

Short abstract
A detailed analysis of the anatomy and distribution of L1 elements in the human genome was carried out using a new computer program, TSDfinder, designed to identify transposon boundaries precisely. Conclusions: The sequence data give no indication that the creation of L1 structural variants depends on characteristics of the insertion locus.
Abstract
Background
As the rough draft of the human genome sequence nears a finished product and other genome-sequencing projects accumulate sequence data exponentially, bioinformatics is emerging as an important tool for studies of transposon biology. In particular, L1 elements exhibit a variety of sequence structures after insertion into the human genome that are amenable to computational analysis. We carried out a detailed analysis of the anatomy and distribution of L1 elements in the human genome using a new computer program, TSDfinder, designed to identify transposon boundaries precisely.
Results
Structural variants of L1 elements shared similar trends in the length and quality of their target site duplications (TSDs) and poly(A) tails. Furthermore, we found no correlation between the composition and genomic location of the pre-insertion locus and the resulting anatomy of the L1 insertion. We verified that L1 insertions with TSDs have the 5'-TTAAAA-3' cleavage site associated with L1 endonuclease activity. In addition, the second target DNA cut required for L1 insertion weakly matches the consensus pattern TTAAAA. On the other hand, the L1-internal breakpoints of deleted and inverted L1 elements do not resemble L1 endonuclease cleavage sites. Finally, the genome sequence data indicate that whereas singly inverted elements are common, doubly inverted elements are almost never found.
Conclusions
The sequence data give no indication that the creation of L1 structural variants depends on characteristics of the insertion locus. In addition, the formation of 5' truncated and 5' inverted L1s are probably not due to the action of the L1 endonuclease.
Background
Transposable elements are a prominent component of the human genome, accounting for approximately 45% of the initial draft sequence [1]. This is probably an underestimate, because the heterochromatic and other regions that are difficult to assemble were excluded from these estimates. Nevertheless, this large fraction is a testament to the efficiency with which these elements, now mostly fossil sequences [2,3,4,5], have been able to propagate, and it is clear that they played a large part in determining the structure and organization of our genome. Transposable elements mobilize either directly through DNA or through an RNA intermediate. Retrotransposons mobilize through an RNA intermediate, and are classified as either having long-terminal repeats (LTRs) or not (non-LTR). L1s are one of the most abundant non-LTR retrotransposons, comprising 17% of the human genome [1]. Also, L1-encoded proteins were almost certainly involved in the insertion of most of the Alu elements and processed pseudogenes in the genome [6,7,8,9,10]; thus, L1s are probably responsible, directly or indirectly, for the genesis of most of the transposed fraction of human DNA.
A full-length, active human L1 is approximately 6,000 nucleotides long and consists of a 5' UTR with an internal promoter, two open reading frames (ORFs) separated by a 63-nucleotide intergenic region, and a 3' UTR terminating in a poly(A) tail [11] (Figure 1a). The functional significance of ORF1 is not clear, whereas ORF2 contains three domains critical for L1 propagation: endonuclease [12], reverse transcriptase [7,13], and a 3'-terminal Zn finger-like domain [14].
Figure 1.

Anatomy of the L1 element and its structural variants found in the human genome. (a) A full-length L1 transcript is approximately 6,000 nucleotides long. It has a 5' UTR, two ORFs separated by 63 nucleotides, and a 3' UTR followed by a poly(A) tail. An L1 insertion in the genome is flanked by TSDs; the 3' TSD is immediately preceded by a poly(A) tail. (b) Variations in the structure of L1 insertions are shown. Arrowheads indicate the orientation of the L1 sequence. Most L1s are 5' truncated. In addition, during the process of insertion, a 5' segment of L1 may become inverted with respect to the 3' end of the L1 (5' inversion). Alternatively or additionally, a weak poly(A) signal in the L1 transcript can result in a portion of the 3' flanking DNA being transposed to another locus in the genome along with the L1 element; this process is called 3' transduction. In this case, the 3' TSD can be located hundreds of nucleotides downstream from the end of the L1 element. The numbers in parentheses represent the percentage of all L1s with TSDs that fall into each category. (c) TSD sequences flanking an L1 insertion are underlined. The poly(A) tail has a line over it. Although the poly(A) tail could potentially be extended, this would require the length (and potentially the score) of the TSDs to be reduced; TSDfinder always finds the longest possible TSDs.
Full-length L1s are capable of autonomous retrotransposition. They propagate by being transcribed from an internal RNA polymerase II promoter [15,16], and then use their endonuclease and reverse transcriptase respectively to nick a target site and reverse transcribe L1 RNA, integrating the L1 into a new genomic locus [12,17,18]; this process is known as target-site-primed reverse transcription (TPRT). A new L1 insertion is usually flanked by short direct repeats derived from the target DNA locus upon L1 integration [19]; these repeats are referred to as target site duplications (TSDs), and can range from just a few to more than 200 nucleotides in length [14].
Relatively recent retrotransposition events can be recognized in genomic DNA with computational sequence analysis tools; however, the precise determination of the boundaries of L1 elements is complicated by the highly variable sequence and anatomy of L1 insertions. Thus, single L1 insertions can be mistakenly annotated as several separate L1 segments by RepeatMasker (A.F.A. Smit and P. Green [20]) and similar repeat-finding algorithms. The most variable features of L1s are the poly(A) tail, which has a variable length and can contain simple repeats, the polyadenylation signal, and the 3' UTR, but many changes have also been reported in the coding regions of young L1s, especially in a segment of ORF1 [21,22,23,24]. Relatively little is known about 5' UTR sequences, especially of the older elements, as the majority of L1s are 5' truncated [25]. Sequence differences in these highly variable components of L1 elements confound precise definition of the TSDs that define L1 insertion boundaries.
In addition to sequence variability, the structure of L1 elements is also quite diverse. Many L1 elements are 5' truncated or both 5' truncated and 5' inverted (Figure 1b). Some L1s have an extra 131 nucleotides in their 5' UTR, starting at position 777 [26]. Also, 3' transduction events associated with L1 insertions add an additional structural variation [27,28]; such elements are generated when the element's transcript includes some downstream flanking sequences that become transduced, along with the L1 sequence, to a new genomic locus. Consequently, the 3' TSD of the resulting L1 insertion event can be located hundreds of nucleotides downstream of the L1 sequence itself [29,30]. Finally, target site deletions can occur; therefore, sequence boundaries for some L1 insertions cannot be determined computationally in the absence of the pre-integration target [14,31,32].
The above variations provide a formidable bioinformatic challenge to accurate and automated identification of L1 termini. Nevertheless, we developed an algorithm that refines the coordinates of L1 insertions found by the existing DNA sequence analysis tool RepeatMasker. The basis of our algorithm is the identification of the flanking TSDs and poly(A) tails of 3' intact L1 insertions. Using this algorithm, we collected from the human genome sequence those L1s with recognizable TSDs. This enabled us to carry out a large-scale study of L1 sequence features related to the molecular mechanisms involved in retrotransposition. We present here some primary structural features of these L1s and a detailed analysis of their TSDs. We also investigated the chromosomal location at which these L1s were found. Studying L1 insertion events in our genome can provide new insights to both L1 biology and the mechanisms by which L1s and the smaller, gene-encoding portion of the human genome have reached an equilibrium.
Results
TSDfinder program
To identify the location of L1s in the human genome, we ran RepeatMasker using default settings with a custom library (see below). For each L1 found, the program TSDfinder [33] was used to refine the location coordinates in the following ways. First, it tested whether adjacent L1 fragments could be merged (see Materials and methods). In the set of 3' intact L1s studied here, approximately 9% were originally annotated by RepeatMasker as several separate L1 segments. This merging step was important for maximizing the yield of correct TSDs; without this merging, we found nearly 10% fewer TSDs. Next, TSDfinder found the poly(A) tail and TSDs, critical components of the insertion signature of an L1. In addition, the algorithm for TSD recognition allowed detection of both 'standard' insertion events (in which the 3' TSD immediately follows the poly(A) tract associated with the retrotransposon) and putative 3' transduction events (in which the 3' TSD follows a poly(A) tail further downstream from the 3' end of the retrotransposon) (Figure 1b). This program also detected any 5' inverted elements and identified the inversion breakpoints.
An important step for the validation of TSDfinder results was the analysis of the TSDs identified by the algorithm. As seen in Figure 2, the majority of TSDs identified by TSDfinder had high TSD scores (the scoring system is described in Materials and methods), with an average score of 117 out of a maximum possible score of 140. The distribution of TSD lengths (Figure 2) correlates well with TSDs observed in biological experiments [12,14,34,35]. The peak of nine-nucleotide TSDs, the minimum TSD length allowed by TSDfinder, is likely to be enriched in false positives because they have a relatively low average TSD score of 98; there is obviously a higher likelihood of finding these shorter matches by chance. The second major peak in Figure 2 indicates that the majority of TSDs with a score greater than 100 were 15 nucleotides long. This finding, combined with generally high TSD scores and manual inspection of hundreds of L1s recovered, gives us confidence that the sequences identified by the TSDfinder algorithm represent actual TSDs. The longest TSD found was 60 nucleotides even after we relaxed and extended the TSDfinder parameters to enable the identification of long TSDs. Although TSDs longer than this have been found in new insertions [14,32], it is possible that long TSDs are eliminated or unstable in the human genome. Alternatively, long TSDs may be an artifact of transposition assays in cultured cells.
