

Abstract
Activation of the epidermal growth factor receptor (EGFR) family is thought to play a critical role in both embryogenesis and oncogenesis. The diverse biological activities of the EGFR family are achieved through various ligand-receptor and receptor-receptor interactions. One receptor that has been found to play a central role in this signaling network is ErbB-2/Neu, and it is considered the preferred heterodimerization partner for other members of the EGFR family. To assess the importance of the catalytic activity of ErbB-2 in embryonic development, we have generated mice expressing a kinase-dead erbB-2 cDNA under the transcriptional control of the endogenous promoter. Here, we show that mice homozygous for the kinase-dead erbB-2 allele die at midgestation and display the same spectrum of embryonic defects seen in erbB-2 knockout mutants. These observations suggest that the catalytic activity of ErbB-2 is essential for normal embryonic development.
The epidermal growth factor receptor (EGFR) family of growth factor receptor tyrosine kinases, including ErbB-1/EGFR, ErbB-2/Neu, ErbB-3, and ErbB-4, have been implicated in breast cancer as well as several other human cancers (2). Recently, gene targeting studies have demonstrated specific roles for each of the EGFR family members in normal mammalian development. For example, erbB-2 (12) and erbB-4 (6) knockout mice die at midgestation due to deficient cardiac function associated with a lack of myocardial ventricular trabeculation and display abnormal development of the peripheral nervous system. Cardiac rescue of the defects seen in erbB-2 null mice revealed additional roles for ErbB-2 at the developing neuromuscular junction (13, 17). ErbB-3 mutant mice have less severe defects in the heart and consequently are able to survive several days later through embryogenesis. However, sensory and motor neurons in these animals show signs of degeneration due to a lack of proper Schwann cell development (1, 22).
Although the structures of the EGFR family receptors have been described in detail, their individual roles in and contributions to these developmental processes remain to be investigated. To achieve the observed diversity in signaling potential, a coordinate array of ligand-receptor and receptor-receptor interactions are possible. Following activation of the various EGFR family members with one of several EGF family ligands, both homodomeric and heterodimeric combinations of receptors are induced, and their intrinsic catalytic tyrosine kinase activity is stimulated (30). Upon receptor dimerization, specific tyrosine residues residing in the terminal tail of the receptor dimer become phosphorylated and serve as important potential binding sites for various intracellular signaling proteins.
Although activated ErbB receptors may partake in any particular combination of homodimerization or heterodimerization complexes, it is important to note that a hierarchical order of preference, stability, and signaling potential for each receptor combination is in effect (7). In particular, there is generally a greater preference and likely an advantage for dimerization complexes to include ErbB-2 as the partner because of its potent intrinsic kinase activity. Since no identified ligand is known to bind to and activate ErbB-2 alone, it is considered an orphan receptor. Thus, stimulation of ErbB-2 kinase activity may be mediated through normal ligand activation of another ErbB receptor first, which subsequently engages in a specific heterodimer complex (3, 18, 19). In contrast, ErbB-3, which can bind to and become activated by the ligand neuregulin, is naturally kinase inactive and therefore must depend on a heterodimerization partner for phosphorylation of its tyrosine residues (8, 26). Indeed, the ErbB-2-ErbB-3 complex is very stable and transmits a strong mitogenic signal (11). These observations strongly suggest that ErbB-2 plays a central role in the EGFR family signal transduction.
Although genetic ablation studies demonstrate the importance of a receptor to a biological function, they do not address precisely how and which of the individual functional domains of the receptor contribute to the phenotype. It is also unclear within this complex array of receptor dimerization whether the loss of ErbB-2 results in direct or indirect consequences of a lack of the receptor and its interactions with other proteins.
To assess exactly how ErbB-2 may act as the central mediator of the EGFR family of receptors, we investigated whether the kinase activity of ErbB-2 is indeed essential for its complete biological effects. To accomplish this, we generated mice expressing a kinase-dead erbB-2 cDNA under the transcriptional control of the endogenous erbB-2 promoter. Mice homozygous for the kinase-dead erbB-2 mutation died at embryonic day 10.5 (E10.5) due to a lack of cardiac trabeculation and displayed defects in neural development. These observations argue that the catalytic activity of ErbB-2 is absolutely required for normal embryonic development and cannot be compensated for by other members of the EGFR family.
