

Abstract
Trypanosoma cruzi infection causes cardiomyopathy and vasculopathy. Previous studies have demonstrated that infection of human umbilical vein endothelial and smooth muscle cells resulted in activation of extracellular signal-regulated kinase (ERK). In the present study, smooth muscle cells were infected with trypomastigotes, and immunoblot analysis revealed an increase in the expression of cyclin D1 and proliferating cell nuclear antigen (PCNA), important mediators of smooth muscle cell proliferation. Interestingly, after infection, the expression of caveolin-1 was reduced in both human umbilical vein endothelial cells and smooth muscle cells. Immunoblot and immunohistochemical analyses of lysates of carotid arteries obtained from infected mice revealed increased expression of PCNA, cyclin D1, its substrate, phospho-Rb (Ser780), and phospho-ERK1/2. The expression of the cyclin-dependent kinase inhibitor p21Cip1/Waf1, caveolin-1, and caveolin-3 was reduced in carotid arteries obtained from infected mice. There was an increase in the abundance of pre-pro-endothelin-1 mRNA in the carotid artery and aorta from infected mice. The ETA receptor was also elevated in infected arteries. ERK activates endothelin-1, which in turn exerts positive feedback activating ERK, and cyclin D1 is a downstream target of both endothelin-1 and ERK. There was significant incorporation of bromodeoxyuridine into smooth muscle cell DNA when treatment was with conditioned medium obtained from infected endothelial cells. Taken together, these data suggest that T. cruzi infection stimulates smooth muscle cell proliferation and is likely a result of the upregulation of the ERK-cyclin D1-endothelin-1 pathway.
Chagas' disease, caused by Trypanosoma cruzi, results in acute myocarditis and chronic cardiomyopathy (38, 39) accompanied by a vasculopathy (23, 30, 31, 34, 37, 43). The precise etiology of chagasic heart disease is multifactorial, involving parasite persistence (41), inflammation (13, 35), autoimmunity (20), and vascular dysfunction (37). The evidence for vascular dysfunction in Chagas' disease includes infection-associated microvascular spasm, reduction in blood flow, increased platelet aggregation in experimental infection, and increased myocardial inflammation and fibrosis (24, 34, 40).
T. cruzi infects many cell types that comprise the cardiovascular system, including cardiac myocytes, cardiac fibroblasts, endothelial cells, and vascular smooth muscle cells. The interaction of the parasite with endothelial cells is among the first encounters in the host-parasite relationship. Studies from our laboratory and others have demonstrated that infection of endothelial cells with T. cruzi results in the expression of proinflammatory cytokines (35), vascular adhesion molecules (13), and the vasoactive peptide endothelin-1 (ET-1) (27-29).
The cells of the cardiovascular system synthesize ET-1 and have endothelin receptors. ET-1 acts locally on cells such as cardiac myocytes, cardiac fibroblasts, endothelial cells, and smooth muscle cells via two types of receptors, ETA and ETB (6, 10, 17, 21). The primary effects of ET-1 on smooth muscle cells are mediated by the ETA receptor, including vasoconstriction and smooth muscle cell proliferation.
During acute murine T. cruzi infection, there were elevated plasma levels of ET-1, as well as increased expression of ET-1, in the vascular and endocardial endothelia (29). In addition, we demonstrated that infected mice treated with phosphoramidon, an inhibitor of endothelin converting enzyme, had reduced cardiac remodeling (16). There was also extracellular signal-regulated kinase 1/2 (ERK1/2) activation and cyclin D1 upregulation in the myocardium and in cultured endothelial and smooth muscle cells as a result of T. cruzi infection (15, 25). Therefore, we investigated the basis of the vasculopathy underlying T. cruzi infection and examined cell cycle-associated signaling pathways in the vasculature, including ET-1, using in vitro and in vivo approaches.
MATERIALS AND METHODS
Reagents.
Mouse monoclonal antibodies directed against caveolin-2 (clone 65) and caveolin-3 (clone 26) were gifts of Roberto Campos-Gonzalez (BD Pharmingen, Inc., San Diego, CA). Rabbit polyclonal antibodies (PAbs) directed against total ERK-1/2 and activated phospho-ERK-1/2 were obtained from Cell Signaling, Inc. (Beverly, MA). Rabbit polyclonal antibodies to caveolin-1 (N-20) and murine p21Cip1 were purchased from Santa Cruz Biotechnology, Inc., Santa Cruz, CA. Additional antibodies and their sources were as follows: anti-cyclin D1 rabbit PAb (NeoMarkers, Fremont, CA), anti-proliferating cell nuclear antigen (PCNA) mouse monoclonal antibody and anti-ETA rabbit PAb (BD Pharmingen, Inc., San Diego, CA), and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Research Diagnostics, Inc., Concord, MA). All other reagents were of the highest grade available.