Figure 2.

Distribution of TSD lengths and scores. Lengths of the 16,266 TSDs found by TSDfinder for L1s in the genome are shown as a histogram. The line indicates the average TSD score for each TSD length. The x axis displays the range of TSD lengths in nucleotides; the y axis displays the number of TSDs. The bimodal distribution suggests that a significant fraction of the 9-11-nucleotide TSDs may be artifactual.
L1s in the human genome
We limited this analysis to younger L1s, which are more likely to be intact, thereby allowing greater confidence in assigning the 5' and 3' endpoints of each L1 element. In addition, TSDs are generally relatively short, and thus relying on their conservation in the genome is most reasonable for the young L1s. Focusing on older L1 family members is likely to increase the rate of false-negative results.
We used the recently transposed L1.3 sequence [36] as the query sequence with which the RepeatMasker program identified the location of young L1s in the human genome. L1.3 is one of the most active L1s in a cell-culture assay [37] and is a member of the human-specific Ta-1 subfamily of L1s which encompasses the vast majority of active, young L1 elements in the human genome [22]. L1 elements used in this study were identified from non-redundant human sequence contigs (NT_* records) assembled at NCBI by 29 August, 2001 (build 25), which constitute approximately 91% of the euchromatic genome (Table 1). The set of all L1s identified by RepeatMasker averaged 76% (± 9%) identity with the L1.3 reference sequence. A total of 99% of the L1s were from primate-specific classes of L1s, and nearly 80% of them belonged to one of the following younger subfamilies: L1Hs, L1P1, L1P2, and L1P3 [23]. The L1s for which TSDs could be identified exhibited a mean identity with the L1.3 sequence of 88% (7%). This confirms that TSDs are more recognizable in younger elements.
Table 1.
Dataset characteristics and summary of TSDfinder results
| Chromosome | Total Mb sequenced* | Estimated chromosome length (Mb) | Sequence completed | Number of 3' intact insertion events† | Number of 3'-intact per Mb sequenced | Number with TSDs | Standard insertion | 3' Trans duction‡ | Number with TSDs/Mb sequenced | Total number of FL L1s§ | Percentage of 3' intact that are FL | Density of FL L1s/Mb sequenced |
| 1 | 230.3 | 252.5 | 91% | 5,123 | 22.2 | 1,195 | 756 | 439 | 5.2 | 258 | 5% | 1.12 |
| 2 | 226.5 | 238.0 | 95% | 5,597 | 24.7 | 1,302 | 807 | 495 | 5.7 | 279 | 5% | 1.23 |
| 3 | 192.0 | 204.4 | 94% | 5,064 | 26.4 | 1,087 | 680 | 407 | 5.7 | 223 | 4% | 1.16 |
| 4 | 175.9 | 189.6 | 93% | 5,433 | 30.9 | 1,094 | 684 | 410 | 6.2 | 240 | 4% | 1.36 |
| 5 | 169.2 | 180.5 | 94% | 4,852 | 28.7 | 1,049 | 651 | 398 | 6.2 | 259 | 5% | 1.53 |
| 6 | 173.4 | 178.1 | 97% | 4,734 | 27.3 | 1,089 | 692 | 397 | 6.3 | 254 | 5% | 1.46 |
| 7 | 153.8 | 160.4 | 96% | 3,734 | 24.3 | 829 | 499 | 330 | 5.4 | 201 | 5% | 1.31 |
| 8 | 134.8 | 143.4 | 94% | 3,586 | 26.6 | 816 | 519 | 297 | 6.1 | 215 | 6% | 1.59 |
| 9 | 114.7 | 131.7 | 87% | 2,735 | 23.9 | 660 | 418 | 242 | 5.8 | 143 | 5% | 1.25 |
| 10 | 134.7 | 139.7 | 96% | 3,048 | 22.6 | 687 | 422 | 265 | 5.1 | 163 | 5% | 1.21 |
| 11 | 139.1 | 141.2 | 99% | 3,675 | 26.4 | 795 | 515 | 280 | 5.7 | 199 | 5% | 1.43 |
| 12 | 130.2 | 138.4 | 94% | 3,317 | 25.5 | 740 | 462 | 278 | 5.7 | 186 | 6% | 1.43 |
| 13 | 99.1 | 116.7 | 85% | 2,596 | 26.2 | 583 | 360 | 223 | 5.9 | 97 | 4% | 0.98 |
| 14 | 89.0 | 105.4 | 84% | 2,173 | 24.4 | 502 | 307 | 195 | 5.6 | 128 | 6% | 1.44 |
| 15 | 80.7 | 98.9 | 82% | 1,644 | 20.4 | 417 | 253 | 164 | 5.2 | 92 | 6% | 1.14 |
| 16 | 77.6 | 92.6 | 84% | 1,399 | 18.0 | 313 | 188 | 125 | 4.0 | 50 | 4% | 0.64 |
| 17 | 81.3 | 83.3 | 98% | 1,256 | 15.4 | 313 | 196 | 117 | 3.8 | 30 | 2% | 0.37 |
| 18 | 79.8 | 81.3 | 98% | 1,908 | 23.9 | 446 | 278 | 168 | 5.6 | 86 | 5% | 1.08 |
| 19 | 58.6 | 77.1 | 76% | 730 | 12.5 | 189 | 113 | 76 | 3.2 | 32 | 4% | 0.55 |
| 20 | 60.4 | 61.8 | 98% | 1,003 | 16.6 | 255 | 163 | 92 | 4.2 | 46 | 5% | 0.76 |
| 21 | 33.9 | 46.2 | 73% | 821 | 24.3 | 195 | 121 | 74 | 5.8 | 17 | 2% | 0.50 |
| 22 | 34.8 | 47.2 | 74% | 422 | 12.1 | 108 | 57 | 51 | 3.1 | 19 | 5% | 0.55 |
| X | 141.1 | 150.3 | 94% | 6,067 | 43.0 | 1,345 | 811 | 534 | 9.5 | 336 | 6% | 2.38 |
| Y | 22.7 | 59.0 | 39% | 959 | 42.2 | 210 | 108 | 102 | 9.2 | 93 | 10% | 4.09 |
| Un¶ | 8.9 | 0.0 | 272 | 30.5 | 47 | 28 | 19 | 5.3 | 11 | 4% | 1.23 | |
| Total | 2,842.6 | 3,117.8 | 91% | 72,148 | 16,266 | 10,088 | 6,178 | 3,657 |
*The total amount of sequence (nucleotides) available on 29 August 2001 for each chromosome is indicated. †The 3' intact L1s must not have more than a 30-nucleotide deletion from the 3' end. ‡If the sequence between the start of the poly(A) tail and the 3' end of the annotated L1 exceeded 30 nucleotides, the L1 was classified as a 3' transduction candidate. § Full-length (FL) L1s must have an intact 3' end and no more than a 10-nucleotide deletion at the 5' end; we did not require this set of full-length L1s to have TSDs. ¶ Un, unknown chromosome localization as reported in the NCBI contigs resource.
Table 1 summarizes the dataset and some general features of the L1 elements analyzed in this study. After merging, we identified 72,148 L1 insertions (22% of the total L1 segments found) with an intact 3' end (that is, < 31 nucleotides deleted from the 3' end of the 3' UTR). It is likely that those L1s that do not meet this definition of 3' intact are distantly related to L1.3 with divergent 3' UTRs. Only the set of 3' intact L1s was further analyzed for poly(A) tails and TSDs. Of these, 10,088 (14%) had TSDs immediately flanking the L1 element, and thus represented standard insertion events, and 6,178 (9%) had TSDs consistent with 3' transduction. This proportion of 3' transduction events is similar to previously reported estimates [29,30]; however, a majority of the 3' transduction events found are likely to be artifacts of segmental genome duplications (S.T.S, O.K.P, D.L. and J.D.B, unpublished observations). These TSDs were selected by the TSDfinder program in part because the 3' TSD was preceded by a poly(A) tail (see Materials and methods). Nevertheless, it is interesting to note that the proportion of L1s with TSDs nearly doubled when the poly(A) tail requirement was eliminated (data not shown). This may indicate that L1 element poly(A) tails shorten over time, obscuring their identification [24], while TSDs stay intact.
We next investigated the chromosomal densities of L1s. Column 10 in Table 1 shows the calculated average density of L1s with TSDs on each chromosome. These data indicate that the L1s annotated here are positioned much more densely on the X and Y chromosomes at approximately nine L1s per megabase (Mb) of genomic DNA, compared with a mean density of around five L1s per Mb for all autosomes. This observation is consistent with previous reports that the density of L1s on the X chromosome is almost double that on the autosomes [1,38,39]. The density of full-length L1s (with and without TSDs) on the sex chromosomes is also more than twice as high as on the autosomes; this may be a simple consequence of the lower recombination potential on the sex chromosomes as reported by Boissinot et al. [38]. With approximately three L1s per Mb, chromosome 22 has the lowest density of L1 insertions.
