

Short abstract
A genome-wide analysis was carried out on patterns of the classified polytopic membrane protein families, and the distribution of conserved amino acids and motifs in the transmembrane helix regions in these families was also analyzed.
Abstract
Background
Polytopic membrane proteins can be related to each other on the basis of the number of transmembrane helices and sequence similarities. Building on the Pfam classification of protein domain families, and using transmembrane-helix prediction and sequence-similarity searching, we identified a total of 526 well-characterized membrane protein families in 26 recently sequenced genomes. To this we added a clustering of a number of predicted but unclassified membrane proteins, resulting in a total of 637 membrane protein families.
Results
Analysis of the occurrence and composition of these families revealed several interesting trends. The number of assigned membrane protein domains has an approximately linear relationship to the total number of open reading frames (ORFs) in 26 genomes studied. Caenorhabditis elegans is an apparent outlier, because of its high representation of seven-span transmembrane (7-TM) chemoreceptor families. In all genomes, including that of C. elegans, the number of distinct membrane protein families has a logarithmic relation to the number of ORFs. Glycine, proline, and tyrosine locations tend to be conserved in transmembrane regions within families, whereas isoleucine, valine, and methionine locations are relatively mutable. Analysis of motifs in putative transmembrane helices reveals that GxxxG and GxxxxxxG (which can be written GG4 and GG7, respectively; see Materials and methods) are among the most prevalent. This was noted in earlier studies; we now find these motifs are particularly well conserved in families, however, especially those corresponding to transporters, symporters, and channels.
Conclusions
We carried out a genome-wide analysis on patterns of the classified polytopic membrane protein families and analyzed the distribution of conserved amino acids and motifs in the transmembrane helix regions in these families.
Background
Genome-wide structural analyses in terms of patterns of protein folding have been useful in revealing functional and evolutionary relationships [1,2,3,4]. Given the abundance of membrane proteins, it would be highly desirable to have a similar analysis for this major category of structures; however, the number of known membrane protein structures remains small. Here we exploit the fact that membrane proteins can be classified into families on the basis of sequence similarities and topology, and use the family groupings to analyze genomic characteristics of membrane protein families.
Most transmembrane proteins are formed from bundles of helices that traverse the membrane lipid bilayer. It is estimated that 20-30% of the proteins in known genomes are of this type [3,4,5,6]. The most general description of the transmembrane helical regions (TMs) is that they comprise a region of 18 or more amino acids with a largely hydrophobic character. This sequence feature can be identified in primary sequences using hydrophobicity scales [7,8,9]. The most abundant amino acids in transmembrane regions are leucine, isoleucine, valine, phenylalanine, alanine, glycine, serine, and threonine. Taken together, these amino acids account for 75% of the amino acids in transmembrane regions [10,11,12]. Analysis of the distribution of amino acids has revealed patterns in TM regions, for example GxxxG, which are thought to be important in helix-helix interactions [11,12,13,14].
We took advantage of the classification of protein domains provided by others (Pfam-A and Pfam-B) [15], to identify families that appear to be polytopic membrane proteins, and augmented these lists with additional family members based on amino-acid sequence comparisons. Furthermore, we identified additional families on the basis of clustering of amino-acid sequences, resulting in 637 distinct families. We used these families to analyze amino-acid compositions in the helical regions, pair motifs, domain structures, and patterns of families, and arrive at a number of generalizations. Among these are that glycine, tyrosine, and proline appear frequently in conserved locations within family transmembrane helices and that the specific pair motifs are found in families that seem to be transporters, symporters, and channels. The number of kinds of domains and families seems to increase with the number of open reading frames (ORFs) in most genomes. Here we present our analysis and discuss these findings.
Results
Classification of polytopic membrane protein domains
The procedure used to classify polytopic membrane domains is based mainly on family classification schemes (Pfam-A and Pfam-B) and is shown in Figure 1a. We identified families of polytopic membrane domains in Pfam [15] by allocating TM-helices annotated in SWISS-PROT [16] to proteins in Pfam. After conservatively picking 183 Pfam-A and 152 Pfam-B families, we conducted an analysis of loops that connect TM-helices. It was shown that the loops tend to be short, with most of them (> 95%) having fewer than 80 amino acids. We therefore took 80 residues as the maximal intra-domain loop between TM-helices to define polytopic membrane domains. Though the 80-residue cutoff may not apply to a small portion (around 5%) of integral membrane proteins, it diminished the chance of including soluble domains within membrane domains, given that the average soluble domain has about 170 residues [17].
Figure 1.
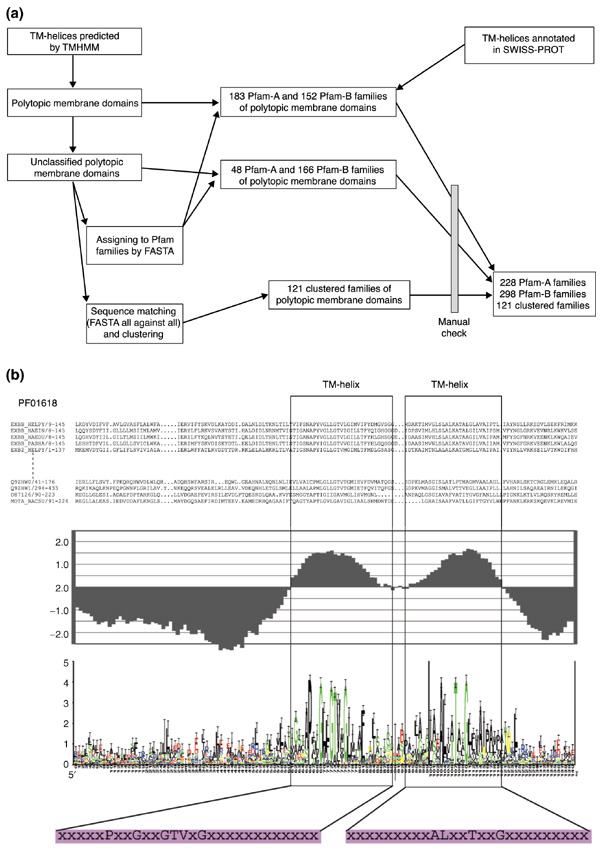
Classification of polytopic membrane domains. (a) Procedure for classifying polytopic membrane domains. Through automatic classification and manual examination, 228 Pfam-A, 299 Pfam-B and 121 clustered families were classified. (b) An example profile (PF01618) of a classified family of polytopic membrane domains consists of (from top to bottom): sequence alignment; an averaged hydrophobicity plot based on GES hydrophobicity value; consensus sequence displayed by sequence logo with conserved residues in hydrophobic regions highlighted; consensus sequences of TM-helices, where only conserved amino acids are shown in the single-letter code (with the remainder represented by "x").
