5% was observed for C. glutamicum ATCC 21543. Due to their lower lysine yields and similar anabolic demands compared to C. glutamicum ATCC 21543, the strains C. glutamicum ATCC 13287, ATCC 21253, and ATCC 21526 showed slightly lower fluxes of 18.8, 21.4, and 19.9%, respectively. Related to the demand for oxaloacetate, mainly for lysine production but also for anabolic purposes, a strong increase in the anaplerotic net flux from 22.4% for C. glutamicum ATCC 13032 to 39.5% for C. glutamicum ATCC 12543 resulted. Since the fluxes entering the pyruvate node from the lower glycolysis were rather similar in all five strains, the increased fluxes into (i) the lysine biosynthetic pathway and (ii) anaplerotic carboxylation caused a significantly decreased flux toward the TCA cycle through pyruvate dehydrogenase. This is exemplified by the flux of 70.8% observed for lysine-producing C. glutamicum ATCC 21543, which was about 30% lower than the value of 100.4% for the wild-type strain, C. glutamicum ATCC 13032 (Fig. 3). The different flux distributions at the pyruvate node in the five generations of the C. glutamicum genealogy examined are depicted by the flux partitioning between pyruvate carboxylase and pyruvate dehydrogenase (φPC) in Fig. 4B. Here, a tight correlation of φPC with the demand for oxaloacetate for lysine production and anabolism becomes obvious. Strain optimization thus resulted in a dramatic redirection of the carbon flux from pyruvate dehydrogenase toward anaplerosis. It is known that C. glutamicum has several enzymes, such as phosphoenolpyruvate carboxykinase and oxaloacetate decarboxylase, that catalyze a C4-decarboxylating backflux from the TCA cycle to glycolysis (14). The anaplerotic net flux in C. glutamicum, therefore, is in fact the result of the concerted action of C3 carboxylation and C4 decarboxylation. This directly raises the question of how forward and backward fluxes contribute to the markedly increased anaplerotic net flux observed. The data clearly show that both forward and backward reactions are involved. The increase of the anaplerotic net flux through the strain genealogy was the combined result of (i) an increased C3 carboxylation flux from pyruvate to oxaloacetate and (ii) a decreased C4 decarboxylation flux in the opposite direction. For example, the 17%-increased net flux in C. glutamicum ATCC 21543 in comparison to C. glutamicum ATCC 13032 is composed of a 9% increase of the forward flux (from 51.0 to 59.9%) and an 8% decrease of the backward flux (28.6 to 20.4%). Accordingly, the reversibility at the pyruvate node, ζPC/PEPCK, decreased significantly with increasing flux through the lysine biosynthetic pathway (Fig. 4C). Isogenic strains of C. glutamicum grown in continuous culture showed a strong correlation between lysine production on one hand and C3-carboxylating and C4-decarboxylating fluxes on the other hand (2). These observed changes in C3 carboxylation and C4 decarboxylation fluxes in relation to lysine production seem to reflect an important regulating phenomenon in C. glutamicum which is active both in batch culture and in continuous culture. Moreover, these findings underline the fact that enzymes catalyzing the fluxes around the pyruvate node are promising candidates for targeted strain optimization. In C. glutamicum, multiple enzymes can catalyze the reactions of C3 carboxylation and C4 decarboxylation (14). Due to the fact that (i) the pools of malate and oxaloacetate in the TCA cycle and (ii) the pools of pyruvate and phosphoenolpyruvate in glycolysis were lumped together, we cannot differentiate on the basis of the present work to which extent the different enzymes contribute to these reactions. With regard to the fact that the flux of anaplerotic carboxylation and the corresponding back reactions exhibit great differences among the strains in the C. glutamicum genealogy, it could be that these flux differences are accompanied by changes in the relative contributions of the different enzymes involved. The previous finding, for lysine-producing C. glutamicum ATCC 21253 grown in chemostat culture, that the ratio of anaplerotic carboxylation to the flux through the lower glycolysis is constant at different lysine yields (4) was not observed in the present work. In contrast, the anaplerotic flux markedly increased through strain optimization, whereas the flux through the lower glycolysis stayed almost constant. This resulted in an increasing ratio of anaplerotic to glycolytic flux. This observation might be due to the facts (i) that metabolic regulation in C. glutamicum might be different under conditions of reduced growth during lysine production in batch culture compared to continuous cultivation and (ii) that mutations affecting the metabolic regulation at the pyruvate node were introduced during random mutagenesis. The statistical analysis of fluxes at the pyruvate node (pyruvate carboxylase, pyruvate dehydrogenase, phosphoenolpyruvate carboxykinase, and the lysine pathway) and the flux parameters calculated from them (φPC and ζPC/PEPCK) showed that the flux parameters at the pyruvate node were determined with high accuracy by the applied MS approach in all of the strains examined, which clearly justifies the conclusions drawn above (Table 5).
TCA cycle.