TSD composition and pre-insertion loci
Sequence patterns of TSDs are likely to reflect the targeting preference of L1 integration machinery. In addition, the target site may influence the resulting L1 anatomy. To investigate this, we segregated our set of L1s with TSDs into the following major classes of structural variants and analyzed their respective TSDs: 5' truncated, 5' inverted, or full-length (L1 start ≥ 10) standard insertions, and 3' transduction candidates (Figure 1b). When the TSDs of each structural variant were analyzed, we observed little difference in the distribution of TSD lengths and scores between these variants (data not shown).
When the A content in all 15-nucleotide TSDs was compared with randomly generated 15-nucleotide sequences based on the human genome percentage A content, it is clear that the majority of TSDs are A-rich (Table 2). This quality is consistent with previous computational [6] and biochemical [12,34] observations of A-rich target sequences of L1s and Alu sequences. Figure 3a shows pre-insertion locus sequence composition displayed as logo graphics for L1s with TSDs between 9 and 18 nucleotides long. It is readily apparent that TSDs equal to or greater than 12 nucleotides long exhibit a TTAAAA top-strand consensus (corresponding to a bottom strand TTTT  AA cleavage site). A comparable profile of L1 endonuclease cleavage sites was seen when the same analysis was performed for each of the L1 structural variants (Figure 1b, and data not shown). Because the 9-11-nucleotide 'TSDs' lack the TTAAAA consensus as a result of contamination by 'fortuitous' TSDs (Figure 2), the scoring scheme for TSDfinder was revised during its development to assign a scoring penalty to TSDs less than 12 nucleotides long.
AA cleavage site). A comparable profile of L1 endonuclease cleavage sites was seen when the same analysis was performed for each of the L1 structural variants (Figure 1b, and data not shown). Because the 9-11-nucleotide 'TSDs' lack the TTAAAA consensus as a result of contamination by 'fortuitous' TSDs (Figure 2), the scoring scheme for TSDfinder was revised during its development to assign a scoring penalty to TSDs less than 12 nucleotides long.
Table 2.
TSD sequences are A-rich
| TSD composition | Observed* | Expected† |
| 0-10% As | 0.1% | 2.7% |
| 10-20% As | 2.5% | 26.6% |
| 20-30% As | 8.6% | 23.8% |
| 30-40% As | 31.5% | 36.8% |
| 40-50% As | 18.0% | 7.4% |
| 50-60% As | 28.5% | 2.7% |
| 60-70% As | 5.2% | 0.1% |
| Over 70% As | 5.5% | 0.0% |
*TSDs of length 15 nucleotides without mismatches were classified according to their A content. †Monte Carlo simulation was used to generate random 15-nucleotide sequences representative of the nucleotide population of the human genome. Only 10% of these randomly generated sequences were equal to or greater than the average A content of the TSDs.
Figure 3.

Nucleotide profile of L1 target loci. (a) Logo graphics were generated for the TSDs of length shown and 10 nucleotides of upstream and downstream flanking sequence. At each position, the highest-frequency base is at the top of the stack, and so the general consensus can be found by reading the top base at every position. The relative height of individual bases at each position is proportional to the frequency of the base at that position. The vertical axis represents the amount of information in the input data. The TSDs included in this analysis did not have any mismatches. Stars indicate the first position of the TSD. The nucleotides are colored as follows: A, green; T, red; C, blue; G, yellow. (b) The percentage of A, T, G, and C nucleotides is shown, calculated for 1,794 15-nucleotide TSDs (without mismatches) and their flanking regions (50 nucleotides on each side). The shaded region represents the TSD sequence.
The target sequence patterns were further analyzed for the set of 1,794 15-nucleotide-long TSDs without mismatches and their 50-nucleotide flanking regions. The percentage of each nucleotide observed at each position in this 115-nucleotide region was calculated and is shown graphically in Figure 3b. Once again, a clear TTAAAA consensus is seen in these data at the boundary between the 5' flanking region and the TSD. Interestingly, symmetrical, but much less dramatic, peaks of T and A nucleotides are seen near the 3' end of the 15-nucleotide-long TSDs.
The initial analysis of the draft human genome reported a GC content of 36-38% in the neighborhood of L1s as opposed to 41% GC across the entire genome [1]. In agreement with these and other data that L1s preferentially reside in AT-rich DNA [24], our pre-insertion loci are only 35% GC. These data suggest that L1s may insert preferentially into pre-existing L1s in the human genome because, excluding the 5' UTR and any poly(A) tail, the L1 element is over 60% AT. To investigate this, we recreated pre-insertion loci by collecting 50 nucleotides of upstream and downstream flanking sequence and the TSD sequence of each L1 insertion for which a high-scoring TSD was found. We then used RepeatMasker to analyze these pre-insertion sequences for the presence of high-copy repeats. L1 insertions were found in unique sequence 61% of the time (Table 3). A robust match with known high-copy repeats (including L1) was found in only 24% of the pre-insertion loci. L1s were found inserted in presumably pre-existing elements of the LINE/L1 family 13.3% of the time and into SINE/Alu just 3% of the time, most probably because Alu sequences have a relatively high GC content (56%). The results for the remaining pre-insertion loci were inconclusive because of limited coverage of the sequence by fragmented repeats.
Table 3.
Insertion of L1s into other repetitive elements in the human genome
| Percent of the pre-insertion loci | Percent of the draft genome sequence | |
| SINEs | 3% | 13.1% |
| LINES | 13.3% | 20.4% |
| LTR elements | 5% | 8.3% |
| DNA elements | 2.2% | 2.8% |
| Satellite/centromeric | 0.5% | ND |
| Repeat did not meet criteria* | 14% | |
| Unique sequence | 61% | 55% |
| Total | 99% | 99.6 |
*The following criteria had to be met in order for a repeat to have been considered a target for L1 insertion: its number of substitutions could be no more than 25% as reported by the RepeatMasker program, and it had to span at least half of the pre-insertion sequence analyzed.
L1 poly(A) tails
During the process of TPRT, whereby a copy of an L1 is inserted into the genome, reverse transcription is believed to be primed on the poly(A) tail of the L1 transcript [12,14,17,27]. To investigate whether there was a correlation between the poly(A) tail of an L1 and its final insertion signature, we analyzed the length and percentage A content of poly(A) tails for each class of L1 structural variant (Figure 1b). We reasoned that the robustness of a poly (A) tail may dictate the fate of the resulting L1 insertion. We found that both standard insertion events and putative 3' transduction events have very similar poly(A) tails of average length 18 nucleotides and 86% A content. All other structural variants had similar values. The range of poly (A) tail lengths found by TSDfinder was 10-85 nucleotides.
The poly(A) tails found in I factors, a transposable element in Drosophila melanogaster similar to L1s, contain several copies of the simple repeat TAA instead of a traditional poly(A) tail [40,41]. We refer to these as patterned repeats. Of the 16,266 L1 poly(A) tails identified in our study, 2,147 (13%) had tails with similar sequence patterns. The most common patterned repeat found in these L1 tails was a TAAA tetranucleotide (mean of 2.8 As per repeat element) with the most common non-A nucleotide being a T (72% of the time), followed by C (16%) and G (12%). The longest patterned tail found was 198 nucleotides and contained almost exclusively the simple repeat GAAA. The presence of patterned repeats in poly(A) tails did not correlate with L1 structural variants; all exhibited a 14% incidence of patterned tails, whereas for putative 3' transduction events we only observed a 9% frequency of patterned tails. Finally, we compared the translated reverse transriptase sequences of L1s with and without patterned repeats. We were unable to detect any predicted amino-acid changes in reverse transcriptase that correlated with patterned tails (data not shown).
Full-length L1 elements
Several instances of disease have been attributed to the disruption of genes by an L1 insertion (for a review see [42]). The progenitor elements of these insertions were always full-length or near full-length L1s with intact ORFs that were active in retrotransposition assays [11,14,35,36,37]. It is estimated that 30-60 full-length active L1s reside in the average human genome [37]. To identify L1s in the human genome that might have retrotransposition potential, we identified L1s that were full-length and had intact coding regions.
We identified 3,657 full-length L1 elements (Table 1). This is a minimum estimate, because it is theoretically possible that some relatively diverged family of L1 element not detected by our threshold criteria (see below) could have active copies. To assemble this set, we allowed no more than a 30-nucleotide deletion at the 3' end and no more than a 10-nucleotide deletion at the 5' end relative to the L1.3 reference sequence, ensuring the presence of an intact L1 internal promoter [16,43,44]. This minimal allowance for a 5' deletion is conservative because the LRE2 element, an L1 element in the same subfamily as L1.3, has a 21-nucleotide 5' deletion, yet retains transposition ability [28]. We did not require our set of full-length L1s to have TSDs. An extra 131 nucleotides are found in the 5' UTR of some L1 elements, starting at nucleotide position 777 [26]. In our set of full-length L1s, 1,687 (46%) lacked this insertion whereas 1,964 (54%) had this sequence in the 5' UTR.
