

Abstract
Urease activity during in vitro growth in the saprobic and parasitic phases of Coccidioides spp. is partly responsible for production of intracellular ammonia released into the culture media and contributes to alkalinity of the external microenvironment. Although the amino acid sequence of the urease of Coccidioides posadasii lacks a predicted signal peptide, the protein is transported from the cytosol into vesicles and the central vacuole of parasitic cells (spherules). Enzymatically active urease is released from the contents of mature spherules during the parasitic cycle endosporulation stage. The endospores, together with the urease and additional material which escape from the ruptured parasitic cells, elicit an intense host inflammatory response. Ammonia production by the spherules of C. posadasii is markedly increased by the availability of exogenous urea found in relatively high concentrations at sites of coccidioidal infection in the lungs of mice. Direct measurement of the pH at these infection sites revealed an alkaline microenvironment. Disruption of the urease gene of C. posadasii resulted in a marked reduction in the amount of ammonia secreted in vitro by the fungal cells. BALB/c mice challenged intranasally with the mutant strain showed increased survival, a well-organized granulomatous response to infection, and better clearance of the pathogen than animals challenged with either the parental or the reconstituted (revertant) strain. We conclude that ammonia and enzymatically active urease released from spherules during the parasitic cycle of C. posadasii contribute to host tissue damage, which exacerbates the severity of coccidioidal infection and enhances the virulence of this human respiratory pathogen.
Coccidioides spp. are airborne molds and respiratory pathogens of humans which grow as a filamentous saprobe in desert and semiarid soils. They are the causative agents of coccidioidomycosis, which is found in areas of endemicity such as the southwestern United States, northern Mexico, and Central and South America, including Guatemala, Honduras, northern Columbia, Venezuela, Paraguay, and Argentina (29). Two species have been identified: Coccidioides immitis, which appears to be biogeographically restricted to the San Joaquin Valley of California, and C. posadasii, which is widespread throughout the regions of endemicity of the Americas (16). The saprobic form of Coccidioides survives under harsh environmental conditions, including meager rainfall, hot summers, and sandy, alkaline soil with high salinity (33). The mycelial phase has been reported to tolerate a broad pH range of pH 2 to 12 (4). In response to incubation in an acidic, sugar-free medium, the saprobe is induced to release ammonium ions and ammonia in quantities which increase the pH of its extracellular environment (8). Parasite-phase cultures similarly respond to acidification of their external environment by secretion of NH4+ and NH3. A source of intracellular ammonia is urea hydrolysis, and Coccidioides has been reported to produce an enzymatically active urease (25, 39). This cytosolic enzyme catalyzes the hydrolysis of urea to yield ammonia and carbamate. The latter spontaneously hydrolyzes to form carbonic acid and a second molecule of ammonia. At physiological pH, the carbonic acid proton dissociates and ammonia molecules equilibrate with water to become protonated, with a resultant increase in pH (26).
Urease has been shown to be an important virulence factor in several bacterial pathogens (5). In Helicobacter pylori, the causative agent of chronic gastritis and duodenal ulcers, urease activity is induced both by acidic conditions of the host microenvironment and the availability of gastric urea (26). This results in production of high amounts of intracellular ammonia, some of which is released and neutralizes the acid in the immediate vicinity of the pathogen. High concentrations of bacterially derived ammonia at sites of infection have been suggested to injure the gastric mucosa as a result of tissue exposure to ammonium hydroxide. Evidence has also been presented that urease stimulates the host gastric epithelial cells to produce proinflammatory cytokines (36). The persistent inflammatory response to H. pylori infection ultimately damages mucosal tissue and exacerbates the ulcerated condition. Recent studies have suggested that engulfment of urease-producing bacteria by macrophages in the presence of exogenous urea results in intraphagocytic release of ammonia, which has an inhibitory effect on macrophage surface expression of major histocompatibility complex class II molecules (34). This, in turn, could have a suppressive effect on cell-mediated immune response to the microbial pathogen. Although urease activity has been reported for several genera of medically important fungi (e.g., Aspergillus, Candida, Cryptococcus, Rhodotorula, and Trichosporon spp.), this enzyme has been suggested to play a role in pathogenesis only in Coccidioides and Cryptococcus spp. (8, 11, 27). In this report we show that disruption of the C. posadasii urease gene results in a major reduction in the ability of the pathogen to kill intranasally challenged BALB/c mice. Survival appears to be related to the host's ability to mount an effective granulomatous response to the pathogen, which leads to clearance of the fungal cells from infected lung tissue. We present evidence that urease activity is an important virulence determinant of Coccidioides during interaction of the pathogen with murine lung tissue.
MATERIALS AND METHODS
Reagents.
Nutrients used in the fungal culture medium were obtained from Difco Laboratories (Detroit, Mich.). All other chemicals and solutions were purchased from Sigma Chemical Company (St. Louis, Mo.), unless otherwise specified.
Fungal strain, media, and growth conditions.
For production of arthroconidia, the saprobic (mycelial) phase of C. posadasii strain C735 was grown on GYE agar (1% glucose, 0.5% yeast extract, 1.5% agar) at 30°C for 3 to 4 weeks. For DNA extraction, parental and transformed strains of the pathogen were grown as the saprobic phase in GYE liquid medium at 30°C. For parasite-phase growth, these same strains were cultured in defined liquid glucose-salts medium at 39°C in the presence of 20% CO2 as previously described (17). Ammonium acetate (16.0 mM) is the sole nitrogen source provided in the glucose-salts medium. Alternatively, arthroconidia were converted to spherules by growth in RPMI 1640 medium supplemented with 10% heat-inactivated calf serum (catalog no. 30-2001 and 30-2020; American Type Culture Collection, Manassas, Va.) plus Na2HPO4, Tamol, and antibiotics as previously reported (30). The supplemented RPMI 1640 growth medium was used to test the effect of the addition of exogenous urea on ammonia production by C. posadasii. For these studies, the culture medium was contained in 24-well, flat-bottom, polystyrene tissue culture plates (Becton Dickinson and Co., Franklin Lakes, N.J.) (1.0 ml/well). Each well was inoculated with 106 arthroconidia. The parasitic cells were incubated with or without the addition of urea (10 mM) at 39°C in the presence of 20% CO2 for 7 days.
Immunolocalization.
In vitro-grown parasite-phase cells were cryofixed, subjected to freeze substitution, and embedded in L.R. White resin at −10°C in preparation for immunoelectron microscopy as previously described (19). Antiserum was raised in BALB/c mice against the purified recombinant urease protein as previously reported (39). The immunoglobulin G (IgG) fraction was isolated from immune sera by protein A affinity chromatography. Thin sections of cultured parasite-phase cells (24-h [first-generation] spherule initials, 72-h segmentation-phase spherules, and 132-h endosporulation-phase spherules; Fig. 1) were reacted with the specific antiurease antibody followed by goat anti-mouse secondary antibody conjugated to colloidal gold (Ted Pella Inc., Redding, Calif.) (20-nm particles) as previously reported (19). Thin sections of resin-embedded cells were examined by transmission electron microscopy. Incubation conditions, poststaining methods, and controls employed were the same as previously reported (19). Thick sections of these same cryofixed and resin-embedded spherules were reacted with antiurease IgG followed by secondary antibody conjugated with fluorescein isothiocyanate (FITC) as previously described (17). The stained sections were examined by fluorescence microscopy.
FIG. 1.

Diagrammatic representation of the in vitro parasitic cycle of Coccidioides posadasii strain C735. The sequence of morphogenetic events is indicated in incubation times (in hours) after arthroconidium inoculation of the parasite-phase culture. SOW, spherule outer wall fraction.
A secreted, spherule outer wall (SOW) fraction, which has previously been described (19), together with intact endospores released from ruptured spherules, was isolated from 132-h parasite-phase cultures. The freshly collected SOW fraction and endospores were reacted with either antiurease serum or preimmune murine serum at a 1:200 dilution in phosphate-buffered saline (PBS). The intact cells and SOW preparation were then washed with PBS, incubated with goat anti-mouse immunoglobulin conjugated with FITC, and examined by fluorescence microscopy as previously described (17).
Immunoblot analysis.
Detection of the C. posadasii urease protein in cell homogenates and culture filtrates of parasitic cells obtained from 24-, 72-, and 132-h cultures was conducted using aliquots of equilibrated total protein of each preparation separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Protein extraction was conducted as previously described (18). Immunoblot analysis with urease-specific murine antiserum was performed as previously reported (19). The SOW fraction, which was isolated by differential centrifugation as previously described (17), was obtained from both 72-h and 132-h parasite-phase cultures and subjected to SDS-PAGE and immunoblot analysis as described above.
Urease activity assay.
Determination of urease activity in parasitic cell homogenate, wall, and culture filtrate preparations was conducted as previously reported (25). One unit of urease activity is defined as the amount of enzyme required to hydrolyze 1 μmol of urea per min at 37°C. Hydrolysis was measured by monitoring the rate of release of ammonium ions (NH4+) plus ammonia (NH3) from the urea substrate as determined by the Bertholet reaction (25).
Isolation of the URE gene.
The original genomic clone of the C. posadasii urease gene (URE) was sequenced by the dideoxy chain termination method (39). The reported genomic sequence was compared to the nucleotide sequence of the same strain now available in the C. posadasii genome database (www.tigr.org), and a suspected error was detected in the former. The original genomic clone was resequenced using an ABI Prism 310 genetic analyzer (Perkin-Elmer, Foster City, Calif.) and examined with MacDNASIS sequence analysis software (version 3.5) (Hitachi, San Bruno, Calif.). The revised genomic sequence of the URE gene matches the sequence in the genome database. The genomic fragment sequenced in this study includes the full-length URE gene plus fragments of the 5′ and 3′ untranslated regions. To clone this genomic fragment, a PCR was first conducted with template genomic DNA of C. posadasii and the following sense and antisense primers: 5′-CTGAGAAAAAGAAAAGGGGAA-3′ and 5′-CTCCACAAGCACTTGAGTA-3′. The primer sequences were based on the C. posadasii genome database. The 5.6-kb PCR product was cloned into the pCR2.1 TOPO plasmid vector (Invitrogen, Carlsbad, Calif.) and sequenced as previously reported (12). The nucleotide sequence of the full-length clone was determined and shown to be identical to the sequence reported in the C. posadasii genome database.
Disruption of the URE gene.
Transformation of C. posadasii for generation of a mutant strain that lacks a functional URE gene was conducted as previously described (19). The pΔure construct was designed as a gene disruption vector (13) and consisted of the pAN7.1 plasmid plus a 1.6-kb genomic fragment of the URE gene. The latter was obtained by digestion of the 5.6-kb genomic clone with BglII and SnaBI (bp 1884 to 3446) and was ligated with BglII and StuI restriction sites of the pAN7.1 plasmid, respectively. Prior to transformation, the pΔure construct was linearized with SplI and then purified by ethanol precipitation. The transformation procedure and method employed to isolate homokaryotic transformants were the same as previously described (19). Hygromycin (Hph)-resistant colonies were selected for examination by PCR, Southern hybridization, and immunoblot analysis as previously reported (19).
Disruption of the URE gene was first confirmed by PCR using the primer pairs listed in Table 1. The HPH gene probe (Pb1), which was used for Southern hybridization to confirm single-site integration of the plasmid construct, consisted of a 354-bp PCR product generated by use of the primer pair e and f (listed in Table 1). A second 524-bp probe, generated with primer pair g and h, consisted of a 5′ fragment of the URE gene (Pb2) and was used for Southern hybridization to confirm that homologous recombination had occurred in the mutant strain and that the transformant was homokaryotic. A restriction map of the genomic locus containing the URE gene and a hypothetical restriction map of the URE gene disruption locus (Δure) were constructed to predict the size of the hybridized fragments in the Southern blots.
TABLE 1.
PCR primers used to screen for disruptants and amplify selected probes for Southern hybridization
| Primer | Nucleotide sequence | Sequence derivationa | Size and use of PCR product |
|---|---|---|---|
| a | 5′-GGCTGCTGCAAGTAGATCG-3′ | URE | 1.9 kb; to screen for wild-type URE gene |
| b | 5′-GTTTGGCTTCTCGACGACG-3′ | URE | |
| c | 5′-AGGTTGAATCATGCAGAAGCG-3′ | URE | 1.9 kb; to screen for disrupted URE gene |
| d | 5′-CCAAATGAAGCTATGCTACCTCC-3′ | pAN7.1 | |
| e | 5′-TGCTTTGCCCGGTGTATGAAACC-3′ | pAN7.1 | 354 bp; probe 1 (Pb1) for Southern hybridation |
| f | 5′-AAGGGATGGGAAGGATGGAGTATGG-3′ | pAN7.1 | |
| g | 5′-TGGACACTCCTTGACCTAGCG-3′ | URE | 524 bp; probe 2 (Pb2) for Southern hybridization |
| h | 5′-TCCGTAGACAATAGGGCGATC-3′ | URE |
Generation of the revertant strain.
To restore expression of the urease gene in the Δure host, the mutant strain was transformed with a plasmid (pUREr), which contained the parental strain-derived 5.6-kb genomic clone of the URE gene in the pCR2.1 TOPO vector. No drug-resistant selection marker was used to screen for transformants in this case. When the parental strain of C. posadasii was grown on Difco Bacto Urea agar base medium containing a phenol red pH indicator, a yellow-to-red color change was clearly visible at the margin and under the surface of the fungal colony within 4 days of incubation. The Δure mutant, on the other hand, failed to show a color change when grown on the same medium. This phenotypic difference was used to select putative transformants which contained the restored wild-type URE gene. In order to distinguish the revertant strain from the parental strain, the 5.6-kb genomic clone used to transform the Δure host was subjected to site-directed mutagenesis using a QuickChange kit (Stratagene, La Jolla, Calif.) according to the manufacturer's protocol. An adenine was added to the genomic sequence between nucleotide positions 2299 and 2300 to create an AflII restriction site (2297CTTAAG2302) within an intron of the URE gene. This introduced restriction site was used for PCR analysis and Southern hybridization to confirm restoration of the wild-type URE gene in the mutant strain and to establish that the 5.6-kb clone had integrated into chromosomal DNA of the Δure host by homologous recombination. Immunoblot analysis and the urease activity assay were used to confirm that the revertant strain (UreR) produced amounts of enzymatically active urease which were comparable to that of the parental strain grown under identical conditions.
Comparison of in vitro growth of the parental, mutant, and revertant strains.
The growth rates of the saprobic phase of the parental, Δure, and revertant (UreR) strains were compared on the basis of dry weight during a 4-day period of incubation in GYE liquid medium. Each culture contained 100 ml of medium and was inoculated with 103 arthroconidia. The results of parasitic cell development of each strain cultured in defined glucose-salts medium (17) were compared by light microscopy to determine the percentages of endosporulating spherules produced after a 6-day period of incubation. Each parasite-phase culture contained 100 ml of medium and was inoculated with 107 arthroconidia. Culture aliquots of each strain were examined by light microscopy.
In an attempt to more closely simulate physiological conditions, we cultured the parasite phase of each strain in supplemented RPMI 1640 medium and examined the change in the extracellular ammonium-ammonia concentration over a 7-day period of incubation. On the basis of our detection of relatively high concentrations of urea at sites of coccidioidal infection reported in this study (an approximately fourfold-higher concentration in Coccidioides-infected mice than in healthy mice), we compared the amounts of NH4+/NH3 released from parasitic cells into the RPMI 1640 medium in the absence or presence of exogenous urea (10 mM). The range of physiological concentration of urea in blood of normal mice is 1 to 3 mM. We also compared the consumption of exogenous urea by the parental versus the Δure mutant strain during the same 7-day period of incubation. Aliquots (50 μl) of the supplemented RPMI 1640 medium plus urea which supported growth of each strain were sampled daily. Each aliquot was diluted 10× with urease assay buffer (25) and incubated with 10 units of purified, commercially available, recombinant jack bean urease for 30 min at 37°C. The ammonium-ammonia content of each aliquot was determined by the Bertholet reaction (25). The concentration of ammonium plus ammonia in each aliquot prior to the addition of urease was subtracted from the estimate of total NH4+/NH3 concentration in the sample, and the final determination of ammonia concentration was divided by a factor of 2 for calculation of the urea content (2 mol of ammonia are produced per mole of urea). All analyses were conducted in triplicate, and standard deviations were determined.
Virulence studies.
To compare the virulence of the parental, mutant, and revertant strains, arthroconidia (50 viable cells) were first obtained from GYE plate cultures of each strain grown for 30 days at 30°C. The conidia were suspended in PBS and used to separately inoculate BALB/c mice (purchased from the National Cancer Institute, Bethesda, Md.) (12 8-week-old female mice per group) by the intranasal (i.n.) route as previously reported (19). The survival plots were subjected to Kaplan-Meier statistical analysis as previously described (19).
Three separate groups of BALB/c mice were challenged intranasally with the parental, mutant, or revertant strain as described above and then sacrificed at 12, 25, or 50 days postinfection to determine the residual CFU of C. posadasii in homogenates of the lungs and spleen as previously reported (12). Groups of five mice per time point were examined. The CFU counts per organ were expressed on a log scale, and the Mann-Whitney U test was used to compare median numbers as previously described (12). The detection limit of the CFU assay is 100 colonies per organ (log10 CFU = 2.00).
In situ pH measurements.
For this in vivo assay, BALB/c mice were infected either by the intraperitoneal route with 103 viable arthroconidia as previously reported (12) or by the i.n. route as described above. Four groups of mice (five mice per group) were separately challenged via one of these two routes with either the parental or Δure mutant strain. Two weeks after challenge, each mouse was subjected to necropsy; abscesses associated with the mesentery, spleen, and lungs were immediately probed with a MI-408 needle electrode (Microelectrodes, Londonderry, N.H.) to determine the internal pH of the infected tissue.
Estimates of urea concentration at sites of lung infection.
BALB/c mice were infected by the i.n. route with arthroconidia of either the parental or Δure mutant strain as described above. At 12 days postchallenge, the mice were sacrificed, lung abscesses were excised and quick-frozen, and the infected tissue was subjected to cryomicrotomy as previously reported (37). The 8-μm-thick sections from each abscess (10 sections per abscess) were pooled. Three mice infected with the parental or mutant strain were examined in this study. The pooled sections of each abscess were extracted separately with three aliquots of 150 μl of distilled water, and the total extract was assayed for urea concentration by use of commercially available, purified, jack bean urease as described above. Three separate lung abscesses obtained from each mouse were examined. This same procedure was conducted to determine the urea concentration in lung sections obtained from noninfected control mice.
Histopathology.
Separate groups of BALB/c mice challenged intranasally with either the parental or Δure mutant strain of C. posadasii as described above were used for comparative histopathology. Nine mice infected with the Δure mutant strain were sacrificed at 12, 20, or 25 days postchallenge. Their lungs were excised, formalin fixed, and paraffin embedded in preparation for histological examination using standard procedures. Three mice infected with the parental strain were examined only at 12 days postchallenge.
Nucleotide sequence accession number.
The revised genomic sequence of the URE gene has been deposited in GenBank under accession number U81509.3.
RESULTS
Urease is localized in the cytoplasm, vesicles, and central vacuole of spherules and is released during endosporulation.
C. posadasii strain C735 typically completes its first generation of the parasitic cycle in vitro within 5 to 6 days, and the sequence of morphogenetic events appears to be identical to that observed in vivo (35). In addition, first-generation parasitic cells of strain C735 grown in vitro have been shown to be near-synchronous in their development (18). This has permitted isolation of different cell types (e.g., spherule initials, spherules in early stages of segmentation, and mature spherules in the process of endosporulation) for biochemical and molecular studies using cultures incubated for 24 h, 72 h, and 132 h, respectively (Fig. 1). Arthroconidia first undergo isotropic growth to form spherule initials. On the basis of ultrastructural studies of this early stage of parasitic cell differentiation, it appears that a multitude of tiny vesicles first visible in spherule initials later coalesce to form a large central vacuole (9). Segmentation of the spherule cytoplasm occurs by ingrowth of the parasitic cell wall and results in the formation of compartments which surround the still-intact, central vacuole (9). It is evident from examinations of thin sections of this stage of development that all of the cytoplasm of the presegmented spherule is not incorporated into the compartments that later differentiate into endospores. The cytosolic debris, combined with the newly formed endospores and residual contents of the central vacuole, is released upon rupture of the mature spherules (Fig. 1). Another peculiar morphogenetic feature of Coccidioides spp. is that a lipid-rich, membranous SOW layer is produced and shed from the spherule surface during the parasitic cycle (Fig. 1) (17). The SOW material, which can be easily isolated by differential centrifugation from parasite-phase cultures as a cell-free fraction, has been shown to be highly immunogenic (17). In this study we provide evidence that enzymatically active urease, derived from the vacuolar contents and cytosolic debris, is released from ruptured spherules at the time of endosporulation and is transiently associated with the SOW and surface of endospores.
Immunolocalization of urease protein in organelles of different parasitic cell types was conducted by transmission electron microscopy using gold particles conjugated to C. posadasii urease-specific IgG. Thin sections of germinated arthroconidia (24-h spherule initials) which were incubated with antibody-coated gold particles revealed small amounts of gold label in the cytoplasm and within vesicles (not shown). Sections of in vitro-grown spherules which had initiated segmentation but not endospore differentiation (72- to 84-h cultures; Fig. 1) showed labeling in both the cytoplasmic vesicles and central vacuole (Fig. 2A). Gold particles were also observed associated with cytosolic debris trapped between the central vacuole and the newly formed segmentation wall (Fig. 1 and 2A). Thick sections of these same segmented spherules showed concentrated urease protein-specific fluorescent labeling in both the vesicles and the central vacuole (Fig. 2B), while control sections of these cells labeled with secondary antibody-FITC conjugate alone were devoid of labeling (Fig. 2C). As segmentation proceeds, the central vacuole ruptures and the preexisting vacuolar space accommodates the newly differentiated endospores (Fig. 1). Thin sections of endospore initials located within the mature, unruptured spherule (120-h culture) showed antiurease-labeled gold particles concentrated in vesicles (Fig. 2D). Endospores incubated with gold particles which had been conjugated to preimmune mouse serum showed no labeling (Fig. 2E). When the spherule eventually ruptures and releases its endospores (132- to 144-h parasite-phase culture; Fig. 1), the pool of urease protein which had presumably accumulated both in the central vacuole and regions of cytosolic debris is also discharged. The released urease protein associates with both the surface of intact endospores and the membranous sheets of lipid-rich SOW, as revealed by immunofluorescence (Fig. 2F). Control endospore and SOW preparations incubated with preimmune serum followed by secondary antibody-FITC conjugate showed no labeling (Fig. 2G).
FIG. 2.

(A) Thin section of segmented spherule (72- to 84-h culture; see Fig. 1) labeled with antiurease and colloidal gold conjugate, which reveals sites of intracellular localization of urease protein in the cytoplasm, vesicles (Vs), central vacuole (C.V.), and cytosolic debris trapped between spherule compartments and central vacuole. (B and C) Thick sections of segmented spherules stained with antiurease and FITC conjugate (B) or FITC-conjugated secondary antibody alone (C) are shown. Note the comparable results for localization of label in panels A and B. (D) Thin section of antiurease and colloidal gold-labeled endospore (120- to 132-h culture; see Fig. 1), which shows localization of urease protein in vesicles (arrow). Control section (E) of endospore was reacted with preimmune antibody and colloidal gold conjugate. (F) Intact endospores and SOW isolated from endosporulation-phase cultures (132 h; Fig. 1), both of which are labeled with antiurease and FITC conjugate. The control cells and SOW fraction in panel G were incubated with preimmune antibody and FITC conjugate. Mt, mitochondria; Seg. wall, segmentation wall; SW, spherule wall. Bars in panels A, B, D, and F represent 1.5 μm, 60 μm, 0.5 μm, and 5 μm, respectively.
Results of immunoblot-urease activity assays correlate with observations of immunolocalization of urease protein.
Immunoblot analyses of total protein preparations obtained from parasitic cell homogenates, SOW, and culture filtrates were conducted using urease-specific antibody (Fig. 3A). The estimated size of the denatured urease protein in SDS-PAGE gels is 101 kDa (25). Aliquots of equal amounts of total protein isolated from parasitic cell homogenates were first separated by electrophoresis and then immunoblotted. A single, faint 101-kDa band was revealed in the 24-h spherule initial preparation. Single bands of the same size but of much higher intensity were detected in preparations obtained from segmented spherules (72-h culture) and endosporulating spherules (132-h culture). The total protein fraction of SOW isolated from pre-endosporulating spherules (72 h) revealed no urease in the immunoblot, while the SOW derived from endosporulating cultures showed a distinct 101-kDa band. The total protein fractions of concentrated culture filtrates obtained from 24-h, 72-h and 132-h parasite-phase cultures had no detectable urease protein in the respective immunoblots.
FIG. 3.

(A) Antiurease immunoblot and (B) corresponding assay of urease activity of samples obtained from parasitic cell homogenates, isolated SOW fraction, and culture (Cult.) filtrates. The isolates were derived from first-generation cultures in near-synchronous isotropic growth phase (24 h), segmentation phase (72 h), and endosporulation phase (132 h). Std., standard.
Total equilibrated protein aliquots isolated from cell homogenates, SOW fractions, and culture filtrates examined (Fig. 3A) were also tested by the urease assay for the presence of active enzyme. The data obtained from these assays correlated well with results of the immunoblot analyses (Fig. 3B). A comparison of units of urease activity in each of the preparations revealed that the highest amounts of active enzyme were present in homogenates of the 72-h and 132-h spherules (Fig. 3B). Relatively low amounts of urease activity were detected in homogenates of the spherule initials (24-h parasite-phase cultures) and the SOW fraction isolated from 132-h cultures of endosporulating spherules. No urease activity was detected in the SOW fraction isolated from pre-endosporulating (72-h) cultures, and only minor activity was observed in the concentrated filtrate obtained from the 132-h culture. The other culture filtrates tested were devoid of urease activity.
Transformation and generation of the URE-deficient mutant (Δure) and URE-reconstituted (revertant) strain (UreR).
The plasmid construct pΔure (not shown) which was used for transformation of C. posadasii was designed to integrate into chromosomal DNA by a single crossover event (13) that resulted in disruption of the URE gene. This latter process is characterized by generation of two mutated copies of the target gene separated by the HPH gene cassette (Fig. 4A). Transformation of approximately 107 germ tube-derived protoplasts with 3 μg of SplI-linearized plasmid DNA yielded 30 to 50 hygromycin-resistant colonies. To obtain homokaryons, individual colonies were serial transferred to fresh GYE plates containing 75 μg/ml of hygromycin. Previous C. posadasii gene disruption experiments have shown that at least three such transfers of putative transformants, each followed by 10 days of incubation, are necessary to obtain homokaryotic mutants (19, 32). One of the candidate transformants, referred to as the Δure strain, was selected for further analysis by Southern hybridization. The Δure transformant was used as the host for generation of a revertant strain. A transformation construct, pUREr (not shown), was designed to integrate into chromosomal DNA of the Δure strain by a double crossover event (12), resulting in replacement of the entire disruption locus of the host strain with the wild-type URE gene. The 5.6-kb transformation construct contained an introduced AflII restriction site within intron 4 of the URE gene downstream from its start codon (Fig. 4B). Putative transformants were initially screened by growth on Bacto Urea agar containing phenol red as a pH indicator. Transformation of 5 × 106 protoplasts with 3 μg of the 5.6-kb plasmid DNA yielded 15 colonies which generated a color change (yellow to red) after 4 days of incubation at 30°C on the agar plates. DNA was separately isolated from 4 of the 15 putative transformants and used for PCR amplification of the 1.9-kb URE gene fragment by use of primer pair a and b (Fig. 4B; Table 1). Template genomic DNA from the parental strain was subjected to the same PCR amplification. Each 1.9-kb amplicon derived from the putative transformants and parent strain was separately digested with AflII, and the products were subjected by agarose gel electrophoresis. The AflII-digested amplicons of the four transformants selected from the GYE-phenol red plates each revealed 1.1-kb and 0.8-kb bands in the ethidium bromide-stained agarose gels, as predicted (Fig. 4B). The 1.9-kb amplicon derived from the parental strain was not digested by AflII. One transformant, referred to as the UREr (revertant) strain, was selected from this PCR-restriction digest screening for further evaluation by Southern hybridization.
FIG. 4.

(A) Urease gene (URE) structure and integrated disruptant (Δure). Disruption of the URE gene resulted from the chromosomal integration of pΔure by a single crossover event. The 1.6-kb internal fragment of URE was cloned into the pAN7.1 plasmid (construct not shown). Homologous recombination between the plasmid construct and chromosomal DNA generated two incomplete copies of the URE gene, separated by the linearized pAN7.1 sequence. Primers used to screen by PCR for the wild-type gene or disruptant are a and b or c and d, respectively. Primers used for PCR amplification of probes (Pb1 and Pb2) employed in Southern hybridization are labeled e and f and labeled g and h, respectively. (B) Structure of disrupted urease gene (Δure) which was replaced by a double crossover event with the modified wild-type gene (UREr) that contained an engineered AflII restriction site in an intron. Primers a and b were used for PCR amplification of the reconstituted URE gene fragment (1.9 kb) that contained the AflII restriction site. (C) Restriction map of the 7.0-kb chromosomal fragment containing the wild-type URE gene. (D and E) Hypothetical restriction maps of chromosomal fragments containing the urease disruption construct (Δure) and reconstituted urease gene (UREr), respectively.
A restriction map was constructed for a 7.0-kb nucleotide sequence identified in the C. posadasii genome database (Fig. 4C), which included the 5.6-kb genomic clone of the URE gene deposited in GenBank (accession no. U81509.3). Restriction maps were also constructed for the hypothetical structures of the chromosomal fragments which contained the integrated pΔure disruption plasmid (Fig. 4D) and the pUREr plasmid used to generate the revertant strain (Fig. 4E). On the basis of these maps, two Southern hybridization probes (Pb1 and Pb2) were generated by PCR using the primer pairs listed in Table 1. Comparison of results of hybridization of Pb1 with SpeI- and XbaI-restricted genomic DNA and ClaI- and SacII-restricted genomic DNA from the parental versus the Δure strain (Fig. 5A) provided evidence for single-site integration of the pΔure construct into the host chromosomal DNA. As indicated, the Pb1 probe failed to hybridize with DNA from the parental strain but did hybridize with bands of the predicted size in the case of the Δure strain. The Pb2 probe hybridized with SpeI- and StuI-restricted genomic DNA, SpeI- and SnaBI-restricted genomic DNA, SpeI- and NheI-restricted genomic DNA, and BstEII- and XhoI-restricted genomic DNA fragments from the parental and putative transformant (Fig. 5B). The sizes of the hybridization fragments of genomic DNA from the parental and mutant strains matched the sizes predicted using their respective restriction maps (Fig. 4C and D). These data provide evidence that homologous recombination occurred at a single chromosomal locus and argue that the Δure strain is homokaryotic. The Pb2 hybridization probe shown in Fig. 4E was predicted to identify a 1.5-kb SpeI- and AflII-restricted genomic DNA fragment of the revertant strain, which is absent from the restricted DNA of both the parental and Δure strains. The results of Southern hybridization in Fig. 5C provide evidence that homologous recombination occurred between the pUREr transformation plasmid and host chromosomal DNA by a double crossover event at a single locus.
FIG. 5.

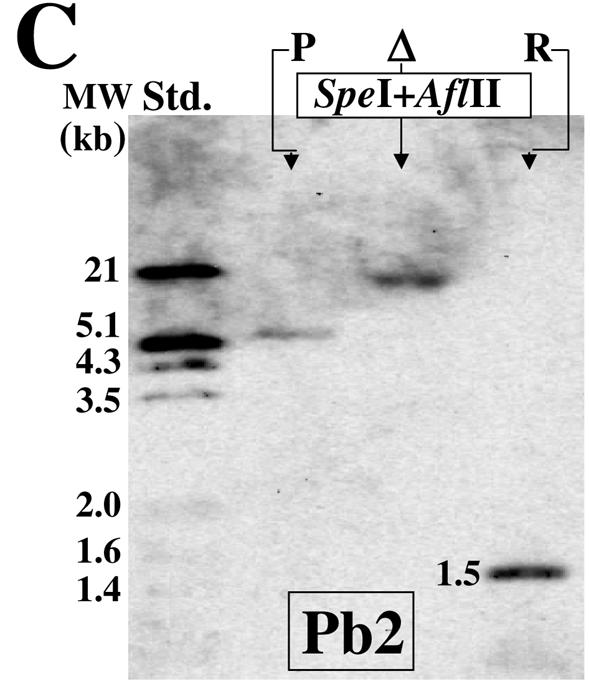
Results of Southern hybridization of restricted genomic DNA derived from the parental, Δure mutant (Δ), and revertant (R) strains with the Pb1 or Pb2 probe. The sizes of the restricted fragments correlated with the predicted sizes (see Fig. 4C to E). Std., standard.
Phenotypic comparison of the parental, mutant, and revertant strains.
The saprobic phase of the three strains grown in GYE liquid medium revealed identical growth curves (not shown). Comparison of parasitic cell development in defined glucose-salts medium demonstrated that the three strains each gave rise to endosporulating spherules at 5 to 6 days after the cultures were inoculated with equal numbers of arthroconidia. The percentages of parasitic cells which had ruptured and released their endospores in cultures of the parental, Δure mutant, and revertant strains after 6 days of incubation as revealed by microscopic examination were 89.2% ± 4.8%, 87.3% ± 2.9%, and 83.5% ± 6.3%, respectively. No significant difference in growth and development results between the three strains was observed.
Equal aliquots of total protein obtained from mycelial homogenates of 4-day-old cultures of the three strains were equilibrated and separated by SDS-PAGE (8% gel) in preparation for immunoblot analysis using antiserum raised against the recombinant urease (Fig. 6A and B). The results showed that the parental and revertant strains each contained a prominent 101-kDa band, which correlates with the molecular size of the purified urease protein (25). The immunoblot of the Δure homogenate, on the other hand, showed no seroreactive protein band. The same equilibrated total protein samples were used to conduct separate urease activity assays. The reaction mixtures (50 μl total volume), which consisted of 50 mM HEPES buffer (pH 7.75), 50 mM urea, and 0.5 mM EDTA plus an equal amount of total protein from each homogenate (10 μg), were incubated at 37°C. Urease activity was measured by monitoring the rate of NH4+/NH3 release per minute in the presence of the urea substrate over a 20-min incubation period (data not shown). The parental and revertant strains revealed comparable levels of urease activity, while the Δure mutant strain showed no detectable ammonia released into the reaction mixture. Control assays were conducted with boiled homogenates (100°C, 10 min) as well as with reaction mixtures without the urea substrate. In both cases, the control reactions showed no ammonia released during the period of incubation.
FIG. 6.

(A) SDS-PAGE separation of equal amounts of total protein obtained from mycelial homogenates (4-day cultures) of the parental (P), Δure mutant (Δ), and revertant (R) strains. (B) Immunoblot of protein separation shown in panel A produced using antiurease antibody. Note the absence of the 101-kDa band in the preparation of the mutant strain (Δ). (C) Comparative analysis of amounts of NH4+/NH3 released into the culture medium during incubation of each strain in the presence or absence of exogenous urea. The concentrations of extracellular ammonia were determined during 7 days of incubation of the parental (▾), Δure mutant (•), and revertant (○) strains in supplemented RPMI medium plus 10 mM exogenous urea. The changes in concentration are plotted as first-order linear regression lines. (Inset) The change in concentration of ammonia in the absence of exogenous urea is plotted point to point. Std., standard.
The amount of ammonium and ammonia released into the culture medium during growth of the parasitic phase of each strain was also measured over a 7-day period of incubation. Equal numbers of arthroconidia (106 cells) from agar plates of the three strains were used to separately inoculate RPMI 1640 medium with or without the addition of 10 mM urea (Fig. 6C). The parental and revertant strains showed comparable amounts of ammonia released into the culture filtrate when incubated in the presence of exogenous urea, achieving NH4+/NH3 concentrations of approximately 9.5 to 10 mM. In contrast, when the same strains were incubated in RPMI medium without exogenous urea, the NH4+/NH3 concentrations peaked at about 1.0 to 1.1 mM (Fig. 6C, inset). Ammonia levels detected in the culture filtrate of the Δure mutant in the absence of exogenous urea showed a sharp increase over the first 2 days of incubation but then began to decrease. With the addition of 10 mM urea to the culture medium, levels of ammonia detected in the culture filtrate of the mutant strain revealed only a slight increase over the 7-day period of incubation. Aliquots of the culture medium supplemented with 10 mM urea were also sampled daily during incubation of the parental, revertant, and mutant strain to test for consumption of urea as described above in the Materials and Methods section. The urea concentration in the filtrate obtained from cultures of the parental and revertant strains showed a near-linear decrease from 10 mM to 4.2 mM over the 7-day period of incubation. The amount of urea consumed correlated approximately with the amount of NH4+/NH3 released into the medium supporting growth of the two strains. The culture filtrate of the mutant strain, on the other hand, showed a slight increase in urea content from 10 mM to 12 mM between days 5 and 7 of incubation.
Comparison of the virulence of the parental, mutant, and revertant strains.
Equal numbers of arthroconidia (50 viable cells) isolated from the parental, mutant (Δure), and revertant (UreR) strains were used to separately inoculate three groups of BALB/c mice by the intranasal route (Fig. 7A). Mice challenged with arthroconidia derived from the parental and revertant strains showed comparable high rates of mortality. All the animals had died by 22 days postchallenge. On the other hand, animals infected with the mutant strain showed a slower rate of mortality over the first 3 weeks after intranasal inoculation, and approximately 55% of the mice survived beyond 50 days postchallenge. The difference between the survival plots for mice infected with the mutant versus the parental or revertant strain was highly significant (P = 0.001).
FIG. 7.

(A) Survival plots of BALB/c mice challenged intranasally with equal numbers of arthroconidia isolated from the parental, Δure mutant, or revertant strain. (B and C) pH of infected host tissue (abscesses) at 2 weeks after intraperitoneal challenge with 103 arthroconidia (B) or after intranasal challenge with 50 arthroconidia (C) of either the parental or Δure mutant strain. Mesen., mesentery; Spl. spleen. (C) Urea concentration in extracts of frozen sections of lung tissue obtained from normal, noninfected BALB/c mice (Cont.), mice infected with the parental strain (P), or mice infected with the mutant strain (Δure) of C. posadasii and sacrificed at 12 days postchallenge. Equal numbers of sections (10 per abscess) were collected, pooled, and extracted with the same total volume (450 μl) of distilled water.
The CFU in homogenates of the lungs and spleen of BALB/c mice infected intranasally with the parental or mutant strain of C. posadasii were determined at 12, 25, and 50 days postchallenge. Comparable numbers of CFU were detected after 12 days in the lungs of mice infected with the parental strain (median of 6.41 [log10]; range of 5.85 to 6.87) or mutant (5.83; 5.70 to 5.94). On the other hand, the spleens of mice challenged with the parental strain were infected (2.85; 2.10 to 3.55), but no CFU were detected in spleens of mice challenged with the Δure strain. The lungs of mice challenged with the mutant strain showed a significant reduction in CFU at 25 days postchallenge (2.78; 2.70 to 3.78) compared to the results seen with mice infected with the parental strain at 12 days postchallenge (P = 0.002). The spleen was still uninfected. None of the mice challenged with the parental strain survived to 25 days. At 50 days postchallenge, no CFU were detected on culture plates inoculated with undiluted homogenates of the lungs or spleen of mice which survived challenge with the Δure mutant strain.
In situ determinations of pH at infection sites.
BALB/c mice challenged via the intraperitoneal route with 103 arthroconidia of either the parental or mutant strain, and sacrificed 2 weeks later, responded by production of visible abscesses in their lungs and spleen, on the surface of the diaphragm, and in association with the mesenteric tissue. Immediately after the mice were sacrificed and internal organs were exposed, pH measurements of abscesses were performed in situ by penetration of the host tissue surface layer with a needle electrode. The results in Fig. 7B show significantly lower pH values for abscesses produced in response to the Δure mutant strain compared to the host response to the parental strain (P < 0.02). BALB/c mice were also inoculated via the intranasal route with 50 arthroconidia of either the parental or mutant strain and sacrificed at 2 weeks postchallenge, and their lungs were examined as described above. The average pH of lung abscesses in mice infected with the parental strain was approximately 7.7. In contrast, the average pH of lung abscesses in mice infected intranasally with the Δure strain was approximately 7.2 (Fig. 7C). This difference in pH of lung abscesses in the intranasally challenged mice is also statistically significant (P = 0.001).
Detection of high concentrations of urea at sites of infection with the parental strain.
On the basis of the substantial increase in the in vitro production of ammonia by the parental strain in the presence of 10 mM urea, we speculated that if exogenous urea is present at sites of lung infection it may also influence the levels of NH4+/NH3 production and the pH of the microenvironment. Figure 7D shows the results of a comparison of the amounts of urea detected in aqueous extracts of frozen sections of normal, noninfected murine lung tissue versus sectioned abscesses from the lungs of mice at 14 days after intranasal challenge with either the parental or Δure mutant strain of C. posadasii. The approximate fourfold increase in urea concentration at sites of infection with the parental strain may at least partly account for the elevated pH values recorded for lungs of mice infected with this strain (Fig. 7B and C). On the other hand, a significantly lower concentration of urea was detected at sites of infection with the mutant strain compared to the parental strain results (P = 0.002).
Comparative histopathology.
Two groups of BALB/c mice were challenged via the intranasal route with equal numbers of arthroconidia produced by either the parental or Δure mutant strain. Mice infected with the parental strain at 12 days postchallenge showed multiple abscesses in their lungs. Histological sections of these abscesses suggested that an intense inflammatory response had occurred without evidence of granuloma differentiation. Large numbers of spherules in pre- and post-endosporulation stages of development were visible, and apparent host tissue damage had occurred at sites of infection (Fig. 8A). Inflammatory cells (see “Inf. cells” in Fig. 8A) appeared to have responded in a chemotactic manner to the released contents of ruptured spherules. Host phagocytes were frequently visible within the lumen of the parasitic cells (arrow at right in Fig. 8A). Sections of murine lung tissue infected with the mutant strain after 12 days showed comparable high numbers of the pathogen but also an indication of a more organized host inflammatory response at sites of lung infection than observed in lung tissue infected with the parental strain (not shown). At 20 days postchallenge, mice infected with the parental strain were moribund or dead, while the surviving animals which had been challenged with the Δure mutant showed visible signs of recovery (activity, weight gain, smooth rather than ruffled fur) and histological evidence of early differentiation of granulomas which had largely contained the pathogen (arrows in Fig. 8B). At 25 days after challenge, mice infected with the mutant strain showed development of well-differentiated granulomas (Fig. 8C). Spherules contained within the granuloma were either destroyed or surrounded by host inflammatory cells, and few endospores were visible. Results of these studies correlate with our observations of a marked reduction in CFU in the lungs of BALB/c mice at 25 days after intranasal challenge with the Δure mutant strain.
FIG. 8.

(A) Histopathology of lung tissue from BALB/c mouse infected intranasally with the parental strain of C. posadasii at 12 days postchallenge. Note the high concentration of host inflammatory cells (Inf. cells), some of which surrounded and entered a ruptured spherule (arrow at right). (B) Sectioned lobe of lung tissue from a BALB/c mouse infected intranasally with the Δure mutant strain of C. posadasii and sacrificed at 20 days postchallenge. Note early development of granulomas (arrows). (C) Sectioned granuloma in lung of mouse infected with the Δure mutant strain and sacrificed 25 days postchallenge. Partially decomposed spherules are surrounded by inflammatory cells. The bar in panel A represents 40 μm, and the bars in panels B and C represent 2 mm.
DISCUSSION
In vitro growth of the saprobic phase of Coccidioides spp. for 8 days at 30°C in liquid medium containing only glucose and yeast extract resulted in a final extracellular NH4+/NH3 concentration of 7.5 mM and an increase in pH from 6.7 to 7.8 (25). Growth of the parasitic phase in defined glucose-salts medium has also been reported to result in release of ammonia from the fungal cells, with a concomitant increase in the extracellular pH (8). Urease activity is a major source of ammonia and an important virulence determinant for microbial pathogens (6). However, little is known about the function of fungal ureases compared to the wealth of information on their homologs in plants and bacteria. The most extensively studied urease is that of jack bean (Canavalia ensiformis); it was the first enzyme to be crystallized and is the first example of a nickel [Ni (II)] metalloproteinase (2). The native plant urease is a hexamer composed of identical polypeptide subunits, each with an estimated molecular size of 91 kDa. The molecular size of the monomer of the C. posadasii urease, whose amino acid sequence is 76% homologous to that of the jack bean urease, has been estimated to be 101 kDa (25). The approximate size of the native fungal enzyme is 450 kDa, and it is apparently composed of four subunits. The C. posadasii urease demonstrates biochemical similarities to other fungal ureases, including a pH optimum of 8.0 and thermostability up to 60 to 65°C (39).
Although urease is a cytosolic enzyme that lacks a signal peptide, evidence derived from studies of both plant and bacterial ureases suggests that the active enzyme may also be cell surface associated (14, 24). In the case of H. pylori, the extracellular urease has been suggested to be essential for the bacteria to resist exposure to acid (22). Host exposure to bacterial urease has been reported to elicit an inflammatory response (36), which may lead to tissue damage and exacerbation of the microbial infection when this response is persistent and intense (26). Recombinant urease of Coccidioides has been shown to be highly immunogenic in BALB/c mice (23). Both the recombinant fungal protein and a mammalian plasmid vector containing the URE gene have been used to vaccinate BALB/c mice and demonstrated significant protective efficacy against coccidioidal infection. These latter results further argue that the fungal urease is presented to the host during the course of a natural infection (23).
The mechanism by which urease becomes associated with the surface of H. pylori involves initial release of the enzyme from cells that undergo autolysis, followed by adsorption of the enzymatically active protein to intact, viable bacteria (15). A comparable sequence of events is suggested to occur during the parasitic cycle of C. posadasii. The urease protein has been immunolocalized to the spherule cytoplasm and vesicles and the large central vacuole. Active urease escapes from spherules when the parasitic cells rupture and release their contents. The enzyme subsequently associates both with the surface of intact endospores and the membranous, SOW fraction which is produced in abundance during in vitro growth of the parasitic phase (17, 19). Positive correlation of data obtained from immunoblot analyses and enzymatic assays of parasitic cell homogenates suggested that most of the active urease is present in the spherule cytosol during the pre-endosporulation (72 h) and endospore differentiation (132 h) stages of development. However, a significant amount of active enzyme was also detected in association with the cell-free, SOW fraction. Preliminary studies have demonstrated that the urease located by immunofluorescence at the surface of endospores is also enzymatically active (not shown).
The results of these initial investigations suggested that urease activity of C. posadasii in vivo may contribute to the generation of an alkaline microenvironment at the surface of the fungal pathogen as well as stimulate host inflammatory response to the extracellular protein. Together, these conditions could lead to host tissue damage and exacerbation of the severity of coccidioidal infections. On the basis of this hypothesis, we designed a strategy to disrupt the URE gene. Targeted disruption of Coccidioides genes by homologous recombination (13) is now well established in our laboratory (19, 32). Application of the protoplast method to transform C. posadasii (32) has proved to be superior to alternative approaches, such as electroporation, biolistic transformation (38), and Agrobacterium-facilitated DNA transfer (1). Results of PCR analysis and Southern hybridization have demonstrated that chromosomal integration of the gene disruption construct occurred at a single site by homologous recombination. In order to satisfy “Koch's postulates,” we have successfully developed a gene reconstitution strategy for Coccidioides spp. (19). We have shown that this process of gene-targeted replacement also occurred by single-site integration of the wild-type gene into chromosomal DNA. We have confirmed that the normal function of the targeted URE gene was restored and that the parental and revertant strains have identical phenotypes.
Acetohydroxamic acid is a reversible urease inhibitor and was previously shown to completely block in vitro activity of the purified Coccidioides enzyme when added to mycelial homogenates at a concentration of 10 mM (25). The addition of acetohydroxamic acid at the same concentration to live mycelial cultures coincubated for 12 h resulted in a dramatic reduction in urease activity compared to control culture results, but NH4+/NH3 continued to be released into the growth medium (25). Metabolic sources of intracellular ammonia synthesis other than urease activity have been previously proposed (6) but have not yet been explored for Coccidioides spp. Nevertheless, disruption of the URE gene of C. posadasii resulted in a significant difference between the amounts of extracellular ammonia detected in cultures of the parental and revertant strains versus the Δure mutant strain cultures. This difference was most clearly demonstrated upon addition of 10 mM urea to the culture medium. The pH change over the 7-day period of incubation of the parental strain in RPMI medium plus exogenous urea was not dramatic (6.6 to 7.2), presumably because of the buffering capacity of the medium. Generation of an alkaline, extracellular microenvironment by bacteria that release ammonia has been suggested to occur by diffusion rather than active transport of NH4+/NH3 (3). This would require that the intracellular concentration of ammonia exceeds that of extracellular ammonia. Although we have not measured intracellular NH4+/NH3 concentrations of the parasitic cells of C. posadasii, indirect evidence suggests that they are high. We have shown that the concentration of urea in the culture medium which supported growth of the parental strain decreased (10 mM to 4.2 mM) over 7 days of incubation, suggesting that the parasitic cells are capable of uptake and consumption of the exogenous substrate. Intracellular urea most likely undergoes hydrolysis immediately after it enters the parasitic cells of C. posadasii because of the toxicity of this substrate. Assuming that the intracellular pH of spherules and endospores is controlled, it is likely that high ammonium-ammonia concentrations resulting from urea uptake and urease activity generate a diffusion gradient for release of NH4+ and NH3 from the cells. However, it is also possible that more than one NH4+/NH3 transport system exists in C. posadasii, as has been proposed for certain ammonia-releasing bacteria (3).
Loss of urease activity from the Δure strain of C. posadasii resulted in a marked reduction in the pathogenicity of the organism in the lungs of BALB/c mice. Approximately 55% of the animals survived an arthroconidial challenge which is typically lethal to this strain of mice, and all the survivors cleared the infection between 25 and 50 days postchallenge. The significance of this partial loss of virulence is reinforced by the fact that BALB/c mice represent a host which is exceptionally susceptible to disseminated coccidioidomycosis following intranasal challenge with arthroconidia (21). Inoculation of BALB/c mice with the same number of spores of the parental or revertant strain resulted in 100% mortality after 22 days. We used both intraperitoneally and intranasally infected mice to generate coccidioidal tissue abscesses which were large enough to allow measurement of the internal pH. We recorded a significantly higher pH at sites of infection with the parental strain (7.4 to 7.7) than with the urease knockout strain (6.8 to 7.1). We also showed that abscesses from BALB/c mice resulting from infection with the parental strain contained higher concentrations of urea (approximately 630 μM) than abscesses produced in response to infection with the mutant strain. Our estimation of the concentration of urea detected in sections of lung tissue obtained from normal, noninfected mice was 160 μM.
We have observed that murine macrophages respond in vitro to infection with the wild-type strain of C. posadasii by an approximately threefold increase in the level of expression of the host arginase I gene compared to that in noninfected macrophages (unpublished data). A sharp rise in arginase I production in vivo during Leishmania infection is indicative of a Th2 pathway of immune response (20), which in both leishmaniasis and coccidioidomycosis is an indicator of a poor disease outcome (10). Arginase I competes with inducible nitric oxide synthase in macrophages for the common substrate, l-arginine, which results in reduction of the basal level of nitric oxide and increased production of ornithine and urea (20). Expression of the arginase gene of C. posadasii (28) during in vitro growth of the parasitic phase in the presence of exogenous urea is constitutive (unpublished data). On this basis, we suggest that the high amounts of urea detected at sites of coccidioidal infection are host derived. The availability of exogenous urea would provide substrate for both extracellular and intracellular urease activity, result in elevated production and accumulation of NH4+/NH3 at infection sites, and cause a pH increase that could be damaging to host tissue (31). Results of histopathological studies of lung tissue infected with the parental strain suggested that extensive tissue damage occurs during the course of disease; this damage may be mediated both by the pathogen and the host (7). Persistently high levels of proinflammatory cytokine production (e.g., interleukin-6) during coccidioidal lung infection may result in additional host tissue damage due to localized and intense inflammatory cell response (10). On the other hand, BALB/c mice infected with the urease knockout strain showed a robust, protective response to intranasal challenge characterized by early formation of granulomas and clearance of the pathogen from animals which survived to 50 days postchallenge. In summary, these data suggest that elevated levels of intracellular and extracellular urease activity occur at sites of coccidioidal infection with virulent strains of C. posadasii and result in localized high concentrations of NH3/NH4+, together with persistent and intense inflammatory response to the pathogen. These combined features of coccidioidomycosis, which are attributed at least in part to urease production, contribute significantly to the pathogenesis of Coccidioides spp.
Acknowledgments
Support for this study was provided by Public Health Service grants AI19149 and AI37232 from the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
We are grateful to Kalpathi Seshan for his assistance in conducting the ultrastructural studies.
Editor: T. R. Kozel
REFERENCES
- 1.Abuodeh, R. O., M. J. Orbach, M. A. Mandel, A. Das, and J. N. Galgiani. 2000. Genetic transformation of Coccidioides immitis facilitated by Agrobacterium tumefaciens. J. Infect. Dis. 181:2106-2110. [DOI] [PubMed] [Google Scholar]
- 2.Blakeley, R. L., and B. Zerner. 1984. Jack bean urease: the first nickel enzyme. J. Mol. Catal. 23:263-292. [Google Scholar]
- 3.Brewin, B., P. Woodley, and M. Drummond. 1999. The basis of ammonium release in nifL mutants of Azotobacter vinelandii. J. Bacteriol. 181:7356-7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bump, W. A. 1925. Observations on growth of Coccidioides immitis. J. Infect. Dis. 36:561-565. [Google Scholar]
- 5.Burall, L. S., J. M. Harro, X. Li, C. V. Lockatell, S. D. Himpsl, J. R. Hebel, D. E. Johnson, and H. L. T. Mobley. 2004. Proteus mirabilis genes that contribute to pathogenesis of urinary tract infection: identification of 25 signature-tagged mutants attenuated at least 100-fold. Infect. Immun. 72:2922-2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bury-Mone, S., J.-M. Thiberge, M. Contreras, A. Maitournam, A. Labigne, and H. D. Reuse. 2004. Responsiveness to acidity via metal ion regulators mediates virulence in the gastric pathogen Helicobacter pylori. Mol. Microbiol. 53:623-638. [DOI] [PubMed] [Google Scholar]
- 7.Casadevall, A., and L.-A. Pirofski. 2003. The damage-response framework of microbial pathogenesis. Nat. Rev. Microbiol. 1:17-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cole, G. T. 1997. Ammonia production by Coccidioides immitis and its possible significance to the host-fungus interplay, p. 247-263. In H. vanden Bossche, D. A. Stevens, and F. C. Odds (ed.), Host-fungus interplay. Plenum Press, New York, N.Y.
- 9.Cole, G. T., D. Kruse, K. R. Seshan, S. Pan, S. J. Szaniszlo, J. Richardson, and B. Bian. 1993. Factors regulating morphogenesis in Coccidioides immitis, p. 191-212. In H. Van den Bossche, F. C. Odds, and D. Kerridge (ed.), Dimorphic fungi in biology and medicine. Plenum Press, New York, N.Y.
- 10.Cole, G. T., J.-M. Xue, C. N. Okeke, E. J. Tarcha, V. Basrur, R. A. Schaller, R. A. Herr, J.-J. Yu, and C.-Y. Hung. 2004. A vaccine against coccidioidomycosis is justified and attainable. Med. Mycol. 42:189-216. [DOI] [PubMed] [Google Scholar]
- 11.Cox, G. M., J. Mukherjee, G. T. Cole, A. Casadevall, and J. R. Perfect. 2000. Urease as a virulence factor in experimental cryptococcosis. Infect. Immun. 68:443-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delgado, N., J. Xue, J.-J. Yu, C.-Y. Hung, and G. T. Cole. 2003. A recombinant β-1,3-glucanosyltransferase homolog of Coccidioides posadasii protects mice against coccidioidomycosis. Infect. Immun. 71:3010-3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.d'Enfert, C., G. Weidner, P. C. Mol, and A. A. Brakhage. 1999. Transformation systems of Aspergillus fumigatus. New tools to investigate fungal virulence. Contrib. Microbiol. 2:149-166. [DOI] [PubMed] [Google Scholar]
- 14.Dunn, B. E., and M. G. Grutter. 2001. Helicobacter pylori springs another surprise. Nat. Struct. Biol. 8:480-482. [DOI] [PubMed] [Google Scholar]
- 15.Dunn, B. E., and S. H. Phadnis. 1998. Structure, function and localization of Helicobacter pylori urease. Yale J. Biol. Med. 71:63-73. [PMC free article] [PubMed] [Google Scholar]
- 16.Fisher, M. C., G. L. Koenig, T. J. White, and J. W. Taylor. 2002. Molecular and phenotypic description of Coccidioides posadasii sp. nov., previously recognized as the non-California population of Coccidioides immitis. Mycologia 94:73-84. [PubMed] [Google Scholar]
- 17.Hung, C. Y., N. M. Ampel, L. Christian, K. R. Seshan, and G. T. Cole. 2000. A major cell surface antigen of Coccidioides immitis which elicits both humoral and cellular immune responses. Infect. Immun. 68:584-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hung, C.-Y., J.-J. Yu, P. F. Lehmann, and G. T. Cole. 2001. Cloning and expression of the gene which encodes a tube precipitin antigen and wall-associated β-glucosidase of Coccidioides immitis. Infect. Immun. 69:2211-2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hung, C.-Y., J.-J. Yu, K. R. Seshan, U. Reichard, and G. T. Cole. 2002. A parasitic phase-specific adhesin of Coccidioides immitis contributes to the virulence of this respiratory fungal pathogen. Infect. Immun. 70:3343-3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iniesta, V., L. C. Gomez-Nieto, I. Molano, A. Mohedano, J. Carcelen, C. Miron, C. Alonso, and I. Corraliza. 2002. Arginase I induction in macrophages, triggered by Th2-type cytokines, supports the growth of intracellular Leishmania parasites. Parasite Immunol. 24:113-118. [DOI] [PubMed] [Google Scholar]
- 21.Kirkland, T. N., and J. Fierer. 1983. Inbred mouse strains differ in resistance to lethal Coccidioides immitis infection. Infect. Immun. 40:912-916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krishnamurthy, P., M. Parlow, J. B. Zitzer, N. B. Vakil, H. L. Mobley, M. Levy, S. H. Phadnis, and B. E. Dunn. 1998. Helicobacter pylori containing only cytoplasmic urease is susceptible to acid. Infect. Immun. 66:5060-5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li, K., J.-J. Yu, C.-Y. Hung, P. F. Lehmann, and G. T. Cole. 2001. Recombinant urease and urease DNA of Coccidioides immitis elicit an immunoprotective response against coccidioidomycosis in mice. Infect. Immun. 69:2878-2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Millanes, A. M., B. Fontaniella, M. L. Garcia, M. T. Solas, C. Vicente, and M. E. Legaz. 2004. Cytochemical location of urease in the cell wall of two different lichen phycobionts. Tissue Cell 36:373-377. [DOI] [PubMed] [Google Scholar]
- 25.Mirbod, F., and R. A. Schaller. 2002. Purification and characterization of urease isolated from the pathogenic fungus Coccidioides immitis. Med. Mycol. 40:35-44. [DOI] [PubMed] [Google Scholar]
- 26.Mobley, H. L., M. D. Island, and R. P. Hausinger. 1995. Molecular biology of microbial ureases. Microbiol. Rev. 59:451-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olszewski, M. A., M. C. Noverr, G.-H. Chen, G. B. Toews, G. M. Cox, J. R. Perfect, and G. B. Huffnagle. 2004. Urease expression by Cryptococcus neoformans promotes microvascular sequestration, thereby enhancing central nervous system invasion. Am. J. Pathol. 164:1761-1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pan, S., M. Zhang, and G. T. Cole. 1995. Isolation and characterization of the arginase-encoding gene (arg) from Coccidioides immitis. Gene 154:115-118. [DOI] [PubMed] [Google Scholar]
- 29.Pappagianis, D. 1980. Epidemiology of coccidioidomycosis, p. 68-85. In D. A. Stevens (ed.), Coccidioidomycosis: a text. Plenum Press, New York, N.Y.
- 30.Petkus, A. F., L. L. Baum, R. B. Ellis, M. Stern, and D. L. Danley. 1985. Pure spherules of Coccidioides immitis in continuous culture. J. Clin. Microbiol. 22:165-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prusky, D., J. L. McEvoy, B. Leverentz, and W. S. Conway. 2001. Local modulation of host pH by Colletotrichum species as a mechanism to increase virulence. Mol. Plant-Microbe Interact. 14:1105-1113. [DOI] [PubMed] [Google Scholar]
- 32.Reichard, U., C.-Y. Hung, P. W. Thomas, and G. T. Cole. 2000. Disruption of the gene which encodes a serodiagnostic antigen and chitinase of the human fungal pathogen Coccidioides immitis. Infect. Immun. 68:5830-5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saubolle, M. A. 1996. Life cycle and epidemiology of Coccidioides immitis, p. 1-8. In H. Einstein and C. Catanzaro (ed.), Coccidioidomycosis. National Foundation for Infectious Diseases, Washington, D.C.
- 34.Sendide, K., A.-E. Deghmane, J.-M. Reyrat, A. Talal, and Z. Hmama. 2004. Mycobacterium bovis BCG urease attenuates major histocompatibility complex class II trafficking to the macrophage cell surface. Infect. Immun. 72:4200-4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun, S. H., G. T. Cole, D. J. Drutz, and J. L. Harrison. 1986. Electron-microscopic observations of the Coccidioides immitis parasitic cycle in vivo. J. Med. Vet. Mycol. 24:183-192. [PubMed] [Google Scholar]
- 36.Tanahashi, T., M. Kita, T. Kodama, Y. Yamaoka, N. Sawai, T. Ohno, S. Mitsufuji, Y. P. Wei, K. Kashima, and J. Imanishi. 2000. Cytokine expression and production by purified Helicobacter pylori urease in human gastric epithelial cells. Infect. Immun. 68:664-671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xue, J., C.-Y. Hung, J.-J. Yu, and G. T. Cole. 2005. Immune response of vaccinated and non-vaccinated mice to Coccidioides posadasii infection. Vaccine 23:3535-3544. [DOI] [PubMed] [Google Scholar]
- 38.Yu, J.-J., and G. T. Cole. 1998. Biolistic transformation of the human pathogen Coccidioides immitis. Microbiol. Methods 33:129-141. [Google Scholar]
- 39.Yu, J.-J., S. L. Smithson, P. W. Thomas, T. N. Kirkland, and G. T. Cole. 1997. Isolation and characterization of the urease gene (URE) from the pathogenic fungus Coccidioides immitis. Gene 198:387-391. [DOI] [PubMed] [Google Scholar]