

Abstract
Although there is a growing body of evidence that different amyloidoses may have a similar molecular mechanism in common, the many details of this mechanism are not understood. In this study, we propose that there is a common molecular structure of the primary agents of these diseases, namely a small oligomer of Perutz's cylindrical double-β-stranded subunit for polyglutamine and that this structure, which contains a central water-filled core, can spontaneously integrate into the bilayers of membranes to form aqueous pores. We suggest that this ability to produce permeable channels in appropriate neuronal membranes is a key element in the toxicity of the β-amyloids. One strong criterion for the stability of the Perutz structure for an amyloid is that it contain ≈40 or more amino acid residues. We show here that the neurotoxic Aβ amyloids 1-40 and 1-42, related to Alzheimer's disease, spontaneously enter the membranes of intact erythrocytes and cause their lysis but that Aβ 1-38 and Aβ 1-35, which are not neurotoxic, have no observable effects on erythrocytes, supporting our proposal. Other aspects of the proposed mechanism of cytotoxicity of the β-amyloids are explored.
Keywords: Alzheimer's disease, neurodegeneration, Parkinson's disease
A large and continually increasing number of diseases, including the neurodegenerative syndromes Alzheimer's disease (AD), Parkinson's disease, and Huntington's disease as well as the spongiform encephalopathies (prion diseases) such as mad cow disease are all associated with the formation of characteristic proteinaceous components that can form aggregates and/or fibrils under appropriate conditions in water. These components are collectively called β-amyloids. In these fibrils and aggregates, the β-amyloids are all largely in the β-conformation and can be stained with Congo red, which reveals that they contain a hydrophobic environment that bonds the dye. These extensively aggregated β-amyloid structures are present at equilibrium with small water-soluble species containing from one to a dozen or so of the repeating subunits of the β-amyloid, each bonded noncovalently to its neighboring subunits. It is widely accepted that some form of the particular β-amyloid is critically involved in the neurotoxicity of its associated disease, but the detailed mechanisms for this involvement have not been elucidated. In previous years, it was vigorously disputed whether the larger aggregates or one or more of the smaller soluble species of β-amyloids are the primary toxic components, but in the last few years, several studies (see refs. 1-4) have clearly implicated the smaller soluble species in the toxic process. The fibrils and aggregates may serve as storage depots of longer β-amyloid chains rather than of the shorter soluble forms.
Close Similarities in the Structures and Functions of Different β-Amyloids
One of the most remarkable of these recent studies is that of Kayed et al. (3). Working with Aβ 1-40, which is a β-amyloid associated with AD, Kayed et al. reported the production of a MAb that binds to, and inhibits the toxicity of, water-soluble intermediate-sized oligomers of Aβ 1-40, but the MAb does not bind to the water-soluble monomer, the large-molecular-weight oligomers, or to Aβ 1-40 fibrils, none of the last three being neurotoxic. In addition, with five other β-amyloids of different amino acid sequences and associated with a wide range of different neurodegenerative diseases, the same MAb that binds specifically to the intermediate-sized water-soluble oligomer of Aβ 1-40, also reacts with similar intermediate-sized watersoluble oligomers of each of the other five amyloids but not with their monomers, larger oligomers, or their fibrils or aggregates. Although the antigenic epitope specific to this MAb is not known, these results strongly suggest that the different β-amyloids, despite their considerable differences in amino acid composition and in mechanisms of production, form intermediate-sized oligomers in the water-soluble state that have closely similar structures that all generate this epitope and, further, suggest that the mechanisms by which the different β-amyloids produce their particular neurotoxicity are closely similar.