The BLASTX program [45] was used to translate our set of full-length L1 DNA sequences in all three reading frames and compare them to the L1.3 ORF1 and ORF2 amino-acid sequences. The translation for each full-length L1 sequence was analyzed for the presence of nonsense mutations, and for missense mutations that inhibit retrotransposition activity by destroying the catalytic sites of the endonuclease or reverse transcriptase or any other critical residues on which retrotransposition depends (see Materials and methods for a complete list) [12,14,46]. We identified 85 full-length L1 elements that, if translated, would yield full-length ORF1 and ORF2 proteins that did not contain any missense mutations known to destroy their retrotransposition potential (Table 4a). On average, these full-length L1s were 99% identical in their amino-acid sequences. The most divergent protein sequence in this group was only 4% different from the L1.3 ORFs. None of these full-length L1s was found on chromosomes 17, 19, or 21, whereas the highest concentration (11% of the total) was found on the X chromosome. One of the most commonly altered codons compared to the L1.3 ORF1 was codon 251 (V→A) in 93% of these L1s. In addition, codon 760 (V→T) in ORF2 differed from the L1.3 ORF2 sequence for 96% of these L1s (Table 4b). Neither of these codon changes represents a conservative amino-acid change; however, these alternative codons have been documented in GenBank records for active ORF1 and ORF2 proteins (pids 5070621 and 483916, respectively). Moreover, the preponderance of these codons suggests that they are, in fact, the consensus, ancestral codons from which L1.3 has diverged.
Table 4.
Analysis of full-length L1s
| (a) Chromosome | Number of L1s with intact ORF proteins* | Average number of codon changes† | Number of L1s with intact ORF proteins/Gb |
| 1 | 8 | 23 | 35 |
| 2 | 7 | 27 | 31 |
| 3 | 7 | 22 | 36 |
| 4 | 6 | 7 | 34 |
| 5 | 2 | 33 | 12 |
| 6 | 5 | 17 | 29 |
| 7 | 3 | 14 | 20 |
| 8 | 3 | 5 | 22 |
| 9 | 1 | 9 | 9 |
| 10 | 5 | 25 | 37 |
| 11 | 8 | 15 | 58 |
| 12 | 6 | 38 | 46 |
| 13 | 1 | 51 | 10 |
| 14 | 2 | 18 | 22 |
| 15 | 2 | 20 | 25 |
| 16 | 1 | 10 | 13 |
| 17 | 0 | NA | 0 |
| 18 | 5 | 21 | 63 |
| 19 | 0 | NA | 0 |
| 20 | 2 | 20 | 33 |
| 21 | 0 | NA | 0 |
| 22 | 1 | 10 | 29 |
| X | 9 | 28 | 64 |
| Y | 1 | 19 | 44 |
| Un | 0 | NA | NA |
| Total: | 85 | 21 | |
| (b)‡ | |||
| Occurrences in this set | |||
| ORF1 | |||
| Codon 168, V → G | 24% | ||
| Codon 251, V → A | 93% | ||
| ORF2 | |||
| Codon 485, M → K | 87% | ||
| Codon 755, G → S | 47% | ||
| Codon 760, V → T | 96% | ||
| Codon 970, K → R | 66% | ||
| Codon 1006, D → A | 62% | ||
| Codon 1182, N → K | 48% | ||
| Codon 1183, E → D | 69% | ||
| Codon 1241, I → M | 46% | ||
*All full-length L1s with intact ORFs, with and without TSDs, were collected. Full-length was defined as ≤ 30 nucleotides deleted from the 3' end and ≤ 10 nucleotides deleted from the 5' end. † The predicted protein translation from the L1.3 sequence was used as a reference. ‡Some codons were more likely to be diverged from the L1.3 reference than others. These codons, their changes, and the frequency with which they were found in our set are shown.
Importantly, all of these putative retrotransposition-competent L1s lacked a common 131-nucleotide insertion in the 5' UTR, supporting the assertion that this is a signature of an older, inactive subfamily [26]. Indeed, we found the average percent identity to the L1.3 DNA sequence of the full-length L1s containing this insertion to be 93%, whereas those full-length L1s lacking this insertion averaged 97% identity. Furthermore, of the 1,687 full-length L1 elements that lack this insertion, 1,233 (73%) have TSDs, whereas only 1,071 (55%) of those that have the extra sequence have recognizable TSDs; the reduced frequency of TSDs further suggests that L1s with the 131-nucleotide insertion in their 5' UTR represent an older class of insertions.
Characteristics of 5' truncated L1 elements
The majority of L1s in the human genome are significantly 5' truncated [25]; indeed, only 5% of the 3' intact L1s in our study were full-length (Table 1). The histogram of start positions of all L1s with TSDs is shown in Figure 4. There is a clear dominance of significantly truncated L1 elements whose 5' end lies in the last 1,000 nucleotides of the L1.3 reference sequence. Although most L1 elements are severely 5' truncated, a significant fraction are full length (Figure 4). Of the full-length elements, 63% have TSDs, whereas only 23% of all L1s do. This may result from imprecision in the assignment of the 5' breakpoint. We investigated the L1 sequence composition at the breakpoint in 5' truncated L1s by collecting 15 nucleotides of L1 sequence upstream and downstream of the position at which the L1 had been truncated; we were unable to detect any consensus nucleotides at these breakpoints (data not shown).
Figure 4.
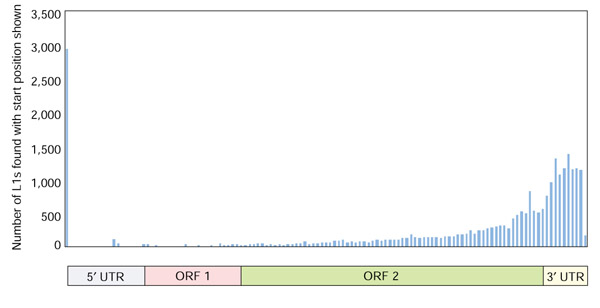
Histogram of L1 start positions for L1 insertions complete at the 3' end. The L1 start positions of the 16,266 elements with TSDs were placed into 50-nucleotide bins. The schematic of L1 along the x axis indicates the L1 start position represented by each bin. The y axis shows the number found in each bin.
Rearranged L1s
An additional variable of L1 structure is the presence of rearranged elements; typically, these consist of two L1 segments, of which the 5' segment is inverted (Figure 1b). Previous reports have estimated that 10% of all L1 insertions in the genome are internally rearranged [42]. We identified a total of 6,063 (8% of all 3' intact L1s) inverted L1 segment pairs, and 3,157 (52%) of these have TSDs. Like other L1s found in the genome, most inverted elements are also 5' truncated, although we identified 11 full-length, 5' inverted L1s.
The annotation of the breakpoint between two segments that make up a 5' inverted L1 element is rarely perfect (Figure 5). Ostertag and Kazazian [47] suggested that this imperfection is a consequence of a twin-priming mechanism by which 5' inverted elements are formed. The vast majority of the time, we found either a small (mean = 12 nucleotides) overlap or deletion in the L1 sequence at the breakpoint location. Furthermore, a five-nucleotide microhomology in the contig sequence occurred nearly 80% of the time at the junction where the 5' ends of the two L1 segments meet (Figure 5).
Figure 5.
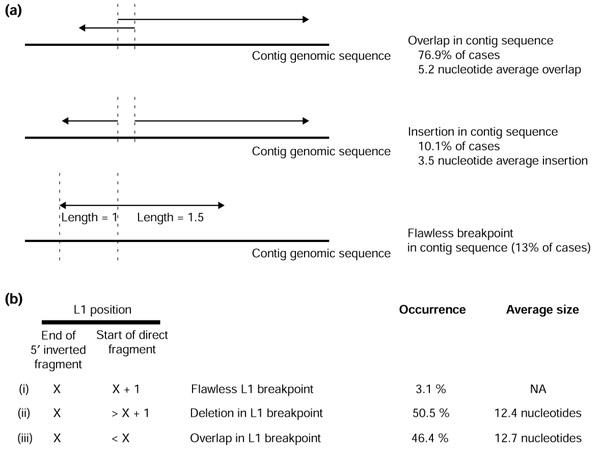
L1 5' inversion structures. For each 5' inverted L1 element for which TSDs were found, the structure at the junction between the two L1 segments was analyzed. Arrows represent the orientation of L1 segments in the genome; an unrearranged L1 would consist of two tandem arrows. Vertical dotted lines indicate breakpoints in the L1 sequence and the alignment of the L1 segments on the genomic sequence. (a) The annotation of the two segments may result in a slight overlap or gap in the contig sequence. Alternatively, the genomic contig sequence may be flawless at the junction of the two fragments. The numbers on the figure for the flawless junction represent the median relative lengths of all of the 5' inverted and 3' direct segments. For those cases in which the 3' direct segment is longer than the 5' inverted segment, the median ratio was 2.3 (70% of cases). (b) In the L1 sequence itself, the two segments that make up the inverted element may overlap (iii) or suggest a small deletion in the L1 sequence (ii). 'X' represents any coordinate in the L1 sequence.