MATERIALS AND METHODS
Generation of targeting construct and mutant mice.
Oligonucleotide-directed PCR mutagenesis with the following primers was used to create the mutant (K757 M) kinase-dead ErbB-2 receptor: AB11151, GGA AGT ATA CGA TCG CTA GGC; AB10015, CAC CAT GAT AGC CAC GGG GAT TTT CAC; AB 10014, C GTG GCT ATC ATG GTG TTG AGA GAA AAC; and AB 11152, CGA CCT CGG TGT TCT CGG AC. The underlined nucleotides identify the sequence substituted to create the desired mutation. PCR products were subsequently subcloned into a wild-type erbB-2 cDNA and then cloned into the final targeting vector. This plasmid was electroporated into R1 embryonic stem (ES) cells, and G418- or geneticin (Gibco-BRL)-resistant colonies were picked and subsequently screened by Southern blot analysis for correctly targeted mutants. Mutant mice were generated from the positive ES cell clones by the method of blastocyst injection into BALB/c-derived blastocysts (10). Subsequent generations were maintained in an SV129/BALB/c background.
RNase protection assays.
RNA was extracted and purified from individual embryos by the guanidine isothiocyanate-cesium chloride method as previously described (27). For RNase protection assays, as described previously (24), 30 μg of total RNA was used and hybridized to an antisense erbB-2 riboprobe (23). The protocol was modified by lowering the overnight hybridization temperature to 45°C and only T1 RNase (450 U) was used in the digestion reactions for 20 min at 37°C.
Western blot analysis.
Fresh or flash-frozen embryos were lysed in TNE lysis buffer (24). Cleared lysates were electrophoresed through sodium dodecyl sulfate (SDS)-polyacrylamide gels, and the proteins were transferred to polyvinylidene difluoride membranes (Immobilon-P; Millipore). The membranes were blocked in 10% skim milk-Tris-buffered saline (TBS) for 1 h at room temperature and then incubated with the appropriate antibody. For ErbB-2 immunoblots, the membranes were incubated with anti-ErbB-2 antibodies (1:1,000; AB-3, Oncogene Science) overnight at 4°C. Immunoblots for Grb2 were performed with rabbit anti-Grb2 polyclonal sera (1:2,500; C-23, Santa Cruz). After the primary antibody incubations, membranes were subjected to four 15-min washes in TBS-1% Tween 20 (Bio-Rad). Subsequently, horseradish peroxidase-conjugated anti-mouse or anti-rabbit immunoglobulin secondary antibodies (1:5,000; Jackson Laboratories) were incubated with the membranes for 1 h at room temperature and then washed twice for 15 min each in TBS-1% Tween-20 and twice for 15 min each in TBS alone. Immunoblots were visualized by enhanced chemiluminescence (Amersham) as specified by the manufacturer.
Histology.
Embryos from timed matings were dissected free from the placenta and cleared of extraembryonic tissues. A small piece of the visceral yolk sac was retained and placed in tail lysis buffer (100 mM Tris-HCl [pH 8.5], 5 mM EDTA, 0.2% SDS, 200 mM NaCl, 100 μg of proteinase K per μl), and the DNA was isolated and subsequently used for genotyping the embryos. Dissected embryos were quickly rinsed in ice-cold phosphate-buffered saline (PBS) and transferred directly into 4% paraformaldehyde, fixed overnight at 4°C, washed twice in 70% ethanol, and stored at 4°C in 70% ethanol.
For standard hematoxylin and eosin (Fisher Scientific) staining, samples were embedded in paraffin, and 8-μm serial sections were cut and mounted. For whole-mount in situ hybridizations, embryos were fixed in 4% paraformaldehyde-0.2% glutaraldehyde (Fisher Scientific), dehydrated through a graded series of methanol-PBT (1× PBS, 0.1% Tween 20; Sigma) baths and stored in 100% methanol at −20°C until needed. Whole-mount in situ hybridizations were carried out as previously described (29).
In vitro transcription of Phox2a riboprobes.