The wild-type strain, C. glutamicum ATCC 13032, exhibited a high flux of 82.7% through the TCA cycle (Fig. 3). Significantly lower values were found for C. glutamicum ATCC 21253 (71.5%), C. glutamicum ATCC 21526 (68.0%), and C. glutamicum ATCC 21543 (59.9%) (Fig. 3). The reduced growth during lysine production observed for these strains due to the depletion of essential amino acids is therefore directly reflected by a reduced flux through the TCA cycle, thus adapting ATP formation to the reduced demand. Moreover, the obviously reduced TCA cycle flux during lysine production results in a decreased flux catalyzed by isocitrate dehydrogenase with NADPH as a cofactor. Therefore, the TCA cycle cannot be the source responsible for an increased NADPH supply. The 90% confidence intervals for the flux through the TCA cycle are exemplified by isocitrate dehydrogenase (Table 5). The intervals obtained were rather small, underlining the fact that the observed differences were due to the different metabolic properties of the studied strains. The strain C. glutamicum ATCC 13287 showed slightly different characteristics (Fig. 4D). It had a lysine yield similar to those of C. glutamicum ATCC 21253, ATCC 21526, and ATCC 21543 but exhibited a higher flux through the TCA cycle. This might be due to the fact that this strain produced a major part of the lysine during the initial hours of the production phase (Fig. 2 B). This initial production phase is linked to certain growth due to the availability of intracellular threonine and methionine and therefore probably also to an increased TCA cycle flux. The reactions of fumarate hydratase and succinate dehydrogenase were found to be reversible in all of the strains studied (Fig. 3). This is in accordance with previous NMR approaches for C. glutamicum (6, 7). In the present work, ζTCA was limited to values between 0 and 25, because the sensitivity of the determination of this flux parameter was rather low. The relatively large confidence intervals for ζTCA show that the determined values are linked to relatively high uncertainty (Table 5). Taking this into account, at least the following tendency can be extracted from the data. The differences for ζTCA observed for the examined strains could be the result of different intracellular levels of the involved intermediates around the oxaloacetate node. The lowest backflux of ζTCA, 1.8, was for C. glutamicum ATCC 13287, which also had the highest net flux through the TCA cycle toward oxaloacetate. Comparing this strain with the other lysine-producing mutants, C. glutamicum ATCC 21253, ATCC 21526, and ATCC 21543, a correlation between the net flux through the TCA cycle toward oxaloacetate and ζTCA can be seen (Fig. 4E). Previously, Kiss and Stephanopoulos observed a rigid regulation of the carbon flux at the oxaloacetate node in lysine-producing C. glutamicum ATCC 21253, demanding a higher TCA cycle flux under conditions of higher demand for oxaloacetate for lysine biosynthesis and anabolism (4). As shown in Fig. 4F, no rigid regulation was identified at this node when the different strains were compared. The flux partitioning at the oxaloacetate node, φOAA, relating the demand for and supply of oxaloacetate, was not constant but became larger with an increasing requirement for oxaloacetate. A higher demand for oxaloacetate was therefore not linked to a higher overall supply. The value of φOAA (26%) obtained for C. glutamicum ATCC 21253 in the present work was slightly lower than the φOAA (30 to 40%) observed in continuous culture for the same strain (4). As described above for regulation at the pyruvate node, there are two possible explanations for the fact that the oxaloacetate node was not rigid among the strains in the genealogy. First, metabolic regulation in C. glutamicum might be different under the conditions of reduced growth during lysine production in batch culture compared to that under continuous cultivation. Second, it also seems possible that mutations affecting metabolic regulation at the oxaloacetate node were introduced during random mutagenesis. For all five strains, the TCA cycle flux from succinyl CoA to succinate was much higher than the flux through the succinylase branch of lysine biosynthesis (Fig. 3), which indicates that (i) the succinylase branch is not limited by a restricted availability of succinyl CoA and (ii) the conversion of succinyl CoA into succinate is mainly carried out by succinyl CoA synthetase in the TCA cycle and thus is used for energy formation by the cells.
Glyoxylate pathway.
The key enzymes of the glyoxylate cycle for the utilization of acetate have been previously found in C. glutamicum (20). The glyoxylate cycle was found to be active in C. glutamicum during the utilization of acetate and coutilization of glucose and acetate in continuous culture (29). In the present work, all of the C. glutamicum strains examined showed inactive glyoxylate pathways (Fig. 3). Considering the 90% confidence intervals for isocitrate lyase, a small flux through this pathway might be possible (Table 5). The upper limit for the wild-type strain of 3.6% and for the other strains of <1%, however, shows that a significant glyoxylic flux can be excluded. During batch cultivation on glucose, the replenishment of the TCA cycle is therefore mainly carried out via anaplerotic carboxylation. Even under conditions of a high demand for TCA metabolites for lysine secretion in C. glutamicum ATCC 13287, ATCC 21253, ATCC 21526, and ATCC 21543, no replenishment of the TCA cycle via the glyoxylate pathway occurred; instead, a marked increase in the anaplerotic carboxylation of pyruvate was observed (Fig. 3). Recycling of acetate, which was secreted by all strains (Table 1) and accumulated in concentrations up to ∼3 mM in the medium, obviously did not take place. As shown previously for the wild-type strain, C. glutamicum ATCC 13032, isocitrate lyase and malate synthase are drastically suppressed when glucose is used as a primary carbon source (28). The presence of high concentrations of glucose during the batch cultivations performed in the present work is therefore a probable explanation for the observed inactivity of the glyoxylate cycle. Under the conditions chosen, the role of the glyoxylate pathway in refilling of the TCA cycle seems negligible. In continuous culture of different lysine-producing strains of C. glutamicum at low concentrations of glucose, the glyoxylate cycle was found to be inactive (7) or present only at a very low flux of 0.2% (6).