To test whether the inversion breakpoints occur at a conserved distance from the 5' or 3' ends of the L1 elements, the start and the end positions of the 5' inverted L1 segment were compared using a scatterplot diagram for each inverted L1 with TSDs (Figure 6a). From this display, it is clear that there is no clear site preference; the lengths of 3' L1 segments are quite variable, and the inversion point itself can be positioned almost anywhere within the L1, although the density of the breakpoints is significantly higher towards the 3' end of the element (as expected, since most L1s are 5' truncated). A similar scatterplot diagram was used to explore any interdependence between the lengths of the inverted and non-inverted segments within each pair (Figure 6b). The distribution of these pairs of lengths is also quite variable, although there may be a tendency for the 5' inverted segment to be shorter than the direct segment. To confirm this observation, we calculated the ratios of the 3' direct segment length to the 5' inverted segment length. For nearly 70% of inverted L1s, this ratio was greater than one, and the median ratio for this group was 2.3. If all length ratios were considered, the median ratio was 1.5 (Figure 5).
Figure 6.

Characteristics of 5' inverted L1 segments. (a) Start and end positions (projected onto L1.3) of 3,157 5' inverted L1 segments is shown for the 5' inverted L1s with TSDs. (b) The 5' (inverted) segment length is plotted along the x axis; the 3' (direct) segment length is plotted along the y axis for all 5' inverted L1s with TSDs. The direction (inverted versus direct) is given relative to that of L1 transcription.
Next we examined the sequence composition of the pre-breakpoint site. This analysis was performed on the subset of the inverted L1 segment pairs that exhibited a 'flawless' breakpoint (no deletion, insertion, or microhomology, Figure 5). We concatenated the first 15 nucleotides of the 3' direct segment with the reverse complement of the 15 nucleotides that were most 3' (with respect to the L1 sequence) in the 5' inverted segment. This pre-inversion breakpoint sequence exhibited a slight tendency for an 'A' (45% of cases) immediately 5' of the inversion breakpoint (data not shown).
We found only two examples of twice-inverted L1s in the genome. The anatomy of these two insertions is shown in Figure 7. These elements lacked TSDs, and in one case, appear to represent coincidental insertion of two elements near each other.
Figure 7.

Twice-inverted L1 elements. L1 segments shown with solid lines were found when we used the L1.3 sequence as our library for RepeatMasker; the percentages indicate the percent identity of each segment to L1.3. L1 segments indicated with dashed lines were found when we further analyzed the sequence using the full set of RepeatMasker libraries; these segments were identified as L1MA9 family members. The 3' UTR of the L1MA9 subfamily of L1s is 160 nucleotides longer than that for the Ta family of L1s [23]; thus, we were unable to determine the percent identity of the L1MA9 segments to L1.3.
Gene neighborhood of L1 insertions
To investigate the distribution of our set of L1s in the genome with respect to genes, we identified the start and end points of coding sequence (CDS) and mRNA annotations defined in the header of each NCBI contig record. Next, the position of each L1 on the same contig was compared with the coordinates of the gene annotations. We found that L1s have inserted within a genomic segment to which an mRNA and/or CDS has been mapped approximately 17% and 13% of the time, respectively (Table 5). The mean length of these L1s was around 1,640 nucleotides, similar to the 1,614-nucleotide mean length of our entire TSD-containing set of L1s. Of the L1s found within an annotated gene segment, 62% were oriented in the opposite direction to the transcription of the gene. This modest majority may reflect a weak selection against L1s in the same orientation; such insertions could result in premature truncation of a gene's mRNA due to the L1 polyadenylation signal [27,48,49]. When we analyzed the groups of structural variants of L1 separately (Figure 1b), we observed little difference among them in their distribution in genes (data not shown).
Table 5.
L1 insertions into annotated genes
| L1 genome location compared to genes* | mRNA | CDS |
| Fully contained in genes | 17% | 13% |
| Same orientation | 38% | 38% |
| Average L1 length within gene annotation (nucleotides) | 1,470 | 1,490 |
| Opposite orientation | 62% | 62% |
| Average L1 length within gene annotation (nucleotides) | 1,815 | 1,780 |
| Average distance from an L1 to a gene (kb) | 282 | 283 |
| Minimum distance 9 nucleotides | ||
| Maximum distance 3,840 kb | ||
| Same orientation as nearest gene | 49% | 49% |
| Opposite orientation as nearest gene | 51% | 51% |
| Annotated genes with L1s within boundaries | ||
| Total number of genes with L1s | 2,864 | 2,352 |
| Average genome extent (kb) | 161 | 141 |
| Minimum genome extent (kb) | 1.9 | 2.5 |
| Maximum genome extent (kb) | 1,971 | 1,281 |
| Average AT composition | 55% | 55% |
| Minimum AT composition | 31% | 31% |
| Maximum AT composition | 69% | 69% |
*The location of all L1 insertions with TSDs was compared to the genomic location of genes. The start-end coordinates of genes were determined from the headers of the GenBank contig (NT_*) files.
We found that 2,864 mRNAs (approximately 9% of all genes) have L1s in their introns or UTRs (Table 5). The average genomic extent of these genes (approximately 150 kb) is five times longer than the 27 kb genome-wide average [1]. The average GC composition of these genes is 45%, which is similar to the 40-45% average GC content of genes in the genome [50]. For L1s that did not insert within the genomic boundaries of a gene, the closest annotated mRNA or CDS was, on average, 282 kb upstream or downstream. No trend in L1 orientation with respect to gene orientation was evident for these L1s (Table 5).
To obtain a more global view of relatively recent L1 insertions and gene location, we plotted the distribution of L1s with TSDs and the annotated genes in each 500 kb bin along the length of select chromosomes (Figure 8). Alongside this distribution, we show the estimated boundaries for the cytogenetic bands on the chromosomes, allowing us to explore whether L1 density is associated with a particular cytogenetic band. It is clear from Figure 8 that L1s are neither concentrated in nor excluded from any particular cytogenetic band. In fact, the proportions of the entire genome defined by each type of cytogenetic band are equivalent to the proportions with which L1s are found in each type of cytogenetic band (data not shown).
Figure 8.

Distribution of L1s with TSDs and annotated genes on chromosomes. Along each 500 kb bin along the chromosomes shown, the total number of L1s with TSDs (blue line) and the annotated genes (pink line) are indicated. Stars represent loci at which the L1 concentration is quite low and gene concentration is high. Among these loci are the histone gene cluster and MHCIII cluster on chromosome 6, a group of genes subject to X inactivation on the X chromosome, and a group of unrelated genes on chromosome 1. The corresponding cytogenetic staining pattern along the length of the chromosomes is shown below the graphs. Centromeric regions are white, and Giemsa-negative bands are indicated by the lightest gray. For the Giemsa-positive bands, the intensity of the grayscale reflects the staining intensity.
Although Figure 8 shows that L1s and genes are generally intermixed, there are regions at which one or the other predominates as reported previously [1] (see peaks with stars in Figure 8). We analyzed chromosomal regions (500 kb bins) for which the difference between L1 number and gene number was maximal, yet the proportion of sequence in the region occupied by genes was less than 60%. Such regions include the class III major histocompatibility complex and histone gene clusters on chromosome 6, the human alpha-globin gene cluster located on chromosome 16, and others. Our analysis of these regions of the genome revealed an enrichment in SINES (approximately 25% of the sequence bin versus 13% overall in the genome) and GC content (approximately 50%), two common properties associated with gene-rich, L1-poor regions of the genome [1].
Interestingly, one of the L1-poor/gene-rich regions identified is on the X chromosome (Figure 8). This particular locus (Xq28) encompasses genes that are believed to be subject to X inactivation. These genes include those for glucose-6-phosphate dehydrogenase, XAP-5, renin-binding protein, N-acetyltransferase (homolog of Saccharomyces cerevisiae ARD1), signal sequence receptor-delta, isocitrate dehydrogenase 3 (NAD+) gamma, biglycan, tafazzin, deoxyribonuclease I-like 1, and emerin [51]. The paucity of L1s identified at this locus suggests that L1s are not critical for the propagation of the X inactivation signal along the length of the X chromosome as has been proposed [39]. Furthermore, the concentration of L1s with TSDs on the p arm of the X chromosome is lower than that on the q arm (Figure 8). Lahn and Page [52] have hypothesized that the distal p arm is the site of the youngest evolutionary strata on the X chromosome, indicating that the X and Y chromosomes actively recombined at this location most recently. Therefore, as proposed by Boissinot et al. [38], it is possible that recombination prevented the accumulation of L1s at this chromosomal region.
Discussion
To gain insight into the mechanisms by which L1 structural variants are created, and to identify trends associated with their insertion, we carried out a comprehensive analysis of the L1 sequences and their surrounding DNA loci. To facilitate this study, we wrote the software TSDfinder, which identifies the TSDs, poly(A) tails, and inversion breakpoints (if any) associated with L1s. We found no correlation between the length and quality of either the TSDs or the poly(A) tails and the resulting anatomy of the L1 element. Thus, the poly(A) tail quality probably does not influence the formation of 5' inverted elements. Furthermore, this suggests that the specificity of the initial step of the retrotransposition is conserved in all classes of elements. It is important to consider, however, that any hallmark sequence structures uniquely associated with the insertion of structural variants may be refractory to analysis after millions of years of evolution. For example, Ovchinnikov et al. reported that the poly(A) tails of L1s may become shorter over time [24].