Infections of cells and mice.
The Tulahuen strain of T. cruzi was maintained by syringe passage in A/J mice (Jackson Laboratories, Bar Harbor, ME), and the Brazil strain was maintained in C3H/HeJ mice (36). Trypomastigotes were maintained and harvested from L6E9 myoblasts as previously described (32). CD1 male mice (Jackson Laboratories) were infected intraperitoneally at 8 to 10 weeks of age with 5 × 104 trypomastigotes of the Brazil strain.
Trypomastigotes of both Brazil and Tulahuen strains were harvested from the supernatants of infected myoblasts, as previously described (32). When grown to near confluence, human umbilical vein endothelial cells or smooth muscle cells were infected with either strain at a multiplicity of infection of 1.5 to 2.0:1. After 48 h of exposure, the parasites were washed off. The approximate percent parasitism at 24, 48, and 72 h was 20, 50, and 80%, respectively, as determined by Giemsa staining. There was no difference in infectivity between the two strains of T. cruzi.
Isolation and cultivation of human cells.
Homogeneous explant smooth muscle cells were obtained from human corporal vascular smooth muscle cells as previously described (1, 2, 24, 46). Cellular homogeneity was verified by immunofluorescent staining using monoclonal antibodies to human smooth muscle myosin. Only passages 2 through 4 were used in all experiments. Human endothelial cells were obtained from umbilical cords as previously described (24, 34). Confluent monolayers were prepared in 100-mm-diameter tissue culture plates coated with 0.2% gelatin. Only passages 2 through 4 were used for this study.
Isolation of the carotid artery.
Carotid arteries were excised from mice that were divided into three groups: (i) uninfected control, (ii) infected mice 20 days postinfection (acute phase), and (iii) infected mice 90 days postinfection (chronic phase). There were eight mice in each group. Mice were anesthetized by intraperitoneal injection of ketamine HCl and xylazine (80 mg/kg and 5 mg/kg body weight, respectively). Both right and left common carotid arteries were dissected and ligated just proximal to their bifurcations with a 6-0 silk ligature, in the aim of identifying their location. The mice were then sacrificed by opening their thoracic cage, and both carotid arteries were collected from the ligation point. After washing with phosphate-buffered saline (PBS), arteries were placed in 10% formalin for morphological and immunohistochemical analysis or snap frozen in liquid nitrogen for immunoblot analysis or RNA detection using PCR. Aortas were prepared in a similar fashion.
Immunoblot analyses.
For cell lysates, smooth muscle cells and endothelial cells were plated at a density of ∼1 × 106 to 2 × 106 cells in complete medium and cultured for 18 to 24 h. Subconfluent cells were then collected in lysis buffer (10 mM Tris, pH 7.5, 150 mM NaCl, 1% Triton X-100, 60 mM octylglucoside) containing protease inhibitors (Roche Applied Science, Indianapolis, IN). For phospho-specific immunoblotting, cells were scraped into boiling lysis buffer to denature endogenous phosphatases. As described above, arteries were collected from infected and control mice and snap frozen in liquid nitrogen. Samples were then homogenized in 5 volumes of boiled lysis buffer (1% sodium dodecyl sulfate [SDS], 1 mM sodium-orthovanadate, 10 mM Tris, pH 7.4). Homogenates were further sonicated and then centrifuged at 16,000 × g at 4°C for 10 min to pellet insoluble material. Protein concentrations were estimated with the bicinchoninic acid protein assay (Bio-Rad). Equal amounts of protein for each sample were loaded and separated on SDS-polyacrylamide gel electrophoresis (10) gels. After transfer to nitrocellulose, the activation state and expression levels of PCNA, ERK1/2, pRb, p21Cip1/Waf1, caveolin-1, and caveolin-3 were examined by using specific antibodies. Antibodies directed against GAPDH were used as a control for equal loading.
Immunohistochemical techniques.
Paraffin (5-μm) sections of blood vessels were immunostained with antibodies directed against cyclin D1, phospho-Rb (pRb-Ser780), and p21Cip1/Wafl using the avidin-biotin peroxidase method. In brief, paraffin sections were dewaxed in xylene for 20 min, rehydrated in alcohol, and washed in PBS, and then all sections were incubated with Triton X-100 (0.2%) for 30 min. After three washes with PBS, the slides were incubated for 30 min with 5% H2O2 to block endogenous peroxidase activity and incubated with 10% normal goat serum for 30 min and then with the corresponding primary antibody for 18 h at 4°C. Next, sections were incubated with biotinylated immunoglobulin G (1:200) for 45 min and stained using the immunoperoxidase technique, according to the manufacturer's instructions (Vectastain ABC Elite kit; Vector Laboratories, Burlingame, CA). The sections were developed using diaminobenzidine hydrogen peroxide, counterstained with hematoxylin, dehydrated, and cleared. Finally, glass coverslips were placed on top of the sections. Negative controls performed to rule out nonspecific staining included secondary antibody alone and the use of nonimmune serum in place of the primary antibody.