Using TMHMM, a membrane protein prediction program based on a hidden Markov model [6], TM-helices of membrane proteins in 26 genomes were predicted. Polytopic membrane domains were identified using the loop size between TM-helices as a guide. These domains were then classified into 231 Pfam-A and 318 Pfam-B families either by direct SWISS-PROT ID matching or by sequence similarity matching using FASTA [18]. Of the aligned domains, most of their TM-helices also aligned well, especially in Pfam-A families, which have alignments based on manually crafted hidden Markov models. Unclassified domains were clustered into 121 families by their sequence similarities. For each family, a profile was constructed, as shown in Figure 1b. This included: an averaged hydrophobicity plot of all members in the family based on the Goldman-Engelman-Steitz (GES) scale [8]; a consensus sequence of the family, represented by a sequence logo plot [19]; and consensus sequences of the TM-helices. By analyzing the hydrophobicity plots, we can locate TM-helices in the aligned sequences in protein families, and assign a number of TM-helices to each family. Some families, including 3 in Pfam-A and 20 in Pfam-B, were eliminated at this step, owing to the ambiguity of TM-helices observed in the plot. From this process, we identified 228 Pfam-A, 298 Pfam-B and 121 clustered families for our analyses, with approximately 95% domains classified in Pfam families.
Analysis of the number of TM-helices in Pfam-A families of polytopic membrane domains
After assigning a number of TM-helices to each family, we conducted a survey of the assigned numbers of TM-helices in 228 Pfam-A families of polytopic membrane domains (Figure 2). Pfam-A families are manually classified families that have well-aligned protein domains, and most of them have a well-defined number for TM-helices. We also picked families in solute transport systems that are annotated as transporters, symporters and channels, and analyzed the number of TM-helices for these families (Figure 2).
Figure 2.
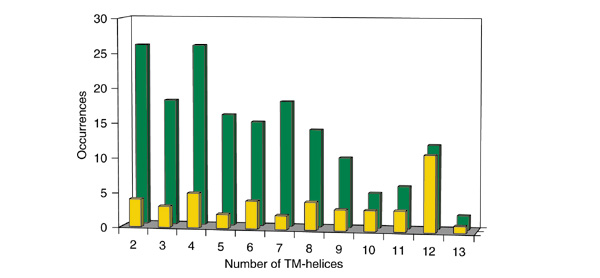
Number of TM-helices in Pfam-A families of polytopic membrane domains. Shown are the number of Pfam-A families of polytopic membrane domains with a given number of TM-helices. Only families with more than 20 members were counted. The green bars indicate numbers from all studied Pfam-A families and the yellow bars those from the Pfam-A families that are annotated as transporters, symporters, and channels.
In general, most Pfam-A families tend to have a small number of TM-helices. For those with seven or fewer TM-helices, the number of families does not vary significantly with helix number, although there are more families with two or four TM-helices than with three, five, six, or seven. For families with more than seven TM-helices, the number of families decreases sharply as the number of TM-helices increases. Families with 12 TM-helices are the exception, however; they have a small peak in numbers against the overall downward slope of the plot. We also carried out the same kind of analysis on Pfam-A families that are annotated as transporters, symporters, and channels, and found that 12-TM-helix families are preferred by transporter-like families. In addition, most (11 out of 12) Pfam-A families with 12 TM-helices are transporter-like families. There seems to be a tendency for the transporter-like families to have an even number of TM-helices, because families with 2, 4, 6, 8, and 12 TM-helices have a relatively higher occurrence than those with a neighboring odd number of TM-helices.
Analysis of amino-acid distribution and pair motifs
We selected 168 families from Pfam-A that had more than 20 members. For each of these families, we then generated consensus sequences with conservation value (Rsequence) using the Alpro program [19]. Relatively conserved amino acids in the consensus sequences (Rsequence value > 3.0, representing the top 15% Rsequence value of all amino acids) and in TM-helical regions were analyzed for their composition as well as for pair motifs.
We compared the amino-acid composition of the TM-helices in general with the composition of only the conserved positions in TM-helices in the 168 families (Figure 3). We noticed that some amino acids are considerably more prevalent in the conserved positions, such as glycine (8% average composition in TM-helices versus 19% composition in conserved positions of TM-helices), proline (4% versus 9%) and tyrosine (3% versus 5%). In contrast, isoleucine (10% versus 4%), valine (8% versus 4%), methionine (4% versus 1%) and threonine (7% versus 4%) are less prevalent in conserved positions.
Figure 3.