The X-Ray Diffraction Structure of Polyglutamine Fibrils Cast from Water Solution
Perutz et al. (5), in 2002, published a subunit (monomer) structure for polyglutamine (poly Q) derived from the x-ray diffraction patterns of fibers of D2 Q15 K2 that had been cast from water solution. Poly Q β-amyloids are involved in Huntington's disease and several other neurodegenerative diseases (6). The subunit structure consists of two turns of a cylindrically wound single β-chain (Fig. 1B) that surround a water-filled channel; the Q side chains extend alternately on either side of the main chain, which makes each turn of the β-chain 5 Å long and the two complete turns 10 Å long, parallel to the chain axis. The two turns are strongly bound to one another by successive main-chain 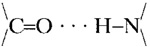 hydrogen bonds on the side of the β-chain facing the bilayer, and, on the side of the β-chains facing the channel, the C—O and N-H dipoles form hydrogen bonds with each other or with H2O molecules. The Q residues are hydrogen-bonded to their neighboring Qs on both sides of the β-sheet (Fig. 1D). Thus, all of the groups capable of forming hydrogen bonds do so, which greatly stabilizes the double-stranded structure in water (and even more so for those hydrogen bonds facing a lipid bilayer, see below). The aqueous central core is 15 Å in diameter, large enough to form an ion-conductance channel. Each subunit contains 40 amino acids, 20 in each turn. As Perutz et al. (5) emphasized, β-chains <40 amino acids, for thermodynamic reasons, would not be able to form stable cylindrical fibrils of repeating subunits in water. It is clear from Fig. 1 that, for example, one turn of a subunit containing 20 residues could not form all of the
hydrogen bonds on the side of the β-chain facing the bilayer, and, on the side of the β-chains facing the channel, the C—O and N-H dipoles form hydrogen bonds with each other or with H2O molecules. The Q residues are hydrogen-bonded to their neighboring Qs on both sides of the β-sheet (Fig. 1D). Thus, all of the groups capable of forming hydrogen bonds do so, which greatly stabilizes the double-stranded structure in water (and even more so for those hydrogen bonds facing a lipid bilayer, see below). The aqueous central core is 15 Å in diameter, large enough to form an ion-conductance channel. Each subunit contains 40 amino acids, 20 in each turn. As Perutz et al. (5) emphasized, β-chains <40 amino acids, for thermodynamic reasons, would not be able to form stable cylindrical fibrils of repeating subunits in water. It is clear from Fig. 1 that, for example, one turn of a subunit containing 20 residues could not form all of the
Fig. 1.

Computer renderings of the Perutz et al. (5) structure of poly Q. (A) Space-filling model of one subunit of 42 Q residues with two turns of β-strands, showing the central pore, here tilted, that is perpendicular to the plane of the membrane. Note the outer convolutions of the surface of the subunit, by the outward-facing Q residues. These might serve to make gear-like associations between β-amyloid subunits in the plane of the membrane; others might serve as regions of attachment of specific integral membrane proteins to the membrane-interior regions of the poly Q amyloid, such as we propose in the text. Red balls are O atoms, and blue ones are N atoms. (B) Stick model of two stacked subunits of poly Q, each subunit containing 42 Q residues, one subunit rendered in dark blue, the other in light blue. (C) Stick model of a short segment of one subunit (enlarged) that faces a region of bonding between one turn and another of the subunit, showing the hydrogen bonds (strings of dots) formed between backbone 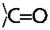 (red) and
(red) and 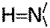 (dark blue) groups. (D) Stick model of a small segment of one subunit, showing (by strings of dots) the hydrogen bonds formed by (i) inter-β-strand bonds between the backbone groups, (the O in red and the N in dark blue) and (ii) of the alternate Q residues extending from one or the other surface of the two-turn helix sheet. This view is parallel to the axis that runs down the pore of the subunit, and perpendicular to the plane of the membrane bilayer.
(dark blue) groups. (D) Stick model of a small segment of one subunit, showing (by strings of dots) the hydrogen bonds formed by (i) inter-β-strand bonds between the backbone groups, (the O in red and the N in dark blue) and (ii) of the alternate Q residues extending from one or the other surface of the two-turn helix sheet. This view is parallel to the axis that runs down the pore of the subunit, and perpendicular to the plane of the membrane bilayer.
main-chain hydrogen bonds with a second turn that had, say, only 16 residues. This would increase the free-energy change accompanying the formation of that two-turn subunit, making its solubility less likely. It is of great interest, therefore, that poly Q repeats with <37 Q are generally not neurotoxic, whereas all those that have 40 or more repeats invariably are (6). In a closely related context, the neurotoxic forms of the Aβ amyloid of AD that are produced in vivo are either 40 or 42 amino acids long (see below). These considerations are of particular relevance to the experimental results obtained with Aβ in this article.