The Phox2a riboprobe plasmid, pKS903 SSN (25), was digested with SstII and transcribed with the T3 RNA polymerase to generate an antisense riboprobe; the sense riboprobe was generated using the same template but linearized with HindIII and transcribed with T7 RNA polymerase. The in vitro transcription reactions were carried out in 20-μl reaction volumes containing 14 μl of distilled water (diethyl pyrocarbonate [DEPC] treated), 2 μl of 10× transcription buffer (Boehringer Mannheim), 2 μl of DIG RNA labeling mix (Boehringer Mannheim), 1 μg of template DNA, 30 U of RNAGuard (Pharmacia), and 30 U of RNA polymerase (Boehringer Mannheim or Gibco-BRL). The reactions were incubated at 37°C for 2 h and stopped with the addition of 20 U of DNase I (RNase-free; Boehringer Mannheim). The riboprobes were precipitated and resuspended in 100 μl of DEPC-treated distilled water (≈100 ng/μl).
RESULTS AND DISCUSSION
A kinase-dead variant of the ErbB-2/Neu receptor was created by oligonucleotide-directed PCR mutagenesis to generate a point mutation affecting lysine residue 757. This K757M alteration ablates the conserved ATP-binding lysine residue in the tyrosine kinase domain, resulting in its inability to phosphorylate its substrates (20, 21, 28). We have confirmed that disruption of this key amino acid results in ablation of ErbB-2-associated kinase activity and also inactivates the potent transforming activity of an oncogenic erbB-2 mutant (data not shown).
To determine the functional importance of the ErbB-2 kinase activity in vivo, we generated a targeting vector in which the first coding exon of the endogenous erbB-2 gene was replaced with either a wild-type erbB-2 cDNA (erbB-2 knock-in [KI]) (Fig. 1A) or a cDNA harboring the K747M mutation (erbB-2 kinase-dead [KD]) (Fig. 1B). To facilitate recovery of targeted recombination events, a PGK-Neo expression cassette was inserted downstream of the inserted cDNA. The constructs were electroporated into R1 ES cells, and independent clonal lines were isolated and subjected to Southern blot analyses with an appropriate external probe to identify successful targeting events (Fig. 1).
FIG. 1.

Targeted erbB-2 cDNA knock-in strategy by homologous recombination. For germ line expression, a targeting vector was constructed in which exon 1 was replaced by either (A) a wild-type erbB-2 cDNA or (B) a cDNA encoding the kinase-dead erbB-2 mutation, followed by a PGK-neomycin (Neo) cassette and targeted to the endogenous erbB-2 locus by homologous 5"- and 3"-flanking arms. Digestion of genomic DNA with HindIII (H) and subsequent Southern blot analysis (inset) with an external probe, as indicated, resulted in a 7.5-kb band for the endogenous allele, whereas the knock-in cDNA alleles introduced a HindIII site and resulted in a 4.0-kb band.
After microinjection of several independently targeted ES cell lines into donor blastocysts, chimeric mice were obtained and bred to identify those that transmitted the mutant alleles through the germ line. erbB-2wt/KI and erbB-2wt/KD mice appeared normal and were fertile. erbB-2KI/KI mice were also viable and were generated at the expected Mendalian ratios (data not shown). Thus, in contrast to the generation of erbB-2−/− mice, in which exon 1 was replaced by a phosphoglycerate kinase (PGK)-neomycin resistance cassette (12), replacement of the first coding exon of erbB-2 with a wild-type erbB-2 cDNA rescued the embryonic lethality associated with disruption and inactivation of the erbB-2 gene. Interestingly, no viable erbB-2KD/KD mutant mice were observed in the litters generated from heterozygous matings that were subsequently genotyped at 3 weeks of age (Fig. 2A).
FIG. 2.

Embryonic lethality at midgestation in kinase-dead mutants. Mendelian ratios from the progeny of heterozygous matings were determined and compared with the frequency of genotypes observed. (A) No homozygous erbB-2KD/KD mutant animals were observed at weaning age (3 weeks old). (B) Observations at E11.5 to E13.5 revealed the expected number of mutant embryos; however, all erbB-2KD/KD (∗∗) embryos were being resorbed, and no heartbeat was detected. (C) At E10.5, all the embryos appeared healthy and were present in proportion with Mendelian frequencies. Solid bars, observed; shaded bars, expected.