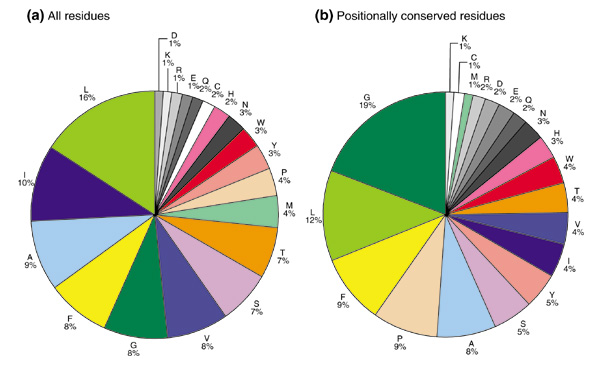
Amino-acid compositions of TM-helices. The amino-acid composition in the TM-helical regions (a) for all sequences and of consensus sequences, and (b) for the 168 Pfam-A families of polytopic membrane domains that contain more than 20 members.
As might be expected, the changes in prevalence of certain amino acids reflect their conservation in the consensus sequence. Therefore, glycine, proline and tyrosine are relatively conserved residues in TM-helical regions, and isoleucine, valine, methionine and threonine have relatively high mutability. This result correlates very well with the mutation data matrix (MDM) for multi-spanning transmembrane regions in membrane proteins [10]. In the MDM of multi-spanning transmembrane α helices, isoleucine, methionine and valine are found to have relatively high mutability as hydrophobic residues, and serine and threonine also rank high in mutability as polar residues. In the matrix, proline appears to be highly conserved. Our results confirm these findings; in addition, we find that glycine and tyrosine are also highly conserved residues in polytopic TM-helices.
We also analyzed the consensus sequences of 168 Pfam-A families for significant amino-acid pair motifs and compared our findings with previous studies. Table 1 shows three pair lists: one includes the top 50 pairs of Senes et al. with their significance [12]; the second includes the top 50 pairs with their occurrences from randomly generated pairs; and the third includes the top 50 pairs with their occurrences using Senes et al.'s top 200 most significant pairs. Of the three lists, the GxxxG pair always ranks first, highlighting its significance in TM-helices [12,13,14]. In the last list, which contains top-ranked pairs in the first two lists, we observed some interesting pair-motif patterns that are associated with glycine. Amino-acid pairs such as ZxxxZ and ZxxxxxxZ (Z represents glycine, alanine, or serine - residues with a small side chain) are highly ranked in the last list. It is known that amino acids are positioned with an average of 3.6 residues per turn in TM-helices [20]. Two residues that are separated by three or six residues are thus oriented in the same direction. Therefore, it was suggested that these motifs are favored for TM-helix packing [12,14]. Our results are in good agreement with the pair motifs that are formed with small residues, but do not favor pairs with β-branched aliphatic residues (isoleucine and valine). This is probably because isoleucine and valine are highly mutable residues in TM-helices.
Table 1.
Top amino-acid pairs in transmembrane helices of the consensus sequences of classified Pfam-A families
| List 1: top 50 pairs and their significance from Senes et al. [12] | List 2: top 50 pairs and their occurrences from random pairs | List 3: top 50 pairs and their occurrences in lists 1 and 2 | |||
| GG4 | 6.35 × 10-34 | GG4 | 46 | GG4 | 46 |
| II4 | 8.36 × 10-24 | GG3 | 32 | GL3 | 28 |
| GA4 | 3.61 × 10-21 | GG1 | 30 | GG7 | 21 |
| IG1 | 4.79 × 10-21 | GG2 | 29 | GL1 | 18 |
| IG2 | 1.29 × 10-16 | GL3 | 28 | AG7 | 18 |
| VG2 | 5.73 × 10-16 | LL1 | 25 | GA7 | 17 |
| IV4 | 2.12 × 10-15 | LG2 | 25 | AG4 | 17 |
| IP1 | 4.52 × 10-15 | GF4 | 24 | PL2 | 16 |
| VV4 | 3.75 × 10-14 | FL3 | 24 | AS4 | 16 |
| VI4 | 1.09 × 10-12 | LL7 | 23 | AL6 | 16 |
| AV1 | 2.17 × 10-12 | GL4 | 23 | LP1 | 15 |
| GL3 | 9.69 × 10-12 | GG6 | 23 | PG9 | 15 |
| AG4 | 9.06 × 10-10 | LL5 | 23 | GA4 | 15 |
| WQ1 | 3.87 × 10-09 | LL3 | 22 | FG1 | 15 |
| IL4 | 4.89 × 10-09 | LG3 | 22 | SL1 | 14 |
| AA3 | 1.33 × 10-08 | LG6 | 21 | SG4 | 14 |
| VG1 | 1.83 × 10-08 | LL8 | 21 | PL1 | 14 |
| GG7 | 2.95 × 10-08 | GG7 | 21 | AA7 | 13 |
| VL4 | 7.71 × 10-08 | GA1 | 21 | AG5 | 12 |
| IS2 | 8.98 × 10-08 | LG10 | 21 | LF8 | 12 |
| SI2 | 1.52 × 10-07 | GG8 | 21 | IA1 | 12 |
| GI1 | 2.93 × 10-07 | LA1 | 21 | GV1 | 12 |
| IY10 | 4.55 × 10-07 | LL2 | 20 | AI1 | 12 |
| YY3 | 6.3 × 10-07 | FG7 | 20 | AA2 | 12 |
| IF10 | 1.63 × 10-06 | FL1 | 20 | GL2 | 12 |
| GI2 | 3.27 × 10-06 | LG4 | 20 | AA3 | 11 |
| PI3 | 3.99 × 10-06 | GA3 | 20 | SL2 | 11 |