More recently, a dispute has arisen about the question whether the x-ray data analyzed in Perutz et al. (5) lead exclusively to the model of the poly Q subunit that they developed or equally well to other models of quite different structure (see ref. 7). In the remainder of this article, therefore, we provisionally regard the Perutz et al. model as a plausible and attractive, but not proven, structure for the poly Q subunit, irrespective of the present state and relevance of the Perutz x-ray data. We present different kinds of data in support of the Perutz et al. structure dissolved in membrane bilayers.
β-Amyloids Form Conducting Pores in Black Lipid Bilayer Films and in Intact Cell Membranes
In early 1994, we became interested in the question of whether Aβ 1-40 might spontaneously solubilize in cell membranes and whether it could cause cytolysis of intact cells. We carried out experiments (unpublished work) with human erythrocytes and Aβ 1-40 and other Aβ-related compounds that were closely similar to experiments that we have performed more recently, some of which are shown in Fig. 2. The results of the two sets of experiments were essentially the same. Aβ 1-40 entered the membrane of the intact erythrocyte spontaneously, and, in ≈25 min, all of the cells had lysed. We never published the earlier results because we could not think of a membrane-permeating structure for Aβ that explained thermodynamically its solubility in membrane bilayers as well as its ability to lyse the cell. Since then, many related experiments have been carried out by others with a number of β-amyloids, usually in conjunction with black lipid bilayers and occasionally with live cells (for reviews, see refs. 8 and 9). These studies yielded generally similar results, including those with poly Q (10, 11); as measured by the onset of conductance, the black lipid bilayers doped with any of a variety of β-amyloids developed ionic (generally cationic) conductances. These data indicated that the amyloids had penetrated the bilayers and created pores across them. However, no thermodynamically satisfactory detailed integral membrane structures were advanced for the β-amyloids to account for their solubility in a bilayer or for their pore formation, nor was a detailed mechanism proposed for the connection between these findings and the neuronal target-specificity of different toxic β-amyloids.
Fig. 2.

Bilayer couple experiments with intact erythrocytes treated with a total of 20 μM of one Aβ oligomer of the size indicated at the left margins of A and B as a function of time after mixing (numbers in min above each row of figures in A and B). Pictures were taken with Nomarski optics in light microscopy. (C) Enlargements of a succession of the major cell forms observed with time (1, crenated; 2, flattened; 3, cup-shaped; and 4, near-spherical). The photographs indicate that Aβ 1-40 and 1-42 (B) show a rapid succession of shapes of the intact erythrocyte (details in text), more rapidly for Aβ 1-42 than for Aβ 1-40. [Control cells (A Upper) without added Aβ exhibit no significant shape changes over the same time interval.] By 25 min, all of the erythrocytes with added Aβ 1-40 or Aβ 1-42 are lysed, leaving membrane ghosts behind. The effects of Aβ 1-38 on erythrocyte shape, by contrast to Aβ 1-40 and Aβ 1-42, are not distinguishable from the unchanged controls (A).
In this article, we propose that the structure of neurotoxic β-amyloids in cell membranes are often, perhaps generally, that of the intermediate aggregates [of the order of trimers (30 Å) or tetramers (40 Å)] of the pore-forming cylindrical double-β-stranded subunit that Perutz et al. (5) ascribed to the oligomeric state of poly Q in water solution or suspension (Fig. 1). In addition, we provide experimental evidence with Aβ oligopeptides and intact erythrocytes that is consistent with this structural proposal for bilayer-bound Aβ subunits.