Our analysis of the pre-insertion loci of L1s with TSDs confirms that the top strand consensus site of L1 endonuclease is TTAAAA. This result is completely consistent with previous studies of the L1 endonuclease targeting preference in vitro and as inferred from the TSDs of Alu elements, which are thought to hijack the L1 machinery for their insertion [6,8,53]. We also observed this pattern, albeit not as robust, at the 3' end of the pre-insertion locus, suggesting that L1 endonuclease may also be responsible for the second-strand cleavage of host DNA during L1 insertion. There is evidence that the related sequence-specific R1 element endonuclease makes both cuts in the target DNA [18].
The frequency with which L1s were found in high-copy repeats indicates that insertion into pre-existing L1s and Alu sequences may be somewhat disfavored. Apart from this, the observed frequencies are roughly similar to those with which these same sequence classifications occur in the human genome [1]; therefore, other than a preference for AT-rich DNA, L1 insertion at a particular genomic locus may be influenced more by local chromatin structure than by specific long-range sequence composition of the DNA. On the other hand, Ovchinnikov et al. [24] found that very recently inserted polymorphic L1s were randomly distributed and their distribution shifted over time. Thus, the current flanking sequences of L1s may be an inaccurate reflection of the target DNA of any given L1 at the time of insertion.
In our analysis of 5' inverted L1s, we failed to reveal any similarity of the breakpoint at the junction between the two fragments with the L1 endonuclease consensus cleavage site. This suggests that L1 endonuclease does not participate in 5' inversion formation by cleaving L1 cDNA. Instead, our findings support the recently proposed twin-priming model for inversion [47]; as we showed above, the 3' end top-strand cleavage product has a modest bias to be a 'T' nucleotide (Figure 3b) and we indeed see a modest preference for a complementary 'A' nucleotide at the corresponding position of the inversion breakpoint (data not shown). On the other hand, whereas Ostertag and Kazazian [47] report that the inversion points are clustered towards the 3' end of inverted L1s, we found no clear bias in the position of the breakpoint position in our set of 5' inverted L1s. This discrepancy may be due to their small sample size of inverted elements, the majority of which were less than 2,000 nucleotides long. Another discrepancy between our datasets is their observation that the length of the non-inverted L1 segment is shorter than the inverted segment. In contrast, we found the 3' segment of 5' inverted L1s to be generally longer than the 5' segment.
As inverted elements are found 8% of the time in 3' intact elements, 462 twice-inverted elements were predicted to occur if such events are permitted (that is, 8% of 8% of the time if the two inversions can occur independently more than once during the same retrotransposition event). This provides a powerful independent test of the twin-priming model which cannot explain multiply inverted elements. The fact that we only found two such events suggests that whereas single inversions are a simple perversion of the normal retrotransposition process such as twin priming, doubly inverted elements require some other extremely rare event to occur.
We found that compared to the other L1 structural variants, a smaller proportion of 3' transduction L1s have a patterned poly(A) tail. This is consistent with the poly(A) tails of 3' transduction events being formed mostly through the action of poly(A) polymerase. On the other hand, the presence of L1s with patterned tails is consistent with the results of Chaboissier et al. [40] who found that elements with patterned tails could be converted to elements with heritable poly(A) tails. We propose that L1 elements also exist as two populations, and that L1s with patterned tails beget other elements with patterned tails. These data suggest that polyadenylation of L1 transcripts by poly(A) polymerase may not be obligatory in the formation of standard L1 insertions. Alternatively, it has been reported that the poly(A) tails of retrotransposons may be a source for the creation of microsatellites through post-insertional mutations [24,54]; the patterned tails we found may therefore exemplify this transition from poly(A) tail to microsatellites.
It has been suggested that the high concentration of L1s on the X chromosome may have a role in X inactivation of select genes [39]. This hypothesis seems unlikely, as we see the same elevated density of L1s on the Y chromosome. Boissinot et al. [38] have hypothesized that the high frequency of full-length L1 elements on the Y chromosome may be due to the inability of the Y chromosome to recombine, which is a process by which potentially harmful full-length L1s are eliminated from the genome. Furthermore, we found that a region of the X chromosome at which many X-inactivated genes are located is quite L1-poor, a finding that is contrary to a direct role for L1s in X inactivation.
Implementation of TSDfinder on a genome-wide scale has provided new insights to a variety of hypotheses about L1 insertion and evolution. Finally, analysis of inverted elements by TSDfinder provides strong, independent support for the twin-priming model.
Materials and methods
Dataset
The dataset of human genomic DNA sequence records used was the set of contigs assembled at NCBI [55] as of 29 August 2001 (build 25). This set of sequences should be non-overlapping, eliminating the problem of multiple sampling of the same L1 elements. The dataset consisted of 3,347 human contigs, amounting to 2.8 gigabases (Gb) of nonredundant genomic sequence and corresponding to approximately 91% of the human euchromatic genome (Table 1). The contigs are a mixture of finished and draft sequence.
Identification of L1s and TSDs
L1 elements in the contigs were annotated using RepeatMasker, version of 9 April 2002 [20], using default settings with a custom library containing only the L1.3 element (GenBank accession number L19088.1) lacking the 37-nucleotide poly(A) tail and therefore ending with the first three nucleotides (AAT) of the putative polyadenylation signal. Within this L1.3 sequence, we also included the variably present 131 nucleotides in the 5' UTR of L1s at the appropriate position [26]. Even though the custom library was limited to the L1.3 element, we expected most young L1 elements to be collected by using this single consensus sequence.
In the TSDfinder program [33] written in Perl [56], L1 coordinates were parsed from the RepeatMasker results in the *.out file. Only L1s intact at the 3' end (RepeatMasker annotation ends < 31 nucleotides from the 3' end) were considered in the TSD-finding analysis. L1s that were fragmented in the RepeatMasker annotation because of the absence of the variably present 131 nucleotides in the 5' UTR were merged. Annotated L1 segments were also merged if intervening insertions were < 31 nucleotides long and deletions of the standard L1 sequence were < 51 nucleotides. By merging adjacent L1 fragments in this way, the actual L1 element was likely to be recreated and positioned the insertion ends with maximal precision. This step was important for maximizing the yield of correct TSDs; without this merging, we found nearly 10% fewer TSDs.
Candidate poly(A) tails were identified in the 3 kb downstream of the 3' UTR and the poly(A) tail start-end coordinates were recorded. Stretches of sequence that qualified as poly(A) tail candidates had a minimum length of 10 nucleotides and at least 73% As. These criteria were previously defined by analyzing the DNA sequences that extended from the end of the 3' UTR to the start of the 3' TSD in a random sampling of 25 L1s from the genome that were collected on the basis of having high-confidence TSDs > 14 nucleotides long. In addition, tails containing more than two adjacent non-A bases were disallowed. These criteria were relaxed if patterned repeats were detected in a poly(A) sequence tract (for example, AAAT).
For each element found by RepeatMasker (following the merge step), 100 nucleotides of upstream sequence and 3 kb of sequence downstream from the L1 boundaries were collected. The longer region downstream is used to detect 3' transduction events. The first and the last 15 nucleotides of the L1 itself are also included in the TSD search to allow for some L1 boundary imprecision. TSDs were identified using bl2seq (parameters: -g F -W 9 -F F -S 1 -d 3000 -e 1000.0) [57,58]. TSDs were required to be at least nine nucleotides long. The algorithm for TSD signature recognition allows for detection of both 'standard' insertion events (in which the 3' TSD immediately follows the poly(A) tract associated with the retrotransposon) and possible 3' transduction events (in which the 3' TSD follows a poly(A) tail further downstream from the retrotransposon 3' end).
TSD scoring scheme
The high-scoring pairs (HSPs) identified by bl2seq were scored heuristically by considering three factors: the HSP position in the 5' flank of the L1; the HSP position in the 3' flank of the L1; and HSP quality score. If the 5' HSP ends within 10 nucleotides from the L1 start, the 5' HSP position score is 100. If the 5' HSP ends further away from L1 start, the score function is described by the formula: P × 80/(FF -10), where FF is the length of the L1 5'-flanking sequence used for TSD finding (100 nucleotides is default) and P is the 5' HSP end position within this flank. The 3' HSP position score was created to give a higher weight to a standard insertion, as opposed to a 3' transduction event, in case both signatures are present for a given L1 element. The 3' HSP position score of 20 is given if the 3' HSP starts within 30 nucleotides of the annotated L1 end. The last scoring function, HSP quality score, assigns a score of 20 for HSPs 11-18 nucleotides long without mismatches, and 10 for all other HSPs with an acceptable number of mismatches (no mismatches allowed for HSPs < 11 nucleotides; one mismatch allowed for HSPs up to 18 nucleotides; one mismatch per 50 nucleotides allowed for HSPs > 18 nucleotides). The final TSD score is the sum of the scores described above. In cases where multiple TSDs were identified, TSDs with the best score and with a candidate poly(A) tail immediately preceding the 3' HSP were defined as the TSD for a particular L1 insertion. When a microhomology existed between the poly(A) tail and the TSD, the microhomologous region was arbitrarily considered part of the TSD by TSDfinder (Figure 1c).