| PV1 | 4.97 × 10-06 | FG4 | 19 | PG5 | 11 |
| PL1 | 5.35 × 10-06 | GG5 | 19 | PG6 | 11 |
| LP1 | 5.35 × 10-06 | GL7 | 19 | IL4 | 11 |
| CG4 | 5.4 × 10-06 | GL1 | 18 | GS5 | 10 |
| VY9 | 5.58 × 10-06 | AG7 | 18 | VL4 | 10 |
| GV2 | 6.04 × 10-06 | FG8 | 18 | GV2 | 10 |
| VP1 | 7.45 × 10-06 | LL4 | 18 | IG1 | 10 |
| IA1 | 7.93 × 10-06 | GV3 | 18 | PG10 | 10 |
| PL2 | 1.13 × 10-05 | AG3 | 18 | LY6 | 10 |
| GN4 | 1.38 × 10-05 | GF1 | 18 | LF10 | 10 |
| GS5 | 1.43 × 10-05 | LA2 | 18 | SA6 | 10 |
| VA2 | 2.51 × 10-05 | AG1 | 17 | LG5 | 10 |
| HQ1 | 2.7 × 10-05 | FL5 | 17 | SA3 | 10 |
| VY10 | 2.95 × 10-05 | AG4 | 17 | PF1 | 10 |
| IQ2 | 3.1 × 10-05 | FG5 | 17 | GS4 | 10 |
| LN2 | 5.74 × 10-05 | FF1 | 17 | IV4 | 9 |
| IM9 | 6.84 × 10-05 | GA7 | 17 | LS1 | 9 |
| PA9 | 8.25 × 10-05 | FG2 | 17 | GY8 | 9 |
| VC5 | 9.87 × 10-05 | AF3 | 17 | IG2 | 9 |
| QD3 | 9.95 × 10-05 | GP2 | 17 | LF9 | 9 |
| LY10 | 1.19 × 10-04 | PL2 | 16 | VF8 | 8 |
| SV2 | 1.24 × 10-04 | FF5 | 16 | VG6 | 8 |
| DE4 | 1.51 × 10-04 | AS4 | 16 | GN4 | 8 |
A pair XYn corresponds to amino acids X and Y separated by (n-1) residues. List 1 shows the top 50 amino-acid pairs and their significances by the TMSTAT method [12]; list 2 shows the top 50 amino-acid pairs generated from random amino-acid pairs and their occurrences in the consensus sequences of Pfam-A families of polytopic membrane domains; and list 3 shows the top 50 amino-acid pairs generated from the intersection of lists 1 and 2 (that is, the top 200 pairs as judged by TMSTAT and their occurrences in the consensus sequences of Pfam-A families of polytopic membrane domains). Pairs of small-side-chain amino acids, such as GG4 and AS7, are in bold.
Of all the 168 Pfam-A families of polytopic membrane domains we studied, 45 are classified as transporters, channels, and symporters, representing 27% of the total families. We studied GxxxG and GxxxxxxG pairs, and found that they tend to be associated within transporter/channel-like membrane proteins (Table 2). When one or both glycines is mutated to a small residue such as serine or alanine, this association is weakened. Therefore, GxxxG and GxxxxxxG pairs are relatively conserved in transporter/channel-like membrane proteins. By comparing the amino-acid composition of conserved residues in the TM-helices of the transporter-like families with that of the rest of the Pfam-A families (Table 3), we found that glycine is two times more conserved in the transporter-like families, reflecting the favored GxxxG and GxxxxxxG pairs in these families. Proline and asparagine are also among the conserved residues favored in transporter-like families, whereas cysteine, histidine, isoleucine, leucine, methionine, and valine are unfavored.
Table 2.
Association of GG4 and GG7 pairs with Pfam-A families annotated as transporters, symporters, and channels
| Pairs | Pfam-A families as transporter/ symporter/channel | All Pfam-A families | Percentage (%) |
| GG4 | 18 | 38 | 47.4 |
| GA4 AG4 AA4 | 11 | 36 | 30.6 |
| GS4 SG4 SS4 | 4 | 25 | 16 |
| GG7 | 7 | 16 | 43.8 |
| GA7 AG7 AA7 | 5 | 18 | 27.8 |
| GS7 SG7 SS7 | 6 | 22 | 27.3 |
| All pairs | 45 | 168 | 26.7 |
A pair XYn corresponds to amino acids X and Y separated by (n-1) residues.
Table 3.
A comparison between amino-acid composition of the conserved residues in the TM-helices of 45 transporter Pfam-A families and that of the other 123 Pfam-A families
| Amino acid | Conserved residues in TMs of transporter families (%) | Conserved residues in TMs of the other families (%) | Ratio |
| G | 31.4 | 15.6 | 2.0 |
| N | 3.2 | 2.5 | 1.3 |
| P | 10.3 | 8.0 | 1.3 |
| D | 2.3 | 1.9 | 1.2 |
| R | 1.8 | 1.5 | 1.2 |
| A | 8.6 | 7.7 | 1.1 |
| Q | 2.3 | 2.1 | 1.1 |
| T | 3.9 | 4.0 | 1.0 |
| W | 3.6 | 3.8 | 0.9 |
| E | 1.9 | 2.1 | 0.9 |
| S | 4.4 | 5.4 | 0.8 |
| F | 7.5 | 9.6 | 0.8 |
| K | 0.9 | 1.2 | 0.8 |
| Y | 3.5 | 5.1 | 0.7 |
| L | 8.4 | 13.1 | 0.6 |
| V | 2.3 | 4.6 | 0.5 |
| M | 0.6 | 1.6 | 0.4 |
| I | 1.9 | 5.2 | 0.4 |
| H | 0.9 | 3.6 | 0.2 |
| C | 0.1 | 1.6 | 0.1 |
Amino-acid composition sorted by ratio of composition in transporter families over that in the other families.