Results
The Detection of Oligopeptide Solubility and of Pore Formation in the Membranes of Intact Erythrocytes. The entry of a small molecule into, and its passage through, the erythrocyte membrane bilayer can be followed with Nomarshi optics on a light microscope by using the “bilayer couple” description of the erythrocyte bilayer (12, 13). If the penetration of a small-molecule substance from the outside medium into the bilayer is sufficiently large and slow enough, its entry, first into the outer half of the lipid bilayer, can be detected in the light microscope by the transient formation of crenated intact cells (e.g., see Fig. 2C1) because of the differential expansion of the total area occupied by the outer half layer relative to the inner half. If the substance then continues to penetrate the bilayer further, into the inner half bilayer, the intact cell flattens and elongates slightly when the total areas occupied by the two half layers are about equal (e.g., Fig. 2C2). If further penetration of the substance occurs, and its concentration in the inner half layer increases, the intact cell is induced to adopt a “cup” morphology (Fig. 2C3), as the area occupied by the inner half of the bilayer exceeds that of the outer half. (Other more irregular shapes are also observed.) In this condition, if the penetrating small molecules now span the thickness of the bilayer and if the substance has a structure that forms a water-filled, ion-conducting channel traversing the bilayer, osmotic equilibration will cause the cell to lyse, releasing the hemoglobin and leaving behind the membrane “ghost.”
The Behavior of Intact Erythrocytes Toward Several Aβ Oligopeptides. As described in the previous section, intact erythrocytes can undergo transient shape changes, detectable in the light microscope, that reflect several stages of insertion of small molecules from the aqueous medium into the erythrocyte bilayer. For example, as shown in Fig. 2B, by 30 sec after immersion in an aqueous medium of 20 μM total concentration of Aβ 1-40, most of the erythrocytes have acquired a crenated shape (as shown enlarged in Fig. 2C1), indicative of entry of the oligopeptide into the outer half of the membrane bilayer, which persists until about 12 min At this time, many of the cells have become flattened and elongated (as shown enlarged in Fig. 2C2), reflecting diffusion of Aβ 1-40 from the outer to the inner half layer, with similar concentrations in each half layer. By 25 min, most of the erythrocytes have lysed to form ghosts, reflecting the formation of pores in the membrane created by Aβ 1-40 molecules, with the cores spanning the bilayer.
With Aβ 1-42 at the same total concentration (Fig. 2B), a similar progression of shape changes and lysis occurs as with Aβ 1-40 only faster. At 6 min, many of the erythrocytes already exhibit cup shapes (shown enlarged in Fig. 2C3), demonstrating that a higher concentration of the amyloid is present in the inner half of the bilayer than in the outer half. By 12 min, significant cell lysis has already occurred and, by 25 min, is complete.
Although we do not know the concentrations of the various oligomers in the Aβ 1-40 and Aβ 1-42 solutions at 20 μM total concentration of the two Aβs, the more rapid membrane entry and formation of membrane pores by Aβ 1-42 than by Aβ 1-40 is correlated with the generally accepted view that Aβ 1-42 is more neurotoxic in AD than is Aβ 1-40, which, in turn, is consistent with the suggestion that pore formation in neuronal membranes by Aβ-amyloids is a critical step in Aβ-induced neurotoxicity.
As discussed in the introduction, the stability of the cylindrically wound double-stranded subunit, either in water or in a membrane bilayer, is expected to undergo a sharp transition as the length of the oligopeptide increases beyond ≈39 amino acids. We therefore synthesized Aβ 1-38 and Aβ 1-35 and examined their effects compared with the erythrocyte shape changes produced by Aβ 1-40 and Aβ 1-42. As shown in Fig. 2 A, 20 μM Aβ 1-38 in aqueous solution produced no significant changes in erythrocyte shape at 20 min compared with a control, by which time the Aβ 1-40- and Aβ 1-42-treated erythrocytes were already undergoing lysis. The results obtained with erythrocytes treated with 20 μM Aβ 1-35 and Aβ 1-28 were indistinguishable from those with Aβ 1-38 (data not shown). Because Aβ 1-38 has a smaller stretch of hydrophobic amino acids than either Aβ 1-40 or Aβ 1-42, this result cannot be attributed to a lower solubility of the small oligomers of Aβ 1-38 than those of Aβ 1-40 and Aβ 1-42 in water solutions. Instead, the result is that predicted by Perutz et al. (5) for the instability in water of the β-stranded subunit structure for β-amyloids of lengths <40 residues. Their reasoning about an unfavorable shortage of hydrogen bonding with Aβs <40 residues in water is even more cogent inside a membrane bilayer.