Since erbB-2−/− embryos died at midgestation due to defects in heart development (12), we assessed whether we could detect viable embryos at E10.5 for our kinase-dead mutants. To accomplish this, timed matings between heterozygous animals were set up, and embryos were dissected from the uterus and observed. Embryo genotypes were determined by analysis of DNA isolated from their visceral yolk sacs. At E10.5, each possible genotype was present at the expected Mendelian frequencies (Fig. 2C), and all embryos were viable, possessed a heartbeat, and appeared normal in size. However, in mutant embryos the hearts were slightly enlarged and had irregular beats. Further observations at E11.5 to E13.5 (Fig. 2B) revealed that homozygous mutant erbB-2KD/KD embryos, although present at the expected frequency, had no heartbeat and showed signs of resorption such as arrested growth, pale color, and soft tissue. Thus, consistent with the embryonic lethality of erbB-2−/− mutations, these observations confirmed that mutant embryos expressing the kinase-dead ErbB-2 receptor were dying in utero at midgestation, between E10.5 and E11.5.
To investigate the cause of embryonic lethality, we performed histological analyses of E10.5 embryos. Both erbB-2KI/KI (Fig. 3A) and erbB-2wt/KD (Fig. 3B) knock-in embryos were completely normal in their development of the heart trabeculae. In contrast, erbB-2KD/KD mutant embryos clearly lacked development of ventricular trabeculae (Fig. 3C) that likely resulted in the observed reduction in embryonic blood flow. These results are strikingly consistent with the defects identified previously in erbB-2−/− embryos as well as in neuregulin−/− and erbB-4−/− embryos (6, 12, 15). Thus, the cardiac defects seen in the kinase-dead mutants were directly attributable to a loss of ErbB-2's enzymatic tyrosine kinase activity.
FIG. 3.
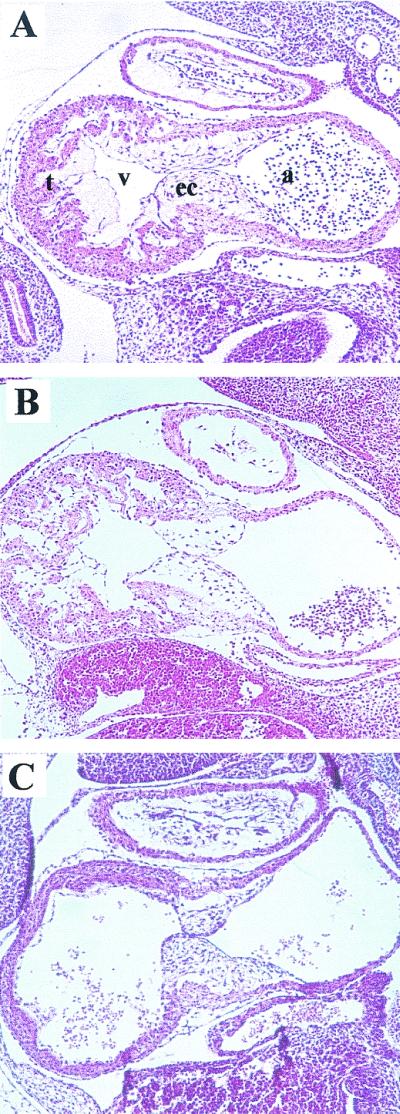
Defects in heart development in E10.5 mutant embryos. Parasagittal sections of (A) erbB-2KI/KI, (B) erbB-2wt/KD, and (C) erbB-2KD/KD embryos at E10.5 were stained with hematoxylin and eosin. Although heartbeats were detected at the time of dissection, histological examinations of the hearts revealed a lack of trabeculae in the ventricles of erbB-2KD/KD mutants but were present in heterozygous littermates and in age-matched erbB-2KI/KI knock-in embryos. t, trabeculae; v, ventricle; ec, endocardial cushion; a, atrium.