Lysine biosynthesis.
C. glutamicum ATCC 13032 exhibited a flux-partitioning ratio (φDH) of 0.22 (Fig. 3). The two alternative routes were therefore both active in the wild-type strain. As previously observed for this strain in NMR studies, the participation of the two branches in lysine synthesis is a natural phenomenon and not introduced only in overproducing mutants (21). The flux-partitioning ratios between the dehydrogenase and the succinylase pathways were relatively similar in the lysine-producing mutants C. glutamicum ATCC 13287 (φDH = 0.20), ATCC 21253 (φDH = 0.21), and ATCC 21543 (φDH = 0.17). A slightly lower value of φDH, 0.10, was found for C. glutamicum ATCC 21526. In contrast to the overall flux into the lysine pathway, which could be quantified with good accuracy, the flux-partitioning ratio revealed a larger confidence interval, indicating that no significant differences among the different strains were identified (Table 5). In summary, the succinylase pathway was the dominant branch for lysine formation in all strains. The drastically increased relative flux through the lysine biosynthetic pathway in C. glutamicum ATCC 21543, which was about eightfold higher than in the wild type, was not linked to significant changes in the relative contributions of the two branches. Similar flux-partitioning ratios in lysine biosynthesis in the wild-type strain, C. glutamicum ATCC 13032, and two lysine-accumulating mutants were previously found by Sonntag et al. (21).
NADPH metabolism.
The following calculations of demand for and supply of NADPH for the investigated strain genealogy provide an outstanding insight into the NADPH metabolism of lysine-producing C. glutamicum. As previously shown for C. glutamicum, the reactions of glucose 6-phosphate dehydrogenase (24), 6-phosphogluconate dehydrogenase (25), and isocitrate dehydrogenase (3) are coupled to the reduction of NADPH. The overall supply of NADPH can therefore be calculated from the estimated fluxes into the PPP and through isocitrate dehydrogenase in the TCA cycle. Due to the high precision for the determination of these flux parameters in the present work, which are revealed by the statistical analysis (Table 5), a precise estimate of the NADPH supply can be given. The excellent accuracy of the estimates of the fluxes involved in NADPH supply is visualized by a phase plane plot of the fluxes through the PPP and through isocitrate dehydrogenase obtained by the Monte Carlo analysis for C. glutamicum ATCC 13032, ATCC 21253, and ATCC 21543 (Fig. 5). Each ellipsoidal zone obtained includes all 250 independent flux estimates for each strain from the Monte Carlo analysis. Although the three strains differ only gradually in the corresponding fluxes, they can be clearly differentiated. The results for C. glutamicum ATCC 13287 and ATCC 21526 revealed similar small ellipsoids in between the displayed strains (data not shown). The achieved high precision is a great advantage of the present MS approach compared (i) to metabolite balancing alone, which provides no information on NADPH but assumes a closed NADPH balance (26), and (ii) to NMR approaches, which result in much higher uncertainty for fluxes into PPP and glycolysis (7, 19).
FIG. 5.

Statistical analysis for estimates of fluxes into the PPP and through isocitrate (ICI) dehydrogenase for C. glutamicum ATCC 13032 (strain 1), ATCC 21253 (strain 2), and ATCC 21543 (strain 3). The data shown for each strain are the results of 250 flux-independent flux estimations by a Monte-Carlo approach.