We have also carried out experiments with these several Aβ oligopeptides acting on neurons. In these studies, mouse hippocampal and cortical neurons in culture were treated with 20 μMAβ 1-40, Aβ 1-42, or Aβ 1-38. After 2 h, the neurons treated with Aβ 1-40 and Aβ 1-42 had undergone marked degeneration, but those treated with Aβ 1-38 for 2 or 24 h were indistinguishable from untreated neurons.
Discussion
The Structure of β-Amyloids in Membranes. Our proposal is that many, if not most, neurotoxic β-amyloids exhibit the same or closely similar cylindrically wound double-β-stranded-subunit structure in cell membranes that Perutz et al. (5) proposed for the structure of poly Q in water (Fig. 1) and that this similar subunit structure accounts, in large part, for the striking similarities of the diverse neurotoxicities of the β-amyloids (3). The two turns of a single β-chain of each subunit surround a water-filled channel of ≈15 Å in diameter, large enough to pass small ions. Because each subunit with its Q residues is ≈10 Å in length along its axis, three or perhaps four subunits linked linearly (A-B · · · A-B · · · A-B) would be required to span a membrane bilayer. The membrane-spanning oligomer could be longer, however, and extend from the bilayer if some additional subunits were attached to the oligomer in the water on either side of the bilayer (in which aqueous solvent the same or closely similar subunit structure would be perfectly stable). Electron microscopic observations of membrane bilayers containing β-amyloid oligomers are required to examine this possibility.
In view of the evidence that β-amyloid monomers are not neurotoxic, whereas certain intermediate-sized oligomers are (3), it is unlikely that monomers are the molecular species that enter the outer half layer of the bilayer and then consecutively join up within the membrane to form the toxic oligomer. (If such were the case, the monomer would register as toxic.) The reason for the absence of monomer toxicity might be that the monomer itself is unstable in the bilayer, perhaps because the two open ends of the water-filled pore of a monomer subunit in transit across the bilayer would have to make a thermodynamically unfavorable contact with the bilayer. It is therefore likely that the appropriate oligomers first form in the aqueous solution and then enter the membrane as such, carrying the central water-filled pore of the oligomer across the membrane bilayer. The water in the oligomer channel would, at most, make direct contact with the bilayer only at the leading region of the first subunit. In this scheme, the water-filled core is never in contact with the bilayer.
These considerations may be the explanation provided by the Perutz et al. model for the observation of Kayed et al. (3) that the monomers of several β-amyloids are not toxic but that their intermediate-sized oligomers are toxic.
Which end of the subunit (residues 1 or 40 in the case of Aβ 1-40) of the intermediate-sized oligomer enters the membrane first is not clear. The orientation of that subunit may, however, be largely determined by an already membrane-intercalated integral protein which specifically binds to the particular β-amyloid oligomer as it enters the membrane (see below).
A singular feature of the Perutz et al. structure is the sharp transition in its stability between chain lengths of 37-38 and 40-42 amino acids, being unstable below that threshold and stable above. Other possible structures proposed for β-amyloids do not predict such a transition (see ref. 7). In the absence of a direct structure determination of the β-amyloid oligomers in the bilayer, we have used this highly restrictive criterion as experimental evidence of the presence of the Perutz et al. structure in the membrane. It has already been remarked above that there is a sharp transition in in vivo neurotoxicity of poly Q β-amyloid at ≈37-40 Q repeats (6), being nontoxic below that threshold, and toxic above it. In our present studies with Aβ oligopeptides, we have shown that, in vitro, there is also a threshold in membrane permeation of ≈40 amino acids in length, Aβ 1-38 being slow to enter the intact erythrocyte membrane and slow to induce cytolysis of erythrocytes compared with Aβ 1-40 or 1-42. It remains to subject differently sized poly Q oligomers and amyloids to these in vitro tests. At this early stage, however, there is already significant evidence for a correlation between the in vitro stability of a β-amyloid exhibiting the Perutz et al. structure (Fig. 1) in membranes, and its neurotoxic activity in vivo.