Genome-wide analysis of families of polytopic membrane domains
Classified polytopic membrane protein domains represent from 40% to 81% of the total polytopic membrane domains in the genomes studied, with an average coverage of 61% (Figure 4a). We kept the family classification relatively conservative instead of aiming for a high overall coverage with a less careful classification. To avoid including falsely predicted families, we based our analysis on families with no fewer than four members. However, a higher proportion of polytopic membrane domains could be classified if smaller families were considered (Figure 4a).
Figure 4.
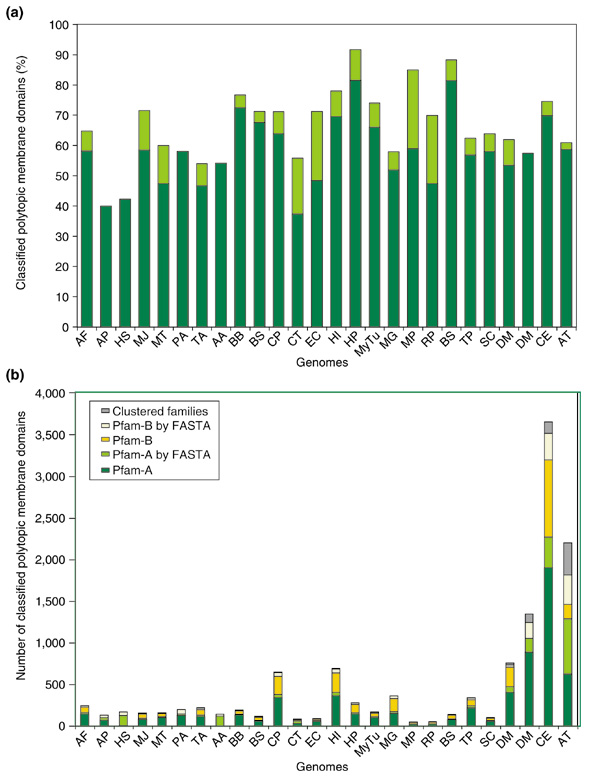
Classified polytopic membrane domains in 26 genomes. (a) The dark-green bars represent the percentage of polytopic membrane domains that are classified in each genome, using only classified families with at least four members. When classified families containing two or three members are included in this analysis, the additional coverage is represented by light-green bars. (b) The proportion of polytopic membrane domains classified by different methods in all genomes studied. Most polytopic membrane domains are identified by direct ID match and sequence-similarity (FASTA) match to members of classified Pfam-A families (green and light-green bars) and Pfam-B families (yellow and light-yellow bars). A small proportion of polytopic membrane domains are clustered on the basis of their sequence similarity (gray bars). For abbreviations for genomes, see Materials and methods.
We classified polytopic membrane domains into Pfam-A, Pfam-B and self-clustered families. Figure 4b shows the distribution of these three kinds of families in all the genomes. Most of the classified polytopic membrane domains belong to Pfam-A and Pfam-B, which cover 95% of classified domains.
Classified polytopic membrane domains and their families were studied in relation to the number of ORFs in each genome. Figure 5a shows the number of classified polytopic membrane domains versus the number of ORFs in all the genomes, and Figure 5b shows the same relation in genomes of single-celled organisms. A rough linear relation seems to exist between the number of classified polytopic membrane domains and the number of ORFs in each genome. However, it is interesting that C. elegans is an obvious outlier in the trend. To try to explain this, we took a closer look at the biggest families of polytopic membrane domains in C. elegans (Figure 5c). The three biggest families in C. elegans are PF01604, PF01461, and PB000009, which are described as 7-TM chemoreceptor families. (The annotation of PB000009 is from PD000148 in Prodom [21].) These families are almost unique to C. elegans, as most of their members in Pfam are from C. elegans. These families contain well-amplified membrane domains, with total numbers of 289, 250, and 216, respectively. Those numbers are more than double the biggest family in Drosophila melanogaster, which is PF00083 (Sugar (and other) transporter) with 108 members. By removing the number of proteins in these three families (a total of 754), we can see a better fit of C. elegans to the trend line. So the unusually large number of polytopic membrane domains is likely to be caused by protein amplification in a few families.
Figure 5.
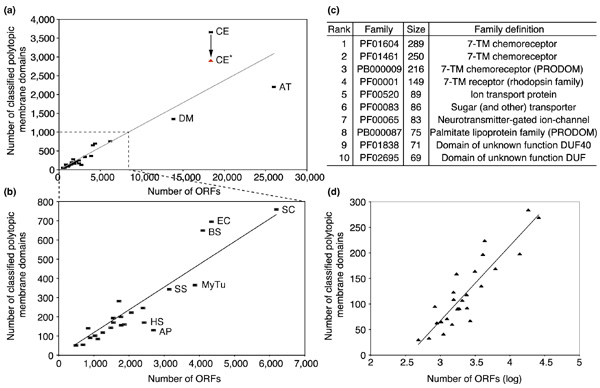
Classified polytopic membrane domains in relation to the number of ORFs in the 26 genomes studied. (a, b) Plots of the number of classified polytopic membrane domains versus the number of ORFs in (a) all the studied genomes and (b) in genomes of single-celled organisms. The trend lines, though generated on the basis of data in each plot, have almost the same slope. CE* in red indicates the number of classified polytopic membrane domains in C. elegans after the three big 7-TM chemoreceptor families are removed (see (c)). (c) The top ten families of polytopic membrane domains, as judged by their occurrence in C. elegans.(d) Plot of the number of classified families of polytopic membrane domains versus the logarithm of the number of ORFs in each genome.
This hypothesis was supported by analysis of Figure 5d, which shows the number of families of polytopic membrane domains in relation to the number of ORFs in studied genomes. The number of families seems to have a logarithmic relation in all studied genomes, including C. elegans. Given that C. elegans has an unusually large number of polytopic membrane domains but a normal number of families, the amplification of polytopic membrane domains is limited to a few families.