NADPH is required for the growth and formation of lysine and by-products, such as alanine, valine, and glycine. The NADPH requirement for growth was estimated from the previously determined NADPH demand of 15.5 mmol of NAPDH g of biomass−1 (7) and the biomass yields for the different strains measured in the present work (Table 1). The amount of NADPH needed for product synthesis was determined from the estimated fluxes into lysine, alanine, valine, and glycine (Table 1) and the corresponding stoichiometric NADPH demand of 4 mol mol−1 for lysine and 1 mol mol−1 for alanine, valine, and glycine (9). The calculated NADPH demand and supply for the examined strain genealogy of lysine-producing corynebacteria is shown in Fig. 6A. Surprisingly, all five strains revealed rather similar fluxes for the NADPH supply of about 190%. Despite the great differences on the level of intracellular metabolic fluxes, all of the strains studied seemed to maintain a constant flux into the supply of NADPH. Interestingly, values similar to the data of the genealogy studied here were previously obtained for batch cultures of C. glutamicum by Sonntag et al. (22), who estimated a molar NADPH supply of 188% during exponential growth of C. glutamicum ATCC 13032 and of 200% during lysine production by an overproducing strain, and by Wittmann and Heinzle (32), who determined a molar NADPH supply of 193% during maximum lysine production for C. glutamicum ATCC 21253. In contrast to the rather constant NADPH supply, a marked increase of the NADPH demand resulted through strain optimization (Fig. 6A). The values ranged from 109% for the wild-type strain, C. glutamicum ATCC 13032, up to 172% for C. glutamicum ATCC 21543, the mutant with the highest lysine yield among the strains studied. All five strains showed an apparent excess of NADPH. In the wild-type strain, C. glutamicum ATCC 13032, there was an enormous apparent NADPH excess of 80%. Moreover, it can be stated that the increased demand for NADPH did not cause an increase in the NADPH supply. The NADPH potential, in fact, became smaller through strain optimization. In contrast to the wild-type strain, the apparent NADPH excess was only 15% in C. glutamicum ATCC 21543. Extrapolating Fig. 6A, it can be speculated that NADPH might indeed become a limiting factor in batch or fed-batch cultures of industrially relevant lysine producers with product yields higher than those of the strains examined here. Despite the relatively constant NADPH supply in all of the different strains, the relative contributions of the PPP and the TCA cycle to NADPH reduction changed significantly during strain optimization, as shown in Fig. 6B. The TCA cycle supplied the major part of the NADPH in the wild-type strain, whereas its contribution to the NADPH supply decreased gradually in the succeeding generations. The opposite effect resulted for the NADPH supply by the PPP. The relative contribution of the PPP thus increased from 55.3 (for C. glutamicum ATCC 13032) to 58.0 (for C. glutamicum ATCC 13287), 61.1 (for C. glutamicum ATCC 21253), 63.3 (for C. glutamicum ATCC 21526), and 68.1% (for C. glutamicum ATCC 21543). In comparison, exponentially growing C. glutamicum ATCC 13032 revealed a higher TCA cycle flux of 108% and a reduced flux of 40% through the PPP (22). Here, the relative contribution of the PPP to the NADPH supply was only 27%. Interestingly, the concerted action of both pathways provided a total NADPH supply of 188%, rather similar to that of the studied genealogy. On the other hand, C. glutamicum ATCC 21253 showed an increased PPP flux of 71% and a decreased flux through isocitrate dehydrogenase of 51% during maximum lysine production (32), resulting in an overall flux of the NADPH supply of 193%, to which the PPP contributed 73.6%. In summary, C. glutamicum seems to be able to compensate for decreased fluxes through isocitrate dehydrogenase due to (i) reduced growth caused by the shortage of essential amino acids and (ii) increased carbon flux into competing fluxes at the pyruvate node for lysine synthesis and for anaplerotic carboxylation by a marked increase of the flux through the PPP. As shown, the relative contributions of the PPP and isocitrate dehydrogenase vary drastically. The decreased apparent NADPH excess, probably linked to decreasing intracellular concentrations of NADPH, might be a reason for the enhanced PPP flux observed, since NADPH is a strong inhibitor of glucose 6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase in C. glutamicum (11). For the depletion of the excess NADPH in C. glutamicum, Marx et al. (6) previously suggested in vivo NADPH regeneration by an oxidase or by malic enzyme. In this context, a potential route for NADPH regeneration was recently identified by Matsushita et al. (8), who showed that the respiratory chain in C. glutamicum is able to oxidize NADPH. Under conditions of normal growth, excess NADPH might therefore be channeled into the respiratory chain. This makes sense, given that isocitrate dehydrogenase, one of the NADPH-producing enzymes, is located in the TCA cycle. The large NADPH potential of 80% for the wild-type strain, C. glutamicum ATCC 13032, could be a key feature to explain the high capacity for amino acid production achieved in mutants derived from this parent strain. Under conditions of reduced growth, as observed for lysine-producing hsd-negative mutants of C. glutamicum in the absence of threonine and methionine linked to decreased respiration activity (26), the enhanced production of NADPH-consuming products such as lysine could act as a valve for the cell to get rid of the large amount of NAPDH.
FIG. 6.

Impact of strain optimization of lysine-producing strains of C. glutamicum on NADPH metabolism. NADPH demand (A) and supply (B) are expressed as molar fluxes related to the glucose uptake flux in five successive generations of a genealogy of lysine producers estimated via metabolic network analysis. The strain numbers represent the different generations C. glutamicum ATCC 13032 (1), ATCC 13287 (2), ATCC 21253 (3), ATCC 21526 (4), and ATCC 21543 (5). ICD, isocitrate dehydrogenase.
Conclusions.