In the case of a β-amyloid containing a large number of different amino acids, such as Aβ (as contrasted with poly Q), certain structural and thermodynamic criteria must exist for such a molecule to stably attain the Perutz et al. structure in a membrane bilayer. For example, proline residues, which would create a large bend in the β-chain, cannot be tolerated in the β-amyloid-subunit sequence for a stable Perutz et al. structure to form. Furthermore, the amino acid residues facing into the hydrophobic bilayer should, on average, be more hydrophobic than those facing into the water-filled core, which is the case for Aβ 1-40 (Fig. 3) and 1-42. The thermodynamic criteria for the amino acid sequences of oligopeptides that might form stable Perutz et al. structures in membrane bilayers has been examined in more quantitative detail. Here, suffice it to say that these criteria are probably not too highly restrictive if account is taken of the likelihood that the interface between the bilayer and the water in the amyloid core is not infinitely sharp, as it is generally implicitly assumed to be. These criteria might be expected to be closely similar to those for the membrane-intercalated portions of the β-strands of the β-barrel proteins, such as the porins (14-16), because the successive amino acids of the β-strands traversing the membrane have environments at the bilayer/water interface that are closely similar for the two structures. Different amino acid sequences for different β-amyloids may also allow some to adopt slightly different Perutz et al. stable structures containing 19 or 21 residues per turn as well as 20 (as with Aβ 1-40 and Aβ 1-42). The facts that (i) β-amyloids invariably aggregate side by side to form fibrils and large aggregates in water attests to the generally hydrophobic nature of the outer surfaces of their subunits, as does (ii) the binding of hydrophobic dyes, such as Congo red, to the fibrils in water.
Fig. 3.

A schematic view of the distribution of the amino acid residues of Aβ 1-40 in projection along the first (A) and second (B) turns of the Perutz et al. subunit, perpendicular to the axis running down the water-filled core. Successive residues (numbered) face into the core or into the membrane bilayer. The residues are colored red if they are predominantly hydrophobic, i.e., A, F, I, L, M, V, and Y); green if, at pH 7, they are predominantly hydrophilic; and yellow if they are in between. For the first turn (A), the numbers of each type of residue facing into the bilayer are six hydrophobic, one hydrophilic (i.e., D, E, K, and R), and three in-between. The residues facing into the water-filled core are two hydrophobic, five hydrophilic (four in a single sequence DERD), and three in between. For the second turn (B), the residues facing into the bilayer are six hydrophobic, two hydrophilic, and two in between. The residues facing into the water-filled core are four hydrophobic, one hydrophilic, and five in between. Generally, therefore, there are more hydrophobic than hydrophilic (plus in between) residues facing the bilayer, and more hydrophilic (plus in between) than hydrophobic residues facing the water-filled core in each turn, as would be expected thermodynamically. The single-letter symbols are used for the amino acid residues. A, alanine; D, aspartic acid; E, glutamic acid; F, phenylalanine; G, glycine; H, histidine; I, isoleucine; K, lysine; L, leucine; M, methionine; N, asparagine; Q, glutamine; R, arginine; S, serine; V, valine; Y, tyrosine.