We also examined whether the same peripheral nervous system abnormalities seen in erbB-2−/− mutants were also similarly affected by expression of the kinase-dead mutation. One important marker for peripheral neural tissues is the Phox2a transcription factor (16). To explore whether neural development was perturbed in the KD mice, embryos derived from E10.5 and E11.5 were subjected to whole-mount in situ analyses using an antisense Phox2a riboprobe (Fig. 4).In both erbB-2wt/wt (Fig. 4A and B) and erbB-2wt/KD (Fig. 4C and D) embryos, the sympathetic chain developed normally. However, in the E10.5 to 11 erbB-2KD/KD homozygous embryos, only a weak Phox2a signal could be detected in the rostralmost regions of the sympathetic chain, a clear indication that this structure had either failed to initiate development or was incapable of developing in the kinase-dead genetic background (Fig. 4E and F).
FIG. 4.

Lack of sympathetic chain ganglia in kinase-dead mutants. Day E10 to E11 embryos were subjected to whole-mount in situ hybridization analysis using an antisense Phox2a riboprobe. Normal sympathetic chain ganglia development was present in (A and B) erbB-2wt/wt embryos and (C and D) erbB-2wt/KD heterozygous embryos, whereas (E and F) erbB-2KD/KD homozygous mutants lacked proper development or they were delayed in (G and H) erbB-2KI/KI embryos. The white arrowheads highlight the developing sympathetic chain ganglia.
Analyses of Phox2a expression in erbB-2KD/KD (Fig. 4E) and erbB-2KI/KI (Fig. 4G) embryos revealed that the sympathetic chain appeared to be absent at E10 to 10.5 compared to age-matched wild-type and heterozygous embryos (compare Fig. 4G with Fig. 4A and C). However, by E10.5 to 11, a lengthy sympathetic chain had developed in the erbB-2KI/KI homozygotes (Fig. 4H) but remained completely absent in the erbB-2KD/KD embryos (Fig. 4F). These observations indicate that the catalytic activity of ErbB-2 is also essential for normal development of the primary sympathetic chain ganglia.
To confirm that these knock-in alleles expressed ErbB-2, we performed Western immunoblot (Fig. 5A)and RNase protection (Fig. 5B) analyses. As shown in Fig. 5B, the wild-type erbB-2 KI allele and the kinase-dead allele expressed similar levels of erbB-2 transcripts. Similarly, immunoblot detection for ErbB-2 also revealed comparable protein levels between the two knock-in mutants at E10.5 (Fig. 5A). The slightly lower levels of ErbB-2 seen in the erbB-2KD/KD embryos relative to erbB-2KI/KI embryos is likely due to and consistent with the loss of tissue structures in the kinase-dead mutants (Fig. 2 and 3), which would normally express significant levels of ErbB-2.
FIG. 5.

Detection of ErbB-2 expression in E10.5 embryos. Expression of ErbB-2 generated from the cDNA insert in erbB-2KIKI and erbB-2KD/KD embryos was determined. For (A) immunoblot analyses, total protein isolated from E10.5 embryos was subjected to SDS-PAGE and incubated with an anti-ErbB-2 antibody. Detection of Grb-2 (lower panel) protein was used to control for equal protein lysate quantification. (B) RNase protection assays with an antisense erbB-2 riboprobe were used to detect erbB-2 transcripts in total RNA isolated from erbB-2wt/wt (lane 1), erbB-2KD/KD (lanes 3 and 4), and erbB-2KI/KI (lanes 6 and 7) embryos. A mouse PGK riboprobe (lower panel) was used as an internal control for equal sample loading in each lane.
Interestingly, both the erbB-2KI/KI and the erbB-2KD/KD knock-in embryos expressed only 10 to 15% of the expected ErbB-2 protein as detected in erbB-2wt/wt embryos (Fig. 5A, compare lanes 1 and 2 to lanes 3 to 6) despite expressing comparable levels of erbB-2 transcripts (Fig. 5B), which may reflect a requirement for splicing events for efficient transport and translation of the mRNA. In this regard it should be noted that embryos expressing both copies of the wild-type knock-in erbB-2 allele display a 24-h delay in development of the sympathetic chain (Fig. 4), suggesting that reduced ErbB-2 levels may have a minor phenotypic consequence.
Our observations have important implications in understanding the role of erbB-2 in promoting both normal cardiac and neural development. Previous studies have demonstrated that the integrity of ErbB-2, ErbB-4, or neuregulin is essential for the development of the cardiac trabecular extensions. Despite the presence of a functional ErbB-4 protein that could potentially transphosphorylate ErbB-2, our results further suggest that the catalytic activity of ErbB-2 alone is required to recapitulate the necessary signal transduction pathways leading to proper trabeculation.