The results of the present work providing precise data on the NADPH metabolism in C. glutamicum clearly show that the demand for and supply of NADPH can differ by more than 80% in relation to the glucose uptake flux. It can be stated that metabolic flux analysis by metabolite balancing relying on a closed balance for NADPH should be treated with great care. Due to the fact that no genomic information is available for the genealogy of lysine-producing corynebacteria examined in the present work, no answer can be given as to which mutations are probably responsible for the observed differences in intracellular fluxes. It can be expected that in addition to the desired mutations, a couple of further mutations are introduced during the various cycles of random mutagenesis performed in strain optimization. The accumulation of side mutations can be clearly deduced from the phenotype of the examined genealogy, where (i) in contrast to preceding generations, C. glutamicum ATCC 21526 and ATCC 21543 were found to be auxotrophic for pantothenic acid and (ii) only the wild-type strain, C. glutamicum ATCC 13032, exhibited yellow pigmentation when grown on agar plates or in liquid culture, whereas cells of the successive generations appeared white to fawn color, which might be linked to a defect in the corresponding biosynthetic pathway. It would be of great interest to investigate whether the observed differences on the metabolic flux level are directly linked to mutations of corresponding enzymes, e.g., in the PPP or at the pyruvate node, or are part of an overall cellular response to only selected key mutations. The impact of the accuracy of 13C labeling analysis by MS on the accuracy of metabolic flux analysis has been shown in this work. For all of the strains, a precise estimation of most intracellular fluxes was achieved. Among the reversibilities, a good quantification of ζPC/PEPCK was achieved. Only ζTCA and ζPGI showed larger confidence intervals. In this context, Wiechert et al. (30) reported that the degree of reversibility can be quantified only with higher uncertainty. It has to be stated that one important factor determining precision in metabolic network analysis is the choice of the tracer substrates. As previously shown, optimal quantification of specific flux parameters demands an appropriate choice of tracer substrates (10, 33). Simulation studies of the experimental design of tracer experiments for C. glutamicum revealed that [1-13C]glucose is optimal for the determination of φPPP and ζPGI, whereas the substrate of choice for measuring fluxes at the pyruvate node, such as φPC and ζPC/PEPCK, is a mixture of naturally labeled glucose and [13C6]glucose (33). In the present case, the combination of two parallel experiments, each with a tracer substrate optimal for a different subset of flux parameters, can be regarded as a useful approach. The larger uncertainty for flux partitioning in lysine biosynthesis could be overcome by the additional choice of, e.g., [4-13C]glucose (33). MS seems to be a favorable method for the determination of flux partitioning between glycolysis and PPP, a key flux parameter in the central metabolism, especially during lysine production by C. glutamicum. This is exemplified by the narrow confidence intervals for fluxes into PPP and glycolysis of <4%. Using NMR, the 90% confidence intervals for these fluxes were substantially (up to 10-fold) larger, even in cases where much more data providing enhanced redundancy were applied, as described for various biological systems (1, 7, 19). Thus, MS offers significantly increased precision for the determination of flux partitioning between glycolysis and PPP compared to NMR. Generally, accuracy and sensitivity make MS a valuable technique in metabolic network analysis. Clearly, metabolic network analysis attains its full potential when applied as a tool for the comparison of different strains or different growth conditions. Taking into account the fact that differences in metabolic fluxes due to mutation or changed environment might be relatively small and that metabolic fluxes can change gradually rather than drastically during repeated improvement of a production strain, precision of flux analysis is of great importance. Moreover, the broad application of metabolic flux analysis on a screening level demands small cultivation and sampling volumes and requires a sensitive method for 13C labeling analysis. The GC-MS and MALDI-TOF MS methods applied in the present work require only about 1 μl of cultivation supernatant for analysis.
The specific glucose uptake rates during lysine production from hours 7 to 25 were rather similar for all strains. Thus, absolute intracellular fluxes calculated on the basis of the corresponding specific glucose uptake rates give a picture similar to the relative flux distributions shown above for the different strains. Therefore, the conclusions drawn above do not significantly change with regard to the overall rates in the different mutants. The approach presented is an integral investigation of the lysine production phase. It provides quantitative information on overall yields, rates, and selectivities, which are important parameters in industrial production processes. For a further elucidation of the optimization potentials of lysine-producing strains, it might be of interest to elucidate time-dependent changes of metabolic flux distributions during production processes, for which experimental strategies and methods have to be developed and refined. In this context, the high specific glucose uptake rate of the wild-type strain, C. glutamicum ATCC 13032, of 2.4 mmol g−1 h−1 in comparison to those of the lysine-producing mutants (1.5 to 1.6 mmol g−1 h−1) during the initial phase of lysine production (approcimately from hours 7 to 16) might indicate a high capacity of metabolic pathways in the central metabolism of the wild-type strain and a potential for a future increase of the corresponding fluxes in the lysine-producing mutants examined.