Discussion
Polytopic membrane domains of integral membrane proteins in 26 genomes have been classified into 637 families, which include 218 Pfam-A, 298 Pfam-B and 121 clustered families. Only families that are reasonably big (≥ 4 members) were selected. The classified families were used for amino-acid distribution and pattern studies for genome-wide analysis.
Our studies on amino-acid distribution and patterns were conducted on Pfam-A families. We also analyzed Pfam-B and the clustered families, but found fewer conservations, probably because the Pfam-B and the clustered families are not as carefully aligned as Pfam-A families. In the analysis of amino-acid positions, glycine, proline and tyrosine were found to be the most conserved residues in TM-helical regions, whereas isoleucine, valine, methionine and threonine were identified as the least conserved residues, relative to average occurrence. This result is mostly consistent with previous results from an MDM [10]. Although hydrophobic residues such as leucine and isoleucine are among the most abundant residues in TM-helices, they are not well conserved in position. The observed conservation in position for residues such as glycine, proline and tyrosine raises the question of whether these residues are associated with the functions of integral membrane proteins.
We also studied amino-acid pair motifs in the conserved sequences in classified families. We show that pairs consisting of a glycine and another small amino acid (glycine, alanine or serine) and facing the same direction in TM α-helices are common in conserved positions. As those pair motifs have been shown to be important for packing of TM-helices [12,13,14], conservation of those motifs probably implies their importance in folding stability of integral membrane proteins, as is the case with hydrophobic residues found in the core regions of soluble proteins.
Our results have some interesting implications for the classified Pfam-A families annotated as transporters, symporters and channels. First, there is a preference for 12 TM-helices among these families. As there is no 12-TM transporter protein structure available, we do not know exactly why a 12 TM-helix bundle is preferred for transport. The structure of MsbA from Escherichia coli [22], an ATP-binding cassette (ABC) transporter homolog, was recently solved. It contains 12 TM-helices in a homodimer of two 6-TM-helical bundles, which form a central chamber to translocate substrates. However, it is unlikely that polytopic membrane domains in the 12-TM Pfam-A families have a structure like that of ABC transporters; as there is no obvious sequence similarity within the sequence containing the 12 TM-helices, it is unlikely to form two 6-TM-helical bundles. By looking at structures of other transport proteins, including the potassium channel [23], the mechanosensitive ion channel [24], the aquaporin water channel [25], and the glycerol facilitator channel [26], it is apparent that 7-10 TM-helices are needed to form a tunnel and transport molecules. This means that proteins with a small number of TM-helices must oligomerize to form a proper tunnel to translocate molecules through the membrane. In addition, families of these proteins tend to have GxxxG and GxxxxxxG instead of related motifs that have one or both glycines changed to alanine or serine. While this preference is interesting, we do not know its origin. Perhaps it reflects especially tight packing among helices in transporters, permitting the Cα-H...O hydrogen bonding that has been discussed [14].
We also studied the distribution of classified families in 26 genomes. Although the classified families of polytopic membrane domains do not provide complete coverage of the total potential polytopic membrane domains, we think they include most membrane proteins that have essential functions in these genomes. The excluded domains are either unique in function for the organism or falsely predicted. In most genomes the number of classified polytopic membrane domains seems to have a linear relation with the number of ORFs. However, C. elegans is an outlier to this trend. By studying the families in C. elegans, we found that it has an exceptional number of 7-TM-helical membrane domains, most of which are annotated as chemoreceptors. As C. elegans cannot see or hear but must search for food, chemosensation is key to survival. C. elegans mediates chemosensation by 32 neurons that are mostly arranged in bilateral pairs on the left and right sides, and it is estimated that there are about 500 G-protein-coupled receptors that act in chemosensation [27]. We have now identified many chemoreceptors (750), classified into three large families. Therefore, classification of polytopic membrane domains into families gives us another way to look at the distribution and functions of integral membrane proteins in genomes.
Materials and methods
Databases
In this study, the following databases were used: SWISS-PROT (release 39 and updated to 19 December, 2000) [16], which contains 91,132 protein entries; Pfam (release 6.1) [15], which contains 2,727 protein families in Pfam-A and 40,230 families in Pfam-B; Proteome Analysis Database [28], where complete non-redundant proteomes were downloaded. We selected eight genomes from archaea: Archaeoglobus fulgidus (AF), Aeropyrum pernix K1 (AP), Halobacterium sp. (HS), Methanococcus jannaschii (MJ), Methanobacterium thermoautotrophicum (MT), Pyrococcus abyssi (PA), Pyrococcus horikoshii (PH), and Thermoplasma acidophilum (TA); 14 genomes from bacteria: Aquifex aeolicus (AA), Borrelia burgdorferi (BB), Bacillus subtilis (BS), Chlamydia pneumoniae strain AR39 (CP), Chlamydia trachomatis (CT), E. coli strain K12 (EC), Haemophilus influenzae (HI), Helicobacter pylori strain 26695 (HP), Mycobacterium tuberculosis (MyTu), Mycoplasma genitalium (MG), Mycoplasma pneumoniae (MP), Rickettsia prowazekii (RP), Synechocystis sp. (SS), and Treponema pallidum (TP); four genomes from eukaryotes: Saccharomyces cerevisiae (SC), D. melanogaster (DM), C. elegans (CE), and Arabidopsis thaliana (AT).