In contrast to myocardial trabecula formation, ErbB-2 and ErbB-3 are thought to mediate the survival of neural crest cells, contributing to the development of the peripheral nervous system, since similar cranial nerve phenotypes were seen in mutant ErbB-2 and mutant ErbB-3 mice as well as in neuregulin mutants (12, 15, 22). Given that ErbB-3 is kinase defective and is completely dependent on its heterodimerization partners for its activation (8), inactivation of ErbB-2 catalytic activity would be expected to have profound effects on neural development. From these data, we can conclude that ErbB signaling is dependent on the kinase activity of ErbB-2 and that a kinase-dead ErbB-2 mutant acts essentially as a functionally null receptor.
In contrast to the embryonic lethality caused by inactivation of the catalytic activity of ErbB-2, mutants carrying a naturally occurring germ line mutation in the kinase domain of EGFR known as Waved-2 are completely viable and display only epithelial defects, such as a wavy hair phenotype (5, 14). Thus, unlike ErbB-2, other EGFR family members can presumably compensate for the severe impairment in EGFR catalytic activity. The difference between these phenotypes may reflect the hierarchical importance of ErbB-2 within the EGFR family signaling network. Alternatively, the difference in the phenotype in these strains may reflect the fact that the Waved-2 mutation has not completely ablated the catalytic activity of EGFR (14) and thus retains a higher degree of biological function.
Given the dominant-negative action of the kinase-dead ErbB-2 receptor expressed in vitro (20) it is surprising that mice heterozygous for the erbB-2 KD allele failed to exhibit any obvious phenotype that might be expected of a trans-dominant inhibition of the remaining wild-type allele. One potential explanation for this observation is that the level of kinase-dead ErbB-2 is insufficient to interfere with the remaining endogenous wild-type ErbB-2 receptor. Indeed, the erbB-2 KD allele produced only 10% of the expected ErbB-2 protein (Fig. 5). To exclude this possibility, we crossed erbB-2 KI mice with erbB-2 KD mice because both of these knock-in alleles expressed similar levels of ErbB-2. Phenotypic analyses of these crosses demonstrated that the erbB-2 KD allele failed to exhibit a dominant-negative effect on the remaining erbB-2 KI allele (data not shown). Thus, like the Waved-2 EGFR kinase mutation, the erbB-2 KD allele fails to exhibit any discernible dominant-negative effect on the remaining intact ErbB-2 receptor.
Studies with other receptor tyrosine kinases have concluded that the catalytic activity of receptor tyrosine kinases is dispensable for normal physiological function of the receptor. For example, the Flt-1 (vascular endothelial growth factor receptor) null mutation resulted in early embryonic lethality at E8.5 with disorganized blood vessels (4). However, mice expressing a kinase-deficient Flt-1 survived and showed normal angiogenesis (9), suggesting that other components of the receptor are more important to its functional role. In contrast to these observations, we have found that the catalytic activity of ErbB-2 is essential for embryonic development.
The embryonic lethality associated with the expression of the kinase-dead ErbB-2 is not likely due to inappropriate localization of the receptor, since it has previously been demonstrated that this mutant receptor is efficiently expressed on the cell surface (20). Given that ErbB-2 is the preferred heterodimerization partner for the other EGFR family members (7), the requirement for ErbB-2 catalytic function in vivo suggests that its catalytic activity is critical for EGFR family signaling. Future studies with these strains should allow identification of downstream targets of ErbB-2.
Acknowledgments
R.C. and W.R.H contributed equally to this work.
We thank Margaret Hibbs and Ashley Dunn for providing genomic clones and Michael Rudnicki and Christo Goridis for providing plasmids used in this study. We also thank Brian Allore and Dinsdale Gooden at the MOBIX Central Facility, McMaster University, for automated DNA sequencing analysis and oligonucleotide synthesis.
This work was supported by a grant awarded to W.J.M. by the Medical Research Council of Canada. W.J.M. is a recipient of a Medical Research Council of Canada scientist award, and R.C. was supported by a studentship from the Cancer Research Society of Canada and a U.S. Army D.O.D. studentship.