Relationship to in Vivo Neurodegeneration. The proposal of a causal connection between the capacity for a β-amyloid to adopt the Perutz et al. structure in a membrane bilayer and the function of that β-amyloid in in vivo neurodegeneration is the subject of this article. However, it is clear that there must be additional factors other than the β-amyloid involved to explain the specificity of the toxic activity of a particular β-amyloid for only one or a few of the many types of neuronal targets in the brain and not for other neurons or other cell types. In one case (poly Q) eight different neurodegenerative diseases, presumably involving eight different cell types of neurons, can be induced by the same β-amyloid (6). All of the β-amyloids themselves, however, appear to be soluble in any bilayer, so the neuronal specificities of their in vivo toxicities may involve one or more other components. For example, each cell type may have in its plasma membrane one or more specific integral proteins that specifically bind to the membrane-intercalated Perutz et al. structure of the particular β-amyloid to which it is vulnerable. Not only would such a specific protein-amyloid interaction discriminate the particular cell target for the β-amyloid, but it would serve to intercalate the toxic amyloid oligomer into the specific membrane at much lower ambient aqueous concentrations than are required for its intercalation into nonspecific bilayers.
If, furthermore, such hypothetical integral membrane proteins specific for the AD-susceptible neurons were localized within the synapses of these neurons (17, 18), and the toxic Aβ 1-42 oligomer with its aqueous pore would become bound and concentrated there, it could account for observations that, in AD, synapses often appear to disintegrate as an early consequence of the disease (19-22). Another problem often encountered with neurodegenerative diseases is their late onset in life. If the β-amyloid involved is produced throughout life, what is responsible for late onset? One among many possible explanations is that the β-amyloid-specific integral proteins that we propose exist may not be expressed in the target cell plasma membrane until late in life, thereby limiting the disease to old age.
Materials and Methods
Human Erythrocytes. Normal human blood was collected into EDTA-coated Vacutainers. The cells were separated by centrifugation and washed several times with freshly prepared 150 mM NaCl. The erythrocytes were stored in citrate-phosphate-dextran (CPD, Sigma) (1.4 volumes CPD per 10 volumes erythrocytes) and washed with 150 mM NaCl before the addition of the Aβ oligopeptides. Erythrocytes were used within 48 h of drawing.
Aβ-Related Oligopeptides. The Aβ oligopeptides 1-28, 1-38, 1-40, and 1-42 were obtained from Bachem (Torrance, CA), and 1-35 was synthesized by Biopeptide (San Diego). These oligopeptides were solubilized according to the manufacturer's instructions as follows: The oligopeptides 1-28 and 1-35 were solubilized in water at 6 mg/ml. To the oligopeptides 1-38 and 1-40 was added dilute acetic acid to help solubilize them. The oligopeptide solutions were then diluted to 1 mg/ml with phosphate-buffered saline without Ca+2 (PBS, pH 7.4) and incubated at 37°C for 3-5 days before use. Oligopeptide 1-42 (at 6 mg/ml in H2O) was diluted to 1 mg/ml with 0.1% NH4OH; this solution was then diluted to 100 μg/ml with PBS and incubated at 37°C for 3-5 days.
Acknowledgments
This work is dedicated to the memory of the late Dr. Max Perutz. We thank Professor Russell F. Doolittle for generating Fig. 1 and for many useful discussions during this work; Dr. Arthur Lesk for providing us with the coordinates for the Perutz et al. structure; Kevin Thompson (Scientific Instrument Company, San Diego) for help with some of our ancient microscopy equipment; and Ms. Michelle Williams (BioMedical Design, San Diego), for help with Figs. 2 and 3. This work was supported by National Institutes of Health Grants 5R01 NS 27580, 5R01 AG 17858, and 5R01 NS044768 (to N.N.D.).
Author contributions: S.J.S. and N.N.D. designed research; N.N.D. performed research; S.J.S. and N.N.D. analyzed data; and S.J.S. and N.N.D. wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviation: AD, Alzheimer's disease.