Classification of polytopic membrane protein domains
Figure 1a shows our complete classification procedure. We extracted 8,301 protein entries in the SWISS-PROT database containing no less than two TRANSMEM annotations in the FT field. In these proteins, a total of 52,636 transmembrane (TM) regions were allocated to proteins in the Pfam database. By analyzing the location of TM regions in protein domains of each Pfam family, we were able to identify families that contain polytopic membrane protein domains. We went through a relatively conservative procedure to identify potential families of polytopic membrane domains. First, a Pfam family needed to have a significant number of proteins containing no fewer than two TM regions to be identified as a polytopic membrane domain family. Second, all families in Pfam-A and some in Pfam-B that have more than seven members are analyzed, as the Pfam-B database is under development and contains thousands of small protein families. Finally, we identified 183 Pfam-A and 152 Pfam-B families. Proteins in these families contain 36,878 TM regions, representing approximately 70% of the total TM regions extracted from SWISS-PROT. We analyzed sizes of the loops between all the TM regions, as shown in the inner chart of Figure 1. By Pfam's protein domain classification, most loops (> 95%) are short peptides, containing less than 80 amino acids.
Proteins from 26 genomes were submitted to TMHMM server for TM-helix prediction [6]. Predicted membrane proteins were searched for polytopic membrane domains, using a rule, generated from the above result, that the intramembrane-domain loop sizes must be less than 80 amino acids. To identify domains that are included in the Pfam families that have been identified, we searched the defined polytopic membrane domains for SWISS-PROT ID matches and regional matches. Unmatched domains are further classified on the basis of Pfam's classification, and additional 48 Pfam-A and 166 Pfam-B families are identified (small size Pfam-B families with no less than four members and no less than three matches are selected). In total, we identified 231 Pfam-A and 318 Pfam-B families as polytopic membrane domains. As not all proteins from the 26 genomes are included in Pfam, we then tried to assign the unclassified polytopic membrane domains to the identified Pfam families by sequence similarity matching to proteins in these families. We used the FASTA program [18] to search for matches, and matches with E-values less than 0.01 were considered positive. Obviously, one can assign Pfam-A domains using the HMMer software [29], which they are closely associated with. However, we chose to take a somewhat simpler tack, using FASTA. This is a somewhat more conservative approach (finding fewer homologs) which has the advantage of using consistent thresholds that can be applied to all the searches. Query domains were assigned to Pfam families that their best matches belong to.
As for those that have not been classified into Pfam families by either ID match or by sequence-similarity match, we tried to cluster these into families on the basis of their sequence similarities. This procedure was done by an all-against-all sequence similarity search (E-value < 0.01) using FASTA, and polytopic membrane domains were clustered by applying a multiple linkage clustering method [30] to the FASTA results. N family members must have more than 0.9N (N-1) links to other members, with tolerance of 10% missing links among members. We selected 121 clustered families that contain no fewer than four members, and aligned protein sequences in each family using the CLUSTAL W program [31]. For a complete list of assigned polytopic membrane domains see Additional data files and [32].
TM-helix identification in the families of polytopic membrane domains
We assume that all protein domains in a classified family have a defined number of TM-helices. To identify the number of TM-helices, we made a hydrophobic plot for each family of polytopic membrane domain. We took the aligned sequences in Pfam's families and in clustered families, and calculated the averaged GES hydrophobic values [8] of all the residues at each aligned position (Deleted and inserted residues, represented by '-' and '.' respectively, are given 0 individual values.) The plot for each family was generated by the averaged GES values along their corresponding aligned positions. Most hydrophobic regions were clearly defined, as most TM-helices aligned well in each family. By identifying hydrophobic regions in the plots, we assigned numbers of TM-helices to classified families of polytopic membrane proteins. We also eliminated 3 Pfam-A and 20 Pfam-B families, as they did not contain multiple hydrophobic regions in their hydrophobicity plots. Therefore, we have 228 Pfam-A, 298 Pfam-B and 121 clustered families for further analysis.
Analysis of amino-acid distribution and pair motifs
We analyzed 168 Pfam-A families with more than 20 members and generated consensus sequences with their sequence logos of all aligned sequences in these families using the Alpro sequence logo program [19]. The selected family size threshold of 20 members is somewhat arbitrary. We chose it because: first, a significant portion (~75%) of the 228 classified Pfam-A families had more than 20 members; and second, the potential bias from small families could be reduced as they tend to have more conserved residues than big families. However, we can show that our results remain unaffected by changing this threshold. In particular, we analyzed Pfam-A families containing more than 25, 30, 35, or 40 members, and got essentially the same results. Amino acids with sequence conservation values (Rsequence) of no less than 3.0 (top 15% of all values) were considered as conserved residues. For all the families, we counted the occurrences of amino acids in the consensus sequences and in all aligned sequences in hydrophobic regions, which are defined to have no fewer than 10 continuous amino acids with GES hydrophobicity value greater than 0.
We used the pair definition from a previous study [12]. For example, a pair XYn (X and Y represent amino acids and n a number) corresponds to amino acids X and Y separated by (n-1) residues. We analyzed occurrences of pair motifs of all combinations of amino acids separated by 1 to 10 residues. This result was compared with a previous study of the 200 most significant over-represented pairs [12,33].
Analysis of the families of polytopic membrane domain in genomes
Using simple cross-referencing based on the above procedure, proteomic entries in each genome were searched for matches of polytopic membrane domains of classified families. Numbers of membrane domains in classified families were counted and analyzed in all genomes studied.
Additional data files
A complete list of assigned polytopic membrane domains is available as additional data and from [32].
Supplementary Material
A complete list of assigned polytopic membrane domains
Acknowledgments
Acknowledgements
M.G. thanks the Keck foundation for financial support. Y.L. is supported by an NLM postdoctoral fellowship. This research was supported in part by NIH grant T15 LM07056 from the National Library of Medicine. We thank Alessandro Senes and Steven Aller for helpful discussions.