REFERENCES
- 1.Britsch, S., L. Li, S. Kirchhoff, F. Theuring, V. Brinkmann, C. Birchmeier, and D. Riethmacher. 1998. The ErbB2 and ErbB3 receptors and their ligand, neuregulin-1, are essential for development of the sympathetic nervous system. Genes Dev. 12:1825-1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burden, S., and Y. Yarden. 1997. Neuregulins and their receptors: a versatile signaling module in organogenesis and oncogenesis. Neuron 18:847-855. [DOI] [PubMed] [Google Scholar]
- 3.Carraway, K. L., and L. C. Cantley. 1994. A neu acquaintance for erbB3 and erbB4: a role for receptor heterodimerization in growth signaling. Cell 78:5-8. [DOI] [PubMed] [Google Scholar]
- 4.Fong, G. H., J. Rossant, M. Gertsenstein, and M. L. Breitman. 1995. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 376:66-70. [DOI] [PubMed] [Google Scholar]
- 5.Fowler, K. J., F. Walker, W. Alexander, M. L. Hibbs, E. C. Nice, R. M. Bohmer, G. B. Mann, C. Thumwood, R. Maglitto, and J. A. Danks. 1995. A mutation in the epidermal growth factor receptor in waved-2 mice has a profound effect on receptor biochemistry that results in impaired lactation. Proc. Natl. Acad. Sci. USA 92:1465-1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gassmann, M., F. Casagranda, D. Orioli, H. Simon, C. Lai, R. Klein, and G. Lemke. 1995. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature 378:390-394. [DOI] [PubMed] [Google Scholar]
- 7.Graus-Porta, D., R. R. Beerli, J. M. Daly, and N. E. Hynes. 1997. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 16:1647-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guy, P. M., J. V. Platko, L. C. Cantley, R. A. Cerione, and K. L. Carraway. 1994. Insect cell-expressed p180erbB3 possesses an impaired tyrosine kinase activity. Proc. Natl. Acad. Sci. USA 91:8132-8136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hiratsuka, S., O. Minowa, J. Kuno, T. Noda, and M. Shibuya. 1998. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc. Natl. Acad. Sci. USA 95:9349-9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Joyner, A. L. 1993. Gene targeting—a practical approach. Oxford University Press, New York, N.Y.
- 11.Karunagaran, D., E. Tzahar, R. R. Beerli, X. Chen, D. Graus-Porta, B. J. Ratzkin, R. Seger, N. E. Hynes, and Y. Yarden. 1996. ErbB-2 is a common auxiliary subunit of NDF and EGF receptors: implications for breast cancer. EMBO J. 15:254-264. [PMC free article] [PubMed] [Google Scholar]
- 12.Lee, K. F., H. Simon, H. Chen, B. Bates, M. C. Hung, and C. Hauser. 1995. Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature 378:394-398. [DOI] [PubMed] [Google Scholar]
- 13.Lin, W., H. B. Sanchez, T. Deerinck, J. K. Morris, M. Ellisman, and K. F. Lee. 2000. Aberrant development of motor axons and neuromuscular synapses in erbB2-deficient mice. Proc. Natl. Acad. Sci. USA 97:1299-1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luetteke, N. C., H. K. Phillips, T. H. Qiu, N. G. Copeland, H. S. Earp, N. A. Jenkins, and D. C. Lee. 1994. The mouse waved-2 phenotype results from a point mutation in the EGF receptor tyrosine kinase. Genes Dev. 8:399-413. [DOI] [PubMed] [Google Scholar]
- 15.Meyer, D., and C. Birchmeier. 1995. Multiple essential functions of neuregulin in development. Nature 378:386-390. [DOI] [PubMed] [Google Scholar]
- 16.Morin, X., H. Cremer, M. R. Hirsch, R. P. Kapur, C. Goridis, and J. F. Brunet. 1997. Defects in sensory and autonomic ganglia and absence of locus coeruleus in mice deficient for the homeobox gene Phox2a. Neuron 18:411-423. [DOI] [PubMed] [Google Scholar]