Reverse transcription-PCR (RT-PCR) technique.
Total RNA was isolated from blood vessels with TRIzol reagent according to the protocol of the manufacturer (Gibco-BRL, Grand Island, NY). First-strand cDNA was prepared by incubation of 1 μg of total RNA with murine leukemia virus reverse transcriptase and oligo(dT)16 primer at 42°C for 15 min. Then, 2 μl of the reaction products was amplified by PCR with 2.5 U of Taq polymerase (Perkin-Elmer, Branchburg, NJ). PCR amplification consisted of 95°C for 30 s for denaturation, 60°C for 40 s for annealing, and 72°C for 2 min for extension, performed for 35 cycles. The primers used for the PCR and for GAPDH, used as controls, were previously published (29). Aliquots of 10 μl of the PCR products were electrophoresed in a 1.6% agarose gel containing ethidium bromide.
Bromodeoxyuridine incorporation in smooth muscle cells.
Confluent smooth muscle cells, cultured in a 100-mm tissue culture dish, were incubated with filtered medium obtained from 72-h-infected endothelial cells for 6 h, followed by addition of bromodeoxyuridine (Sigma-Aldrich, Germany) at a final concentration of 1 mM and incubated for an additional 2 h at 37°C. Cells were washed twice with PBS, fixed in acetone for 5 min, and air dried at room temperature for 10 min. Cells were incubated with PBS for 5 min at room temperature, followed by treatment with 3% H2O2 in PBS for 5 min to block endogenous peroxidase activity. The cells were permeabilized with PBS containing 0.1% Triton X-100 (PBST) for 5 min and blocked with PBST containing 1% bovine serum albumin for 15 min. Immunostaining was performed using mouse antibromodeoxyuridine antibody (1:100; Roche Molecular Biochemicals, Indianapolis, IN) overnight at 4°C. The dishes were washed for 5 min with PBST four times each and then treated with biotinylated rabbit anti-mouse antibody at 1:500 dilutions for 60 min at room temperature. Finally, the staining was detected using the ABC HRP kit from DAKO as directed by the manufacturer. Images were captured using a SPOT Jr charge-coupled device camera mounted on a Nikon DIAPHOT microscope and processed using Adobe Photoshop 7.0.
RESULTS
Cyclin D1 and PCNA in infected smooth muscle cells.
Previously, we demonstrated that cultured smooth muscle cells exhibited activation of ERK1/2 over the course of T. cruzi infection (25). In order to identify the role of other cell cycle regulatory proteins in infected smooth muscle cells, we performed immunoblotting with antibodies directed against cyclin D1 and PCNA. Increased expression of both cyclin D1 and PCNA was observed in infected cells at 24 h postinfection which persisted to 72 h postinfection. These observations demonstrate a persistent activation of the pathway (Fig. 1) and are indicative of the proliferation of infected smooth muscle cells.
FIG. 1.
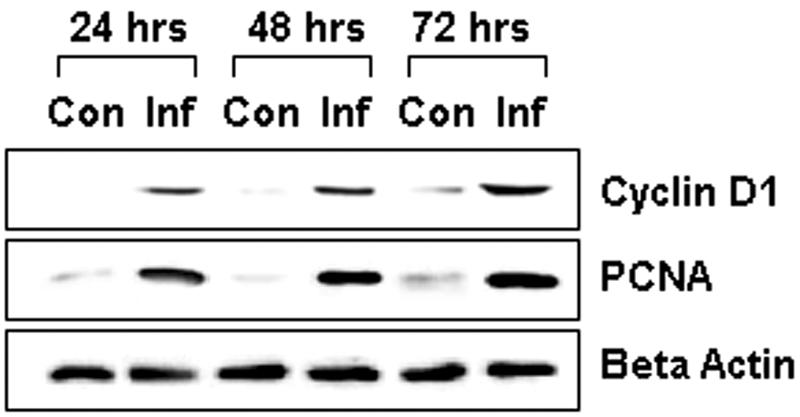
Representative immunoblot demonstrating the expression of cyclin D1 and PCNA in infected smooth muscle cells. Lysates of smooth muscle cells were probed with antibodies directed against cyclin D1 and PCNA. Note that infected cells (Inf), at 24 h, 48 h, and 72 h postinfection, exhibited increased cyclin D1 and PCNA expression compared to control cells (Con). β-Actin was used as a loading marker.
Activation of ERK in carotid arteries of infected mice.