References
- Paulsen IT, Sliwinski MK, Saier MHJ. Microbial genome analyses: global comparisons of transport capabilities based on phylogenies, bioenergetics and substrate specificities. J Mol Biol. 1998;277:573–592. doi: 10.1006/jmbi.1998.1609. [DOI] [PubMed] [Google Scholar]
- Paulsen IT, Nguyen L, Sliwinski MK, Rabus R, Saier MHJ. Microbial genome analyses: comparative transport capabilities in eighteen prokaryotes. J Mol Biol. 2000;301:75–100. doi: 10.1006/jmbi.2000.3961. [DOI] [PubMed] [Google Scholar]
- Gerstein M. A structural census of genomes: comparing bacterial, eukaryotic, and archaeal genomes in terms of protein structure. J Mol Biol. 1997;274:562–576. doi: 10.1006/jmbi.1997.1412. [DOI] [PubMed] [Google Scholar]
- Gerstein M. Patterns of protein-fold usage in eight microbial genomes: a comprehensive structural census. Proteins. 1998;33:518–534. doi: 10.1002/(sici)1097-0134(19981201)33:4<518::aid-prot5>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Wallin E, von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998;7:1029–1038. doi: 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Engelman DM, Steitz TA, Goldman A. Identifying nonpolar transbilayer helices in amino acid sequences of membrane proteins. Annu Rev Biophys Biophys Chem. 1986;15:321–353. doi: 10.1146/annurev.bb.15.060186.001541. [DOI] [PubMed] [Google Scholar]
- von Heijne G. Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J Mol Biol. 1992;225:487–494. doi: 10.1016/0022-2836(92)90934-c. [DOI] [PubMed] [Google Scholar]
- Jones DT, Taylor WR, Thornton JM. A mutation data matrix for transmembrane proteins. FEBS Lett. 1994;339:269–375. doi: 10.1016/0014-5793(94)80429-x. [DOI] [PubMed] [Google Scholar]
- Arkin IT, Brunger AT. Statistical analysis of predicted transmembrane alpha-helices. Biochim Biophys Acta. 1998;1429:113–128. doi: 10.1016/s0167-4838(98)00225-8. [DOI] [PubMed] [Google Scholar]
- Senes A, Gerstein M, Engelman DM. Statistical analysis of amino acid patterns in transmembrane helices: the GxxxG motif occurs frequently and in association with beta-branched residues at neighboring positions. J Mol Biol. 2000;296:921–936. doi: 10.1006/jmbi.1999.3488. [DOI] [PubMed] [Google Scholar]
- Russ WP, Engelman DM. The GxxxG motif: a framework for transmembrane helix-helix association. J Mol Biol. 2000;296:911–919. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- Senes A, Ubarretxena-Belandia I, Engelman DM. The Calpha-H...O hydrogen bond: a determinant of stability and specificity in transmembrane helix interactions. Proc Natl Acad Sci USA. 2001;98:9056–9061. doi: 10.1073/pnas.161280798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Birney E, Durbin R, Eddy SR, Howe KL, Sonnhammer EL. The Pfam protein families database. Nucleic Acids Res. 2000;28:263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28:45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein M. How representative are the known structures of the proteins in a complete genome? A comprehensive structural census. Fold Des. 1998;3:497–512. doi: 10.1016/S1359-0278(98)00066-2. [DOI] [PubMed] [Google Scholar]
- Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc Natl Acad Sci USA. 1988;85:2444–48. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branden C, Tooze J. Introduction to Protein Structure. London: Garland Publishing;; 1991. [Google Scholar]
- Corpet F, Servant F, Gouzy J, Kahn D. ProDom and ProDom-CG: tools for protein domain analysis and whole genome comparisons. Nucleic Acids Res. 2000;28:267–269. doi: 10.1093/nar/28.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang G, Roth CB. Structure of MsbA from E. coli: a homolog of the multidrug resistance ATP binding cassette (ABC) transporters. Science. 2001;293:1793–1800. doi: 10.1126/science.293.5536.1793. [DOI] [PubMed] [Google Scholar]
- Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Chang G, Spencer RH, Lee AT, Barclay MT, Rees DC. Structure of the MscL homolog from Mycobacterium tuberculosis: a gated mechanosensitive ion channel. Science. 1998;282:2220–2226. doi: 10.1126/science.282.5397.2220. [DOI] [PubMed] [Google Scholar]
- Murata K, Mitsuoka K, Hirai T, Walz T, Agre P, Heymann JB, Engel A, Fujiyoshi Y. Structural determinants of water permeation through aquaporin-1. Nature. 2000;407:599–605. doi: 10.1038/35036519. [DOI] [PubMed] [Google Scholar]
- Fu D, Libson A, Miercke LJ, Weitzman C, Nollert P, Krucinski J, Stroud RM. Structure of a glycerol-conducting channel and the basis for its selectivity. Science. 2000;290:481–486. doi: 10.1126/science.290.5491.481. [DOI] [PubMed] [Google Scholar]
- Bargmann C. Neurobiology of the Caenorhabditis elegans genome. Science. 1998;282:2028–2033. doi: 10.1126/science.282.5396.2028. [DOI] [PubMed] [Google Scholar]
- Apweiler R, Biswas M, Fleischmann W, Kanapin A, Karavidopoulou Y, Kersey P, Kriventseva EV, Mittard V, Mulder N, Phan I, Zdobnov E. Proteome Analysis Database: online application of InterPro and CluSTr for the functional classification of proteins in whole genomes. Nucleic Acids Res. 2001;29:44–48. doi: 10.1093/nar/29.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Kaufman L, Rousseeuw PJ. Finding Groups in Data: An Introduction to Cluster Analysis. New York: John Wiley and Sons; 1990. [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Index of genome/tms http://bioinfo.mbb.yale.edu/genome/tms
- TMSTAT: statistical analysis of transmembrane sequences http://engelman.csb.yale.edu/tmstat/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A complete list of assigned polytopic membrane domains