- 17.Morris, J. K., W. Lin, C. Hauser, Y. Marchuk, D. Getman, and K. F. Lee. 1999. Rescue of the cardiac defect in ErbB2 mutant mice reveals essential roles of ErbB2 in peripheral nervous system development. Neuron 23:273-283. [DOI] [PubMed] [Google Scholar]
- 18.Pinkas-Kramarski, R., M. Shelly, B. C. Guarino, L. M. Wang, L. Lyass, I. Alroy, M. Alimandi, A. Kuo, J. D. Moyer, S. Lavi, M. Eisenstein, B. J. Ratzkin, R. Seger, S. S. Bacus, J. H. Pierce, G. C. Andrews, Y. Yarden, and M. Alamandi. 1998. ErbB tyrosine kinases and the two neuregulin families constitute a ligand-receptor network. Mol. Cell. Biol 18:6090-6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pinkas-Kramarski, R., L. Soussan, H. Waterman, G. Levkowitz, I. Alroy, L. Klapper, S. Lavi, R. Seger, B. J. Ratzkin, M. Sela, and Y. Yarden. 1996. Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO J. 15:2452-2467. [PMC free article] [PubMed] [Google Scholar]
- 20.Qian, X., W. C. Dougall, M. E. Hellman, and M. I. Greene. 1994. Kinase-deficient neu proteins suppress epidermal growth factor receptor function and abolish cell transformation. Oncogene 9:1507-1514. [PubMed] [Google Scholar]
- 21.Qian, X., C. M. LeVea, J. K. Freeman, W. C. Dougall, and M. I. Greene. 1994. Heterodimerization of epidermal growth factor receptor and wild-type or kinase-deficient Neu: a mechanism of interreceptor kinase activation and transphosphorylation. Proc. Natl. Acad. Sci. USA 91:1500-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riethmacher, D., E. Sonnenberg-Riethmacher, V. Brinkmann, T. Yamaai, G. R. Lewin, and C. Birchmeier. 1997. Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature 389:725-730. [DOI] [PubMed] [Google Scholar]
- 23.Siegel, P. M., D. L. Dankort, W. R. Hardy, and W. J. Muller. 1994. Novel activating mutations in the neu proto-oncogene involved in induction of mammary tumors. Mol. Cell. Biol. 14:7068-7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siegel, P. M., E. D. Ryan, R. D. Cardiff, and W. J. Muller. 1999. Elevated expression of activated forms of Neu/ErbB-2 and ErbB-3 are involved in the induction of mammary tumors in transgenic mice: implications for human breast cancer. EMBO J. 18:2149-2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valarche, I., J. P. Tissier-Seta, M. R. Hirsch, S. Martinez, C. Goridis, and J. F. Brunet. 1993. The mouse homeodomain protein Phox2 regulates Ncam promoter activity in concert with Cux/CDP and is a putative determinant of neurotransmitter phenotype. Development 119:881-896. [DOI] [PubMed] [Google Scholar]
- 26.Wallasch, C., F. U. Weiss, G. Niederfellner, B. Jallal, W. Issing, and A. Ullrich. 1995. Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. EMBO J. 14:4267-4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Webster, M. A., J. N. Hutchinson, M. J. Rauh, S. K. Muthuswamy, M. Anton, C. G. Tortorice, R. D. Cardiff, F. L. Graham, J. A. Hassell, and W. J. Muller. 1998. Requirement for both Shc and phosphatidylinositol 3" kinase signaling pathways in polyomavirus middle T-mediated mammary tumorigenesis. Mol. Cell. Biol. 18:2344-2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weiner, D. B., Y. Kokai, T. Wada, J. A. Cohen, W. V. Williams, and M. I. Greene. 1989. Linkage of tyrosine kinase activity with transforming ability of the p185neu oncoprotein. Oncogene 4:1175-1183. [PubMed] [Google Scholar]
- 29.Wilkinson, D. G., and M. A. Nieto. 1993. Detection of messenger RNA by in situ hybridization to tissue sections and whole mounts. Methods Enzymol. 225:361-373. [DOI] [PubMed] [Google Scholar]
- 30.Yarden, Y., and A. Ullrich. 1988. Growth factor receptor tyrosine kinases. Annu. Rev. Biochem. 57:443-478. [DOI] [PubMed] [Google Scholar]