Previously, we reported activation of ERK in cultured endothelial cells and smooth muscle cells infected with T. cruzi (25). In the current experiments, we also observed activation of ERK in the carotid arteries obtained from acutely (20 days postinfection) and chronically infected (90 days postinfection) animals (Fig. 2). These results suggest that activation of ERK is associated with the vasculopathy of T. cruzi infection.
FIG. 2.
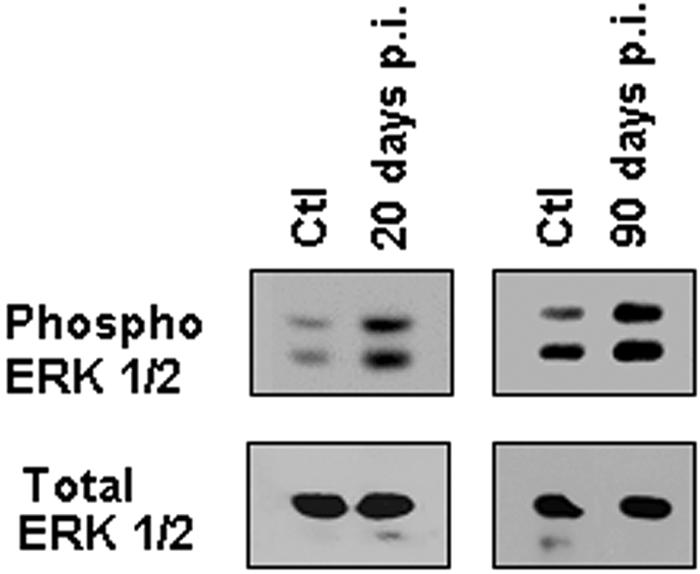
Activation of ERK1/2 in infected carotid arteries. Carotid arteries were isolated from mice 20 and 90 days postinfection (p.i.) and from uninfected control mice (Ctl). A representative immunoblot of pooled carotid artery lysates is shown, demonstrating that phospho-ERK1/2 expression was increased in infected arteries at 20 and 90 days postinfection. Total ERK1/2 was used as a loading control.
Expression of caveolin in infected cultured endothelial and smooth muscle cells.
Previous studies from this laboratory indicated a regulatory relationship between ERK and caveolin-1 (3, 8, 9, 26). Consistent with these observations, in the present study we found that in infected endothelial and smooth muscle cells there was a significant decrease in levels of caveolin-1 and caveolin-2 after infection (Fig. 3). Caveolin-3 expression was not determined in cultured cells because it is typically downregulated under culture conditions (M. P. Lisanti, unpublished observations).
FIG. 3.

Representative immunoblot demonstrating the expression of caveolins in infected cultured endothelial and smooth muscle cells. Human umbilical vein endothelial cells (HuVEC) and smooth muscle cells (HuSMC) were infected with trypomastigotes of the Tulahuen strain for 48 h. Expression of both caveolin-1 (Cav-1) and caveolin-2 (Cav-2) was decreased in infected cells (Inf) compared to controls (Con).
Expression of caveolin in carotid arteries from infected mice.
We next examined the expression levels of caveolins in carotid arteries. During the acute phase of infection (day 20 postinfection), carotid arteries exhibited a decreased level of both caveolin-1 and caveolin-3. The expression of caveolin-1 returned to baseline levels, while the expression of caveolin-3 decreased further in chronically infected arteries (day 90) compared to uninfected arteries (Fig. 4). Caveolin-2 expression was not determined.
FIG. 4.

Representative immunoblot of caveolin-1 (Cav-1) and caveolin-3 (Cav-3) levels in carotid arteries. The immunoblot of pooled lysates of carotid arteries isolated from infected mice showed downregulation of both Cav-1 and Cav-3 expression in the acute phase of the infection (20 days postinfection [p.i.]) compared to the control (Ctl). Cav-1 levels reverted back to control levels 90 days postinfection. Ponceau red staining is shown as a loading marker (lower panels).
Expression of cell cycle regulatory proteins in infected arteries.
To further characterize the role of cell cycle regulatory proteins in the vasculopathy that attends Chagas' disease, we next performed immunoblotting on lysates of infected carotid arteries. Carotid arteries obtained from acutely infected mice exhibited increased expression of PCNA, similar to the increase seen in infected cultured smooth muscle cells (Fig. 1). There was no difference in the levels of PCNA expression observed in arteries obtained 90 days postinfection compared with controls (Fig. 5). Some of the control mice at 90 days exhibited increased expression of PCNA. This may be related to other factors. Consistent with the increased expression of cyclin D1, there was an increase in expression for retinoblastoma protein (Rb) phosphorylated at serine 780, a substrate of cyclin D1, during acute infection, returning to baseline during the chronic phase (Fig. 5). Rb inactivation normally occurs through its phosphorylation on Ser780 by the cyclin D1-cyclin-dependent kinase 4/6 (CDK4/6) complex, thus driving cell cycle progression. These results provide additional evidence of a cyclin D1-dependent mechanism in the infection induced by T. cruzi. Moreover, the expression of the cell cycle inhibitor p21Cip1/Waf1 was decreased in carotid arteries from acutely infected mice. This reduction in expression persisted into the chronic phase of infection (Fig. 5). These data suggest that p21Cip1/Waf1 downregulation may be an additional mechanism implicated in T. cruzi-induced vasculopathy.