References
- 1.Lambert, M. P., Barlow, A. K., Chromy, B. A., Edwards, C., Freed, R., Liosatos, M., Morgan, T. E., Rozovsky, I., Trommer, B., Viola, K. L., et. al. (1998) Proc. Natl. Acad. Sci. USA 95, 6448-6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., Rowan, M. J. & Selkoe, D. J. (2002) Nature 416, 535-539. [DOI] [PubMed] [Google Scholar]
- 3.Kayed, R., Head, E., Thompson, J. L., McIntire, T. M., Milton, S. C., Cotman, C. W. & Glabe, C. G. (2003) Science 300, 486-489. [DOI] [PubMed] [Google Scholar]
- 4.Arrasate, M., Mitra, S., Schweitzer, E. S., Segal, M. R. & Finkbeiner, S. (2004) Nature 431, 805-810. [DOI] [PubMed] [Google Scholar]
- 5.Perutz, M. F., Finch, J. T., Berriman, J. & Lesk, A. (2002) Proc. Natl. Acad. Sci. USA 99, 5591-5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orr, H. T. (2001) Genes Dev. 15, 925-932. [DOI] [PubMed] [Google Scholar]
- 7.Sikorski, P. & Atkins, E. (2005) Biomacromolecules 6, 425-432. [DOI] [PubMed] [Google Scholar]
- 8.Kourie, J. I., Culverson, A. L., Farrelly, P. V., Henry, C. L. & Laohachai, K. N. (2002) Cell Biochem. Biophys. 36, 191-207. [DOI] [PubMed] [Google Scholar]
- 9.Kagan, B. L., Azimov, R. & Azimova, R. (2004) J. Membr. Biol. 202, 1-10. [DOI] [PubMed] [Google Scholar]
- 10.Hirakura, Y., Azimov, R., Azimova, R. & Kagan, B. L. (2000) J. Neurosci. Res. 60, 490-494. [DOI] [PubMed] [Google Scholar]
- 11.Monoi, H., Futaki, S., Kugimiya, S., Minakata, H. & Yoshihara, K. (2000) Biophys. J. 78, 2892-2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sheetz, M. P. & Singer, S. J. (1974) Proc. Natl. Acad. Sci. USA 71, 4457-4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim, G. H. W., Wortis, M. & Mukhopadhyay, R. (2002) Proc. Natl. Acad. Sci. USA 99, 16766-16769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kennedy, S. J. (1978) J. Membr. Biol. 42, 265-279. [DOI] [PubMed] [Google Scholar]
- 15.Weiss, M. S., Wacker, T., Weckesser, J., Welte, W. & Schulz, G. E. (1990) FEBS Lett. 267, 268-272. [DOI] [PubMed] [Google Scholar]
- 16.Jap, B. K. (1989) J. Mol. Biol. 205, 407-419. [DOI] [PubMed] [Google Scholar]
- 17.Lacor, P. N., Buniel, M. C., Chang, L., Fernandez, S. J., Gong, Y., Viola, K. L., Lambert, M. P., Valesco, P. T., Bigio, E. H., Finch, C. E., et. al. (2004) J. Neurosci. 24, 10191-10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lue, L.-F., Kuo, Y.-M., Roher, A. E., Brachova, L., Shen, Y., Sue, L., Beach, T., Kurth, J. H., Rydel, R. E. & Rogers, J. (1999) Am. J. Pathol. 155, 853-862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terry, R. D., Masliah, E., Salmon, D. P., Butters, N., DeTeresa, R., Hill, R., Hansen, L. A. & Katzman, R. (1991) Ann. Neurol. 30, 572-580. [DOI] [PubMed] [Google Scholar]
- 20.Svennerholm, L. & Gottfries, C. G. (1994) J. Neurochem. 62, 1039-1047. [DOI] [PubMed] [Google Scholar]
- 21.Jeffrey, M., Halliday, W. G., Bell, J., Johnston, A. R., Macleod, N. K., Ingham, C., Sayers, A. R., Brown, D. A. & Fraser, J. R. (2000) Neuropathol. Appl. Neurobiol. 26, 41-54. [DOI] [PubMed] [Google Scholar]
- 22.Snyder, E. M., Nong, Y., Almeida, C. G., Paul, S., Moran, T., Choi, E. Y., Nairn, A. C., Salter, M. W., Lombroso, P. J., Gouras, G. K., et. al. (2005) Nat. Neurosci. 8, 1051-1058. [DOI] [PubMed] [Google Scholar]