In summary, the straightforward approach presented here, describing genealogy profiling by metabolic network analysis, can be valuable for exploiting the various strain genealogies deposited in public or industrial strain collections and for tracing the history of strain development in order to increase the standard of knowledge of metabolic functioning and regulation in different biological systems.
Acknowledgments
We appreciate the valuable assistance of Irene Kochems and Michel Fritz in the cultivation experiments.
This work was supported by BASF AG (Ludwigshafen, Germany).
APPENDIX
Flux-partitioning ratios (φ) and reversibilities (ζ) were defined as relative fluxes into one of the two branches and as ratios of backward or exchange flux to the net flux in the forward direction, respectively. Fluxes are represented by ν; the indexing refers to Fig. 1. The following abbreviations and subscripts are used: DH, diaminopimelate dehydrogenase branch in lysine biosynthesis; ICD, isocitrate dehydrogenase; ICL, isocitrate lyase; PGI, glucose 6-phosphate isomerase; PC, lumped reaction of pyruvate and phosphoenolpyruvate carboxylase; PDH, pyruvate dehydrogenase; PEPCK, lumped reaction of phosphoenolpyruvate carboxykinase, oxaloacetate decarboxylase, and malic enzyme; DAP, diaminopimelic acid; OAA, oxaloacetate.
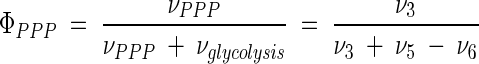 |
(2) |
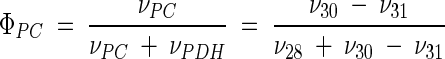 |
(3) |
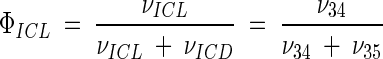 |
(4) |
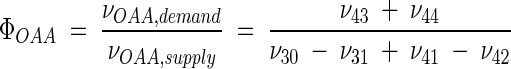 |
(5) |
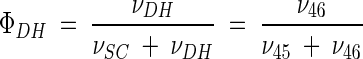 |
(6) |
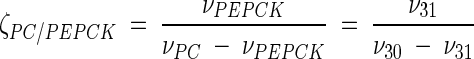 |
(7) |
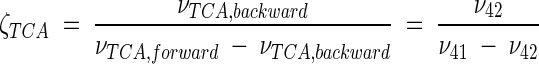 |
(8) |
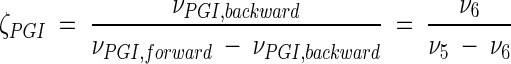 |
(9) |
The following balances around intracellular metabolite pools were formulated for the network of lysine-producing C. glutamicum examined, applying the numbering of fluxes given in Fig. 1.
 |
(10) |
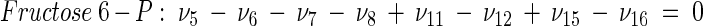 |
(11) |
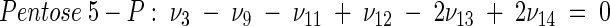 |
(12) |
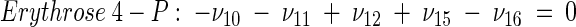 |
(13) |
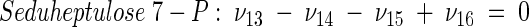 |
(14) |
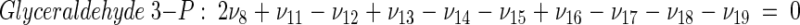 |
(15) |
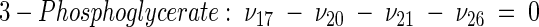 |
(16) |
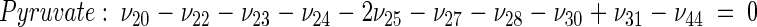 |
(17) |
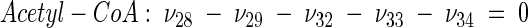 |
(18) |
 |
(19) |
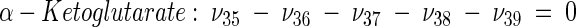 |
(20) |
 |
(21) |
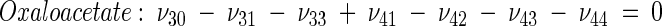 |
(22) |
 |
(23) |
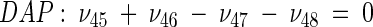 |
(24) |
The rank of the stoichiometric matrix formulated for equations 10 to 24 was 15, as determined with Matlab, indicating that the 15 balance equations were linearly independent. Additional constraints resulted from the fixation of the reversibilities of transaldolase and transketolases in the PPP and from stoichiometric data on the anabolic demand of glucose 6-phosphate (ν4), fructose 6-phosphate (ν7), pentose 5-phosphate (ν9), erythrose 4-phosphate (ν10), glyceraldehyde 3-phosphate (ν19), 3-phosphoglycerate (ν26), pyruvate (ν27), acetyl-CoA (ν29), oxaloacetate (ν43), α-ketoglutarate (ν36), and diaminopimelate (ν47) (Table 4). In total, 29 constraints were obtained (i) from stoichiometric balancing (15 constraints), (ii) from the fixation of the PPP reversibilities (3 constraints), and (iii) from stoichiometric constraints on biomass composition (11 constraints). Accordingly, 19 pieces of information had to be provided by experimental measurements. Fourteen pieces of information were gained from experimental determination of substrate uptake (ν1), product secretion (ν2, ν18, ν21, ν22, ν23, ν24, ν25, ν32, ν37, ν38, ν40, ν48), and cell growth (Table 1). Therefore, five additional pieces of information were required from mass spectrometric labeling measurements to calculate the entire distribution of 48 intracellular fluxes. The relative measurement of three mass isotopomer fractions of a compound provides two independent pieces of information on mass isotopomer ratios. Thus, the quantification of the mass isotopomer fractions m, m + 1, and m + 2 for alanine, valine, and lysine in the tracer experiment with (i) [1-13C]glucose and (ii) a mixture of naturally labeled and [13C6]glucose yields 12 additional pieces of information. With the additional estimation of the mass isotopomer ratio m + 2/m + 1 for trehalose in the tracer experiment with a mixture of naturally labeled and [13C6]glucose, a total of 13 pieces of information obtained from the labeling analysis compared to five pieces of information required for the flux calculation. A least-square approach was therefore possible in the parameter optimization.
REFERENCES
- 1.Dauner, M., J. E. Bailey, and U. Sauer. 2001. Metabolic flux analysis with a comprehensive isotopomer model in Bacillus subtilis. Biotechnol. Bioeng. 76:144-156. [DOI] [PubMed] [Google Scholar]
- 2.de Graaf, A. A., L. Eggeling, and H. Sahm. 2001. Metabolic engineering for l-lysine production by Corynebacterium glutamicum. Adv. Biochem. Eng. Biotechnol. 73:9-29. [DOI] [PubMed] [Google Scholar]
- 3.Eikmanns, B. J., D. Rittmann, and H. Sahm. 1995. Cloning, sequence analysis, expression, and inactivation of the Corynebacterium glutamicum icd gene encoding isocitrate dehydrogenase and biochemical characterization of the enzyme. J. Bacteriol. 177:774-782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kiss, R. D., and G. Stephanopoulos. 1992. Metabolic characterization of a l-lysine-producing strain by continuous culture. Biotechnol. Bioeng. 39:565-574. [DOI] [PubMed] [Google Scholar]
- 5.Kitson, F. G., B. Larsen, and C. N. McEven. 1996. Gas chromatography and mass spectrometry: a practical guide. Academic Press, San Diego, Calif.
- 6.Marx, A., A. A. de Graaf, W. Wiechert, L. Eggeling, and H. Sahm. 1996. Determination of the fluxes in the central metabolism of Corynebacterium glutamicum by nuclear magnetic resonance spectroscopy combined with metabolite balancing. Biotechnol. Bioeng. 49:111-129. [DOI] [PubMed] [Google Scholar]
- 7.Marx, A., B. Eikmanns, H. Sahm, A. A. de Graaf, and L. Eggeling. 1999. Response of the central metabolism in Corynebacterium glutamicum to the use of an NADH-dependent glutamate dehydrogenase. Metabol. Eng. 1:35-48. [DOI] [PubMed] [Google Scholar]
- 8.Matsushita, K., A. Otofuji, M. Iwahashi, H. Toyama, and O. Adachi. 2001. NADH dehydrogenase of Corynebacterium glutamicum. Purification of an NADH dehydrogenase II homolog able to oxidize NADPH. FEMS Microbiol. Lett. 204:271-276. [DOI] [PubMed] [Google Scholar]
- 9.Michal, G. 1999. Biochemical pathways. Spektrum Akademischer Verlag, Heidelberg, Germany.
- 10.Möllney, M., W. Wiechert, D. Kownatzki, and A. A. de Graaf. 1999. Bidirectional reaction steps in metabolic networks, part IV: optimal design of isotopomer labeling experiments. Biotechnol. Bioeng. 66:86-103. [DOI] [PubMed] [Google Scholar]
- 11.Moritz, B., K. Striegel, A. A. de Graaf, and H. Sahm. 2000. Kinetic properties of the glucose-6-phosphate and 6-phosphogluconate dehydrogenases from Corynebacterium glutamicum and their application for predicting pentose phosphate pathway flux in vivo. Eur. J. Biochem. 267:3442-3452. [DOI] [PubMed] [Google Scholar]
- 12.Nakayama, K., and K. Araki. January 1973. U.S. patent 3,708,395.
- 13.Ohnishi, J., S. Mitsuhashi, M. Hayashi, S. Ando, H. Yokoi, K. Ochiai, and M. A. Ikeda. 2002. A novel methodology employing Corynebacterium glutamicum genome information to generate a new l-lysine-producing mutant. Appl. Microbiol. Biotechnol. 58:217-223. [DOI] [PubMed] [Google Scholar]
- 14.Petersen, S., A. A. De Graaf, L. Eggeling, M. Möllney, W. Wiechert, and H. Sahm. 2000. In vivo quantification of parallel and bidirectional fluxes in the anaplerosis of Corynebacterium glutamicum. J. Biol. Chem. 275:35932-35941. [DOI] [PubMed] [Google Scholar]
- 15.Rosman, K. J. R., and P. D. P. Taylor. 1998. Isotopic compositions of the elements 1997. Pure Appl. Chem. 70:217-235. [Google Scholar]
- 16.Rowlands, R. T. 1984. Industrial strain improvement: mutagenesis and random screening procedures. Enzyme Microb. Technol. 6:3-10. [Google Scholar]
- 17.Sahm, H., L. Eggeling, and A. A. de Graaf. 2000. Pathway analysis and metabolic engineering in Corynebacterium glutamicum. Biol. Chem. 381:899-910. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt, K., M. Carlsen, J. Nielsen, and J. Villadsen. 1997. Modeling isotopomer distributions in biochemical networks using isotopomer mapping matrices. Biotechnol. Bioeng. 55:831-840. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt, K., L. C. Norregaard, B. Pedersen, A. Meissner, J. O. Duus, J. Nielsen, and J. Villadsen. 1999. Quantification of intracellular metabolic fluxes from fractional enrichment 13C-13C coupling constraints on the isotopomer distribution in labeled biomass components. Metabol. Eng. 1:166-179. [DOI] [PubMed] [Google Scholar]
- 20.Shiio, I., H. Momose, and A. Oyama. 1969. Genetic and biochemical studies on bacterial formation of glutamate. 1. Relationship between isocitrate lyase, acetate kinase and phosphate acetyltransferase levels and glutamate production in Brevibacterium flavum. J. Gen. Appl. Microbiol. 15:27-40. [Google Scholar]
- 21.Sonntag, K., L. Eggeling, A. A. de Graaf, and H. Sahm. 1993. Flux partitioning in the split pathway of lysine synthesis in Corynebacterium glutamicum. Quantification by 13C- and 1H-NMR spectroscopy. Eur. J. Biochem. 213:1325-1331. [DOI] [PubMed] [Google Scholar]
- 22.Sonntag, K., J. Schwinde, A. A. de Graaf, A. Marx, B. J. Eiksmanns, W. Wiechert, and H. Sahm. 1995. 13C NMR studies of the fluxes in the central metabolism of Corynebacterium glutamicum during growth and overproduction of amino acids in batch cultures. Appl. Microbiol. Biotechnol. 44:489-495. [Google Scholar]
- 23.Stephanopoulos, G., A. A. Aristidou, and J. Nielsen. 1998. Metabolic engineering. Academic Press, San Diego, Calif.
- 24.Sugimoto, S. I., and I. Shiio. 1987. Regulation of glucose 6-phosphate dehydrogenase in Brevibacterium flavum. Agric. Biol. Chem. 51:101-108. [Google Scholar]
- 25.Sugimoto, S. I., and I. Shiio. 1987. Regulation of 6-phosphogluconate dehydrogenase in Brevibacterium flavum. Agric. Biol. Chem. 51:1257-1263. [Google Scholar]
- 26.Vallino, J. J., and G. Stephanopoulos. 1993. Metabolic flux distributions in Corynebacterium glutamicum during growth and lysine overproduction. Biotechnol. Bioeng. 41:633-646. [DOI] [PubMed] [Google Scholar]
- 27.Van Winden, W. A., C. Wittmann, E. Heinzle, and J. J. Heijnen. 2002. Correcting mass isotopomer distributions for naturally occurring isotopes. Biotechnol. Bioeng. 80:477-479. [DOI] [PubMed] [Google Scholar]
- 28.Wendisch, V. F., M. Spies, D. J. Reinscheid, S. Schnicke, H. Sahm, and B. J. Eiksmanns. 1997. Regulation of acetate metabolism in Corynebacterium glutamicum: transcriptional control of the isocitrate lyase and malate synthase genes. Arch. Microbiol. 168:262-269. [DOI] [PubMed] [Google Scholar]
- 29.Wendisch, V. F., A. A. de Graaf, H. Sahm, and B. J. Eikmanns. 2000. Quantitative determination of metabolic fluxes during coutilization of two carbon sources: comparative analyses with Corynebacterium glutamicum during growth on acetate and/or glucose. J. Bacteriol. 182:3088-3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wiechert, W., C. Siefke, A. A. de Graaf, and A. Marx. 1997. Bidirectional reaction steps in metabolic networks: II. Flux estimation and statistical analysis. Biotechnol. Bioeng. 55:118-135. [DOI] [PubMed] [Google Scholar]
- 31.Wittmann, C., and E. Heinzle. 1999. Mass spectrometry for metabolic flux analysis. Biotechnol. Bioeng. 62:739-750. [DOI] [PubMed] [Google Scholar]
- 32.Wittmann, C., and E. Heinzle. 2001. Novel approach for metabolic flux analysis—application of MALDI-TOF MS to lysine-producing Corynebacterium glutamicum. Eur. J. Biochem. 268:2441-2455. [DOI] [PubMed] [Google Scholar]
- 33.Wittmann, C., and E. Heinzle. 2001. Modeling and experimental design for metabolic flux analysis of lysine-producing Corynebacteria by mass spectrometry. Metabol. Eng. 3:173-191. [DOI] [PubMed] [Google Scholar]
- 34.Wittmann, C. 2002. Metabolic flux analysis using mass spectrometry. Adv. Biochem. Eng. Biotechnol. 74:39-64. [DOI] [PubMed] [Google Scholar]