FIG. 5.

Expression of cell cycle regulatory proteins in infected carotid arteries. A representative immunoblot obtained from lysates of pooled carotid arteries isolated from infected mice showed increased expression of phospho-Rb (Ser780) and PCNA and downregulation of p21Cip1/Waf1 20 days postinfection (p.i.). Note that pRb and PCNA levels reverted back to control levels at 90 days postinfection. Ponceau red staining is shown as a loading control (lower panels).
Immunostaining of carotid arteries from T. cruzi-infected mice.
We next stained carotid artery sections of infected and uninfected mice with antibodies directed against cyclinD1, p21Cip1/Waf1, and the pRb-Ser780. As shown in Fig. 6, cyclin D1 was detected in a few minor areas of the medial layer of control uninfected arteries. However, the staining was more intense in arteries obtained during the acute phase, and even more positive in chronically infected arteries. These results are consistent with the observations of infected cultured smooth muscle cells (Fig. 1). Similarly to cyclin D1, its substrate, phospho-Rb (Ser780), which was minimally detected in uninfected arteries, exhibited increased staining in arteries from acutely infected mice. However, unlike that with cyclin D1, the intensity of the staining returned to baseline during chronic infection (Fig. 6). This pattern of staining between the acute and chronic phases of the infection is consistent with the results obtained by immunoblotting (Fig. 5). The CDK inhibitor p21Cip1/Waf1 was detected in the medial layer of uninfected arteries (Fig. 6). Interestingly, the staining intensity was decreased in carotid arteries from acute and chronically infected mice. Although colocalization was not performed, we did stain consecutive sections.
FIG. 6.

Representative immunohistochemical analysis of cyclin D1, phospho-Rb (Ser780), and p21Cip1/Waf1 in carotid arteries obtained from infected and control mice. (A to C) Cyclin D1 immunostaining was increased in infected arteries 20 days postinfection (B) and more intensely 90 days postinfection (C), compared to control uninfected arteries (A). (D to F) phospho-Rb (Ser780). Immunostaining for the retinoblastoma protein phosphorylated at Ser780 was clearly more pronounced at 20 days postinfection in infected carotid arteries (E) than in control arteries (D). At 90 days postinfection (F), pRb staining was similar to control levels. (G to I) p21Cip1/Waf1. Interestingly, the level of p21Cip1/Waf1 immunostaining was less pronounced in infected carotid arteries 20 days postinfection (H) and even less at 90 days postinfection (I) compared to control uninfected arteries (G). Boxed areas (in red) are shown as insets at higher magnification.
Pre-pro-ET-1 mRNA and ETA protein levels in blood vessels of infected mice.
A role for ET-1 in the pathogenesis of chagasic heart disease and its associated vasculopathy has been suggested. Thus, we examined the mRNA expression levels of pre-pro-ET-1 in carotid arteries and the aorta isolated from infected mice, using RT-PCR. As shown in Fig. 7, pre-pro-ET-1 mRNA was significantly increased in infected carotids and aorta, compared with control, uninfected vessels. We next examined the protein expression levels of the ET-1 receptor, ETA, in infected aorta. Notably, there was a significant upregulation of ETA expression in infected carotid arteries, which was persistent over the course of infection (Fig. 8).
FIG. 7.

Increased mRNA expression of pre-pro-ET-1 in the aorta and carotid arteries obtained from infected and control mice. The RT-PCR technique was used on extracts of pooled carotid arteries and aorta isolated from infected mice (I) and showed increased mRNA expression of pre-pro-ET-1 compared to controls (C). GAPDH mRNA was used as an internal control.
FIG. 8.

Increased expression of the ETA receptor in carotid arteries obtained from infected and control (Ctl) mice. A representative immunoblot of carotid artery lysates obtained from pooled arteries isolated from infected mice showed increased expression of the ETA receptor 20 and 90 days postinfection (p.i.) compared to controls. Ponceau red staining is shown as a loading control (lower panels).
Bromodeoxyuridine incorporation in smooth muscle cells.