As in vivo studies of L1 insertions have shown that the average length of TSDs is approximately 14 nucleotides, our scoring function reflected this by giving a higher weight to exact matches of lengths 11-20 nucleotides. To exclude the possibility that the abundance of the 11-20-nucleotide long TSDs in our collection is exclusively a result of this aspect of the scoring scheme, TSDfinder analysis of this dataset was repeated without rewarding a higher value to TSDs of length > 10 nucleotides, and the distribution of TSD lengths remained generally unchanged.
Monte Carlo simulation of TSD nucleotide composition was carried out using an array of 100 nucleotides with the same composition as the human genome (41% GC, 59% AT) [1] to generate 1,800 15-nucleotide random sequences. (Note for comparison that there were 1,794 15-nucleotide-long TSDs without mismatches found in the genome.)
Pre-insertion loci
The pre-insertion locus of each L1 was recreated by collecting 50 nucleotides upstream and downstream from the 5' and 3' TSDs, respectively, and appending them to the TSD sequence. This sequence was used to analyze the nucleotide neighborhood of each L1 insertion for which the TSD score was ≥ 100. These sequences were input to RepeatMasker software for determining the frequency with which L1s had inserted into pre-existing high-copy repeats. To perform this analysis, the *.out file was parsed. In determining whether an L1 had inserted into a high-copy sequence, the repeat must be equal to or greater than 75% identical to a reference repeat, and it must extend across the fused TSD/flank sequence for 25 nucleotides in either direction unless the repeat starts or ends elsewhere in the pre-insertion locus.
DNA sequence logos [59] of TSDs were generated using binary code downloaded from the site [60]. Only TSDs that did not have mismatches were used to generate logos. Ten nucleotides upstream and downstream of the TSDs were included in the logos.
Genome distribution and features of L1 insertion sites
To determine the gene neighborhood of each L1, the header for each chromosomal contig GenBank flatfile (the *.gbs files are available in the individual chromosome files at [55]) was collected and scanned for mRNA or CDS annotations. The name and the absolute start and end positions of each annotation were recorded. The location of each L1 within that particular contig was compared with the locations of the mRNA and CDS annotations. L1s were recorded as either falling entirely within the boundaries of a gene annotation, or the distances to the closest upstream and downstream gene annotations were calculated. Such statistics for certain L1s could not be determined because some contigs with L1s include no annotated CDS or mRNAs. The same approach was used to generate the mRNA and CDS densities along the length of each chromosome; if multiple, mildly variable annotations were assigned to the same gene name, the first annotation listed in the GenBank flatfile header was used as the definitive location.
The cytogenetic banding pattern associated with each L1 was determined from the information included in the seq_contig.md and ISCN800 files [61]. These same files were used to generate the cytogenetic figure; if the orientation of a contig was unknown, it was arbitrarily considered to be positive.
Full-length L1 analysis
All L1 sequences for which there was no more than a 10-nucleotide deletion at the 5' end and no more than a 30-nucleotide deletion at the 3' end were collected. The BLASTX program [46] was used to translate these L1 DNA sequences in all three reading frames and compare them to the ORF1 and ORF2 amino acid sequences of L1.3. We analyzed the highest-scoring translation for each L1 sequence for the extent of the protein alignment, nonsense mutations, and missense mutations that would likely eliminate protein function. In the ORF1 protein, residues critical for retrotransposition include the following conserved blocks of amino acids: REKG (235-238), ARR (residues 260-262), and YPAKLS (282-287) [14]. For ORF2, key residues for endonuclease activity include the residues at positions 12(N), 43(E), 115(Y), 145(D), 147(N), 192(T), 205(D), and 228-230(SDH) [12]. In addition, mutations at the catalytic FADD box in the reverse transcriptase domain (codons 700-703), a cysteine-rich region (1143, 1147, 1160, and the invariant HC at 1155-1156), and the conserved blocks of amino acids 1091-1094(HMKK) and 1096-1098(888) result in reduced L1 transposition activity [14]. Finally, residues 472(G) and 474(D) in the conserved Z motif in ORF2 are required for reverse transcriptase activity [46].
Acknowledgments
Acknowledgements
We are grateful to members of the Boeke lab and Landsman group for helpful discussions during the preparation of this manuscript, especially Liora Strichman-Almashanu, John S.J. Anderson, Wataru Fujibuchi, David Symer, and Greg Cost. We thank Qing Liu for work on Figure 3a. We also acknowledge Joana Silva for critical reading of the manuscript. This work was supported by NIH grant CA16519 to J.D.B.
References
- International Human Genome Sequencing Consortium (IHGSC) Initial sequencing of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Jurka J. Subfamily structure and evolution of the human L1 family of repetitive sequences. J Mol Evol. 1989;29:496–503. doi: 10.1007/BF02602921. [DOI] [PubMed] [Google Scholar]
- Matera AG, Hellmann U, Hintz MF, Schmid CW. Recently transposed Alu repeats result from multiple source genes. Nucleic Acids Res. 1990;18:6019–6023. doi: 10.1093/nar/18.20.6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AF. The origin of interspersed repeats in the human genome. Curr Opin Genet Dev. 1996;6:743–748. doi: 10.1016/s0959-437x(96)80030-x. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Goodier JL, Ostertag EM, Kazazian HH., Jr Rapid amplification of a retrotransposon subfamily is evolving the mouse genome. Nat Genet. 1998;20:288–290. doi: 10.1038/3104. [DOI] [PubMed] [Google Scholar]
- Jurka J. Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc Natl Acad Sci USA. 1997;94:1872–1877. doi: 10.1073/pnas.94.5.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias SL, Scott AF, Kazazian HH, Jr, Boeke JD, Gabriel A. Reverse transcriptase encoded by a human transposable element. Science. 1991;254:1808–1810. doi: 10.1126/science.1722352. [DOI] [PubMed] [Google Scholar]
- Boeke JD. LINEs and Alus - the polyA connection. Nat Genet. 1997;16:6–7. doi: 10.1038/ng0597-6. [DOI] [PubMed] [Google Scholar]
- Dhellin O, Maestre J, Heidmann T. Functional differences between the human LINE retrotransposon and retroviral reverse transcriptases for in vivo mRNA reverse transcription. EMBO J. 1997;16:6590–6602. doi: 10.1093/emboj/16.21.6590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esnault C, Maestre J, Heidmann T. Human LINE retrotransposons generate processed pseudogenes. Nat Genet. 2000;24:363–367. doi: 10.1038/74184. [DOI] [PubMed] [Google Scholar]
- Dombroski BA, Mathias SL, Nanthakumar E, Scott AF, Kazazian HH., Jr Isolation of an active human transposable element. Science. 1991;254:1805–1808. doi: 10.1126/science.1662412. [DOI] [PubMed] [Google Scholar]
- Feng Q, Moran JV, Kazazian HH, Jr, Boeke JD. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell. 1996;87:905–916. doi: 10.1016/s0092-8674(00)81997-2. [DOI] [PubMed] [Google Scholar]
- Dombroski BA, Feng Q, Mathias SL, Sassaman DM, Scott AF, Kazazian HH, Jr, Boeke JD. An in vivo assay for the reverse transcriptase of human retrotransposon L1 in Saccharomyces cerevisiae. Mol Cell Biol. 1994;14:4485–4492. doi: 10.1128/mcb.14.7.4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HH., Jr High frequency retrotransposition in cultured mammalian cells. Cell. 1996;87:917–927. doi: 10.1016/s0092-8674(00)81998-4. [DOI] [PubMed] [Google Scholar]
- Mizrokhi LJ, Georgieva SG, Ilyin YV. jockey, a mobile Drosophila element similar to mammalian LINEs, is transcribed from the internal promoter by RNA polymerase II. Cell. 1988;54:685–691. doi: 10.1016/s0092-8674(88)80013-8. [DOI] [PubMed] [Google Scholar]
- Swergold GD. Identification, characterization, and cell specificity of a human LINE-1 promoter. Mol Cell Biol. 1990;10:6718–6729. doi: 10.1128/mcb.10.12.6718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan DD, Korman MH, Jakubczak JL, Eickbush TH. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell. 1993;72:595–605. doi: 10.1016/0092-8674(93)90078-5. [DOI] [PubMed] [Google Scholar]
- Feng Q, Schumann G, Boeke JD. Retrotransposon R1Bm endonuclease cleaves the target sequence. Proc Natl Acad Sci USA. 1998;95:2083–2088. doi: 10.1073/pnas.95.5.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanning TG, Singer MF. LINE-1: a mammalian transposable element. Biochim Biophys Acta. 1987;910:203–212. doi: 10.1016/0167-4781(87)90112-6. [DOI] [PubMed] [Google Scholar]
- RepeatMasker version 04/09/2000 http://repeatmasker.genome.washington.edu
- Boissinot S, Furano AV. Adaptive evolution in LINE-1 retrotransposons. Mol Biol Evol. 2001;18:2186–2194. doi: 10.1093/oxfordjournals.molbev.a003765. [DOI] [PubMed] [Google Scholar]
- Boissinot S, Chevret P, Furano AV. L1 (LINE-1) retrotransposon evolution and amplification in recent human history. Mol Biol Evol. 2000;17:915–928. doi: 10.1093/oxfordjournals.molbev.a026372. [DOI] [PubMed] [Google Scholar]
- Smit AF, Toth G, Riggs AD, Jurka J. Ancestral, mammalian-wide subfamilies of LINE-1 repetitive sequences. J Mol Biol. 1995;246:401–417. doi: 10.1006/jmbi.1994.0095. [DOI] [PubMed] [Google Scholar]
- Ovchinnikov I, Troxel AB, Swergold GD. Genomic characterization of recent human LINE-1 insertions: evidence supporting random insertion. Genome Res. 2001;11:2050–2058. doi: 10.1101/gr.194701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott AF, Schmeckpeper BJ, Abdelrazik M, Comey CT, O'Hara B, Rossiter JP, Cooley T, Heath P, Smith KD, Margolet L. Origin of the human L1 elements: proposed progenitor genes deduced from a consensus DNA sequence. Genomics. 1987;1:113–125. doi: 10.1016/0888-7543(87)90003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori M, Hidaka S, Sakaki Y. Sequence analysis of a KpnI family member near the 3' end of human beta-globin gene. Nucleic Acids Res. 1985;13:7813–7827. doi: 10.1093/nar/13.21.7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symer DE, Connelly C, Szak ST, Caputo EM, Cost GJ, Parmigiani G, Boeke JD. Human L1 retrotransposition is associated with genetic instability in vivo. Cell. 2002;110:327–338. doi: 10.1016/s0092-8674(02)00839-5. [DOI] [PubMed] [Google Scholar]
- Holmes SE, Dombroski BA, Krebs CM, Boehm CD, Kazazian HH., Jr A new retrotransposable human L1 element from the LRE2 locus on chromosome 1q produces a chimaeric insertion. Nat Genet. 1994;7:143–148. doi: 10.1038/ng0694-143. [DOI] [PubMed] [Google Scholar]
- Pickeral OK, Makalowski W, Boguski MS, Boeke JD. Frequent human genomic DNA transduction driven by LINE-1 retrotransposition. Genome Res. 2000;10:411–415. doi: 10.1101/gr.10.4.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier JL, Ostertag EM, Kazazian HH. Transduction of 3'-flanking sequences is common in L1 retrotransposition. Hum Mol Genet. 2000;9:653–657. doi: 10.1093/hmg/9.4.653. [DOI] [PubMed] [Google Scholar]
- Narita N, Nishio H, Kitoh Y, Ishikawa Y, Minami R, Nakamura H, Matsuo M. Insertion of a 5' truncated L1 element into the 3' end of exon 44 of the dystrophin gene resulted in skipping of the exon during splicing in a case of Duchenne muscular dystrophy. J Clin Invest. 1993;91:1862–1867. doi: 10.1172/JCI116402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symer DE, Connelly C, Szak ST, Caputo EM, Cost GJ, Parmigiani G, Boeke JD. Human L1 retrotransposition is associated with genetic instability in vivo. Cell. 2002;110:327–338. doi: 10.1016/s0092-8674(02)00839-5. [DOI] [PubMed] [Google Scholar]
- Pickeral OK. Bioinformatics of human retrotransposons. In: Baltimore MD, editor. PhD dissertation. The Johns Hopkins University; 2000. [Google Scholar]
- Cost GJ, Boeke JD. Targeting of human retrotransposon integration is directed by the specificity of the L1 endonuclease for regions of unusual DNA structure. Biochemistry. 1998;37:18081–18093. doi: 10.1021/bi981858s. [DOI] [PubMed] [Google Scholar]
- Kimberland ML, Divoky V, Prchal J, Schwahn U, Berger W, Kazazian HH., Jr Full-length human L1 insertions retain the capacity for high frequency retrotransposition in cultured cells. Hum Mol Genet. 1999;8:1557–1560. doi: 10.1093/hmg/8.8.1557. [DOI] [PubMed] [Google Scholar]
- Dombroski BA, Scott AF, Kazazian HH., Jr Two additional potential retrotransposons isolated from a human L1 subfamily that contains an active retrotransposable element. Proc Natl Acad Sci USA. 1993;90:6513–6517. doi: 10.1073/pnas.90.14.6513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassaman DM, Dombroski BA, Moran JV, Kimberland ML, Naas TP, DeBerardinis RJ, Gabriel A, Swergold GD, Kazazian HH., Jr Many human L1 elements are capable of retrotransposition. Nat Genet. 1997;16:37–43. doi: 10.1038/ng0597-37. [DOI] [PubMed] [Google Scholar]
- Boissinot S, Entezam A, Furano AV. Selection against deleterious LINE-1-containing loci in the human lineage. Mol Biol Evol. 2001;18:926–935. doi: 10.1093/oxfordjournals.molbev.a003893. [DOI] [PubMed] [Google Scholar]
- Bailey JA, Carrel L, Chakravarti A, Eichler EE. Molecular evidence for a relationship between LINE-1 elements and X chromosome inactivation: the Lyon repeat hypothesis. Proc Natl Acad Sci USA. 2000;97:6634–6639. doi: 10.1073/pnas.97.12.6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaboissier M-C, Finnegan D, Bucheton A. Retrotransposition of the I factor, a non-long terminal repeat retrotransposon of Drosophila, generates tandem repeats at the 3' end. Nucleic Acids Res. 2000;28:2467–2472. doi: 10.1093/nar/28.13.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucheton A, Paro R, Sang HM, Pelisson A, Finnegan DJ. The molecular basis of I-R hybrid dysgenesis in Drosophila melanogaster: identification, cloning, and properties of the I factor. Cell. 1984;38:153–163. doi: 10.1016/0092-8674(84)90536-1. [DOI] [PubMed] [Google Scholar]
- Kazazian HH, Jr, Moran JV. The impact of L1 retrotransposons on the human genome. Nat Genet. 1998;19:19–24. doi: 10.1038/ng0598-19. [DOI] [PubMed] [Google Scholar]
- Singer MF, Krek V, McMillan JP, Swergold GD, Thayer RE. LINE-1: A human transposable element. Gene. 1993;135:183–188. doi: 10.1016/0378-1119(93)90064-a. [DOI] [PubMed] [Google Scholar]
- Minakami R, Kurose K, Ethoh K, Furuhata Y, Hattori M, Sakaki Y. Identification of an internal cis-element essential for the human L1 transcription and a nuclear factor(s) binding to the element. Nucleic Acids Res. 1992;20:3139–3145. doi: 10.1093/nar/20.12.3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements AP, Singer MF. The human LINE-1 reverse transcriptase: effect of deletions outside the common reverse transcriptase domain. Nucleic Acids Res. 1998;26:3528–3535. doi: 10.1093/nar/26.15.3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag EM, Kazazian HH., Jr Twin priming: A proposed mechanism for the creation of inversions in L1 retrotransposition. Genome Res. 2001;11:2059–2065. doi: 10.1101/gr.205701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AF. Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr Opin Genet Dev. 1999;9:657–663. doi: 10.1016/s0959-437x(99)00031-3. [DOI] [PubMed] [Google Scholar]
- Eickbush T. Exon shuffling in retrospect. Science. 1999;283:1465–1467. doi: 10.1126/science.283.5407.1465. [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Carrel L, Cottle AA, Goglin KC, Willard HF. A first-generation X-inactivation profile of the human X chromosome. Proc Natl Acad Sci USA. 1999;96:14440–14444. doi: 10.1073/pnas.96.25.14440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahn BT, Page DC. Four evolutionary strata on the human X chromosome. Science. 1999;286:964–967. doi: 10.1126/science.286.5441.964. [DOI] [PubMed] [Google Scholar]
- Cost GJ, Golding A, Schlissel MS, Boeke JD. Target DNA chromatinization modulates nicking by L1 endonuclease. Nucleic Acids Res. 2001;29:573–577. doi: 10.1093/nar/29.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcot SS, Wang Z, Weber JL, Deininger PL, Batzer MA. Alu repeats: a source for the genesis of primate microsatellites. Genomics. 1995;29:136–144. doi: 10.1006/geno.1995.1224. [DOI] [PubMed] [Google Scholar]
- Human genome sequences ftp://ftp.ncbi.nlm.nih.gov/genomes/H_sapiens
- NCBI Computational Biology Branch: David Landsman's group http://www.ncbi.nlm.nih.gov/CBBresearch/Landsman/TSDfinder/
- BLAST 2 Sequences http://www.ncbi.nlm.nih.gov/blast/bl2seq/bl2.html
- Tatusova TA, Madden TL. BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol Lett. 1999;174:247–250. doi: 10.1111/j.1574-6968.1999.tb13575.x. [DOI] [PubMed] [Google Scholar]
- Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delila programs: index program http://www.lecb.ncifcrf.gov/~toms/delila
- Human genome sequences: maps ftp://ftp.ncbi.nlm.nih.gov/genomes/H_sapiens/maps/mapview/