In order to determine whether smooth muscle cell proliferation was associated with DNA synthesis, we used a deoxybromouridine uptake assay. We tested the ability of conditioned medium obtained from T. cruzi-infected or uninfected endothelial cells to induce DNA synthesis in smooth muscle cells. These cells were incubated with conditioned medium, obtained from 72-h-infected endothelial cells for 6 h, followed by further incubation with bromodeoxyuridine for 2 h. Immunostaining of the cells revealed a dramatic uptake of bromodeoxyuridine in smooth muscle cells treated with infected conditioned medium compared with conditioned medium obtained from uninfected endothelial cells (Fig. 9). Approximately 65% of the smooth muscle cells treated with infected conditioned medium had deeply brown-stained nuclei, indicating induction of DNA synthesis.
FIG. 9.

Bromodeoxyuridine incorporation in smooth muscle cells. A significant uptake of bromdeoxyouridine was observed in those cells that were treated with infected conditioned medium (B) compared to those treated with uninfected conditioned medium (A).
DISCUSSION
In the present study, we have demonstrated that infected vascular cells exhibit an upregulation of the cell cycle regulatory proteins cyclin D1 and PCNA, as well as a decreased expression of the tumor suppressor protein caveolin-1. Using immunoblotting and immunohistochemical techniques, carotid arteries isolated from infected mice demonstrated an increased activation of ERK1/2, upregulation of cyclin D1, PCNA, and pRb, and decreased levels of the CDK inhibitor p21Cip, as well as a downregulation of caveolin-1 and caveolin-3. In addition, using an RT-PCR technique, our results indicate increased ET-1 and ETA receptor in vasculature obtained from infected mice.
Previously, we suggested that a mechanism by which the parasite could regulate vascular dysfunction is the increased activation of ERK1/2, which is one of three major mitogen-activated protein kinases in mammalian cells. Injury to the cardiovascular system caused by hypoxia, ischemia/reperfusion, restenosis, and other infections results in the activation of ERK (14). We reported previously that the myocardium of T. cruzi-infected mice exhibits activation of ERK (14). Activation of ERK has also been reported in T. cruzi-infected cultured endothelial and smooth muscle cells (25). In this paper, we showed that activation of ERK in carotid arteries isolated from infected mice persisted from the acute into the chronic phase. Thus, these results provide the first in vivo evidence that T. cruzi infection activates ERK in the vasculature and suggest that the ERK pathway is one of the mechanisms implicated in vasculopathy.
Recent evidence indicates that a regulatory relationship exists between ERK1/2 and caveolin-1 (3, 8, 9, 26). For example, caveolin-1 has been shown to interact with and suppress the kinase activity of the epidermal growth factor receptor and several members of the Ras-p42/44 mitogen-activated protein kinase cascade, including MEK and ERK, in vitro (5, 7). Conversely, downregulation of caveolin-1 in NIH 3T3 fibroblasts (using an antisense cDNA approach) results in ERK activation and cellular transformation (9). Moreover, we reported previously that there was activation of ERK in neointimal lesions from caveolin-1 null mice following blood flow cessation, compared to wild-type mice (11). In the present study we demonstrated that infected cultured endothelial cells and smooth muscle cells in culture exhibited a downregulation of the caveolin-1 and caveolin-2 isoforms. A downregulation of caveolin-1 and caveolin-3 was also detected in carotid arteries obtained from acutely infected mice compared to controls. However, in the chronic phase of the infection, the expression of caveolin-1 reverted to baseline levels, while that of caveolin-3 decreased even further. T. cruzi infection caused a reduction in the expression of caveolin-1 in cultured infected cells.
The cyclin D1 protein is the regulatory component of the holoenzyme that inactivates pRb, implying a role for cyclin D1 in cellular proliferation and transformation (15). D-cyclins are involved in controlling cell cycle progression by activating their associated kinases, cdk4 and cdk6. These cyclin-dependent kinases phosphorylate pRb, leading to transition through the G1 phase of the cell cycle (33). We found increased expression of the proliferation markers cyclin D1 and PCNA in cultured smooth muscle cells as well as in carotid arteries obtained from infected mice. In addition, the studies with bromodeoxyuridine uptake underscore the important relationship between endothelial and smooth muscle cells.
Immunohistochemical studies revealed increased staining intensity for cyclin D1 in infected arteries during both the acute and the chronic phases of the infection. This is similar to the results observed in cultured cells (25). Similarly, studies have shown increased expression of cyclin D1 and PCNA in the myocardium of infected mice (14). Furthermore, in this study our results also demonstrated increased levels of the retinoblastoma protein, phosphorylated at Ser780, in arteries obtained from acutely infected mice. The phosphorylation of Rb at Ser780 is induced specifically by the cyclin D1-cdk4 complex, and not the cyclin E-cdk2 complex, and inhibits the binding of pRb to E2F-1 (19). This process induces cell cycle progression. Moreover, we have previously reported enhanced phosphorylation of pRb associated with an overexpression of cyclin D1 in T. cruzi-infected myocardial tissues (14). Therefore, our findings suggest a mechanism involving the cyclin D1-phospho-Rb pathway in the vasculopathy induced by T. cruzi infection. The fact that our results demonstrated unaltered expression levels of Rb phosphorylated at serine 780 between chronically infected arteries and control blood vessels suggests that cyclin D1 might be acting on a substrate other than Rb during the chronic stage of the infection.
In addition to the CDK activator cyclin D1, we investigated the expression levels of p21Cip1/Waf1, a CDK inhibitor. Many studies have reported an inverse relationship between p21Cip/Waf1 and the cyclin D1 pathway. In fact, upregulation of cyclin D1 and pRb expression is associated with the proliferative processes and is accompanied by decreased protein levels of p21Cip/Waf1 (12, 18). Using immunoblotting and immunohistochemical techniques, we demonstrated that the p21Cip/Waf1 expression level decreased in carotid arteries obtained from acute infected mice and even more dramatically in the chronic stage of infection. These data suggest that downregulation of the tumor suppressor p21Cip1/Waf1 could be an additional mechanism implicated in the vasculopathy induced by T. cruzi. The levels of p21Cip1/Waf1 seen in Fig. 5 suggest that there is a general lack of cellular proliferation in the absence of T. cruzi infection, which is consistent with the high levels of pRb (Ser780) phosphorylation. Additionally, while it is well established that p21Cip1/Waf1 binding to PCNA inhibits DNA replication, this molecular interaction apparently does not necessarily interfere with PCNA-dependent DNA repair. The expression of PCNA, such as that observed in the control mice at day 90 postinfection (Fig. 5), may represent a normal role for PCNA in the maintenance of the arterial endothelium.
The relationship between T. cruzi infection and ET-1 is now well known (28, 29, 36, 44). Both treatment of infected mice with phosphoramidon (an inhibitor of endothelin converting enzyme) (16) and infection of mice in which the ET-1 gene was deleted from cardiac myocytes resulted in an amelioration of cardiac remodeling (36). In addition, our laboratory has demonstrated that smooth muscle cells incubated with supernatants from infected endothelial cells exhibited increased ERK activation and cyclin D1 expression and an increase in thymidine incorporation, all of which were abolished by pretreatment of the cells with the ETA antagonist BQ123 (25). In the present study, we provide the first in vivo validation of a possible role for ET-1 in the T. cruzi-induced vasculopathy. Carotid arteries isolated from infected mice exhibited increased mRNA expression of pre-pro-ET-1 (the ET-1 precursor), as well as increased protein levels of the ETA receptor. Taken together, these findings suggest that ET-1 contributes to the pathogenesis of vasculopathies caused by T. cruzi. Previously, we have demonstrated that, in the mouse model of Chagas' disease, there is a significant reduction in blood flow in the cremaster bed as well as in the coronary circulation (37, 38). In primates and humans, there is evidence for vascular dysfunction; however, the presence and amount of vascular dysfunction varies with the stage of the disease (4, 22, 42, 43, 45).
Our results in the mouse model indicate that T. cruzi infection can induce vascular damage and results in the activation of interrelated signaling pathways, including activation of ERK, upregulation of cyclin D1, and decreased expression of the tumor suppressor genes p21Cip1/Waf1 and caveolin proteins (Fig. 10). The increased synthesis of ET-1 and its receptor, ETA, could be the key process activating the above-mentioned pathways and a target for drug development to pharmacologically treat T. cruzi-associated vasculopathies.
FIG. 10.

Schematic diagram summarizing the role of caveolin-1 (Cav-1) in regulating proliferative signaling. Note that Cav-1 functions as an inhibitor of ERK signaling. During infection with T. cruzi, Cav-1 levels are downregulated, leading to the activation of ERK and cyclin D1 and resulting in vascular smooth muscle cell (VSMC) proliferation. Cyclin D1 is a downstream target of both ERK and ET-1. The large bold font indicates major regulatory proteins.
Acknowledgments
This work was supported by NIH grants AI-12770, AI-52739, and HL-073732 (H.B.T.). G.S.H. is a recipient of a postdoctoral fellowship from the Foundation of Health Research, Quebec, Canada. M.P.L. was supported by grants from the NIH and the American Heart Association and by the Hirschl/Weil-Caulier Career Scientist Award. M.S.D. was supported by NIH Training Grant Mechanisms of Cardiovascular Diseases T32 HL 07675 and by the IDSA ERF/NFID Colin L. Powell Minority Postdoctoral Fellowship in Tropical Disease Research sponsored by GlaxoSmithKline. C.J.d.A. was supported by NIH-Fogarty Training Grant Interhemispheric Research and Training in Infectious Diseases D43TW007129.
Editor: W. A. Petri, Jr.
REFERENCES
- 1.Brink, P. R., S. V. Ramanan, and G. G. Christ. 1996. Human connexin 43 gap junction channel gating: evidence for mode shifts and/or heterogeneity. Am. J. Physiol. 271:C321-C331. [DOI] [PubMed] [Google Scholar]
- 2.Christ, G. J., A. P. Moreno, A. Melman, and D. C. Spray. 1992. Gap junction-mediated intercellular diffusion of Ca2+ in cultured human corporal smooth muscle cells. Am. J. Physiol. 263:C373-C383. [DOI] [PubMed] [Google Scholar]
- 3.Cohen, A. W., D. S. Park, S. E. Woodman, T. M. Williams, M. Chandra, J. Shirani, A. P. de Souza, R. N. Kitsis, R. G. Russell, L. M. Weiss, B. Tang, L. A. Jelicks, S. M. Factor, V. Shtutin, H. B. Tanowitz, and M. P. Lisanti. 2003. Caveolin-1 null mice develop cardiac hypertrophy with hyperactivation of p42/44 MAP kinase in cardiac fibroblasts. Am. J. Physiol. Cell Physiol. 284:C457-C474. [DOI] [PubMed] [Google Scholar]
- 4.Consolim-Colombo, F. M., H. F. Lopes, E. A. Rosetto, E. C. Rubira, J. A. S. Barreto-Filho, A. C. A. Baruzzi, N. N. Rocha, C. Mady, M. C. Irigoyen, and E. M. Krieger. 2004. Endothelial function is preserved in Chagas' heart disease patients without heart failure. Endothelium 11:241-246. [DOI] [PubMed] [Google Scholar]
- 5.Couet, J., M. Sargiacomo, and M. P. Lisanti. 1997. Interaction of a receptor tyrosine kinase, EGF-R, with caveolins. Caveolin binding negatively regulates tyrosine and serine/threonine kinase activities. J. Biol. Chem. 272:30429-30438. [DOI] [PubMed] [Google Scholar]
- 6.D'Orleans-Juste, P., J. Labonte, G. Bkaily, S. Choufani, M. Plante, and J. C. Honore. 2002. Function of the endothelinB receptor in cardiovascular physiology and pathophysiology. Pharmacol. Ther. 95:221-238. [DOI] [PubMed] [Google Scholar]
- 7.Engelman, J. A., C. Chu, A. Lin, H. Jo, T. Ikezu, T. Okamotod, D. S. Kohtz, and M. P. Lisanti. 1998. Caveolin-mediated regulation of signaling along the p42/44 MAP kinase cascade in vivo. A role for the caveolin-scaffolding domain. FEBS Lett. 428:205-211. [DOI] [PubMed] [Google Scholar]
- 8.Engelman, J. A., C. C. Wycoff, S. Yasuharai, K. S. Song, T. Okamoto, and M. P. Lisanti. 1997. Recombinant expression of caveolin-1 in oncogenically transformed cells abrogates anchorage-independent growth. J. Biol. Chem. 272:16374-16381. [DOI] [PubMed] [Google Scholar]
- 9.Galbiati, F., D. Volonte, J. A. Engelman, G. Watanabe, R. Burk, R. G. Pestell, and M. P. Lisanti. 1998. Targeted down-regulation of caveolin-1 is sufficient to drive cell transformation and hyperactivate the p42/44 MAP kinase cascade. EMBO J. 17:6633-6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galie, N., A. Manes, and A. Branzi. 2004. The endothelin system in pulmonary arterial hypertension. Cardiovasc. Res. 61:227-337. [DOI] [PubMed] [Google Scholar]
- 11.Hassan, G. S., J.-F. Jasmin, W. Schubert, P. G. Frank, and M. P. Lisanti. 2004. Caveolin-1 deficiency stimulates neointima formation during vascular injury. Biochemistry 43:8312-8321. [DOI] [PubMed] [Google Scholar]
- 12.Hou, Y. Z., J. Yang, G.-R. Zhao, and Y.-J. Yuan. 2004. Ferulic acid inhibits vascular smooth muscle cell proliferation induced by angiotensin II. Eur. J. Pharmacol. 499:85-90. [DOI] [PubMed] [Google Scholar]
- 13.Huang, H., T. M. Calderon, J. W. Berman, V. L. Braunstein, L. M. Weiss, M. Wittner, and H. B. Tanowitz. 1999. Infection of endothelial cells with Trypansoma cruzi activates NF-κB and induces vascular adhesion molecule expression. Infect. Immun. 67:5434-5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang, H., S. B. Petkova, A. W. Cohen, B. Bouzahzah, J. Chan, J.-N. Zhou, S. M. Factor, L. M. Weiss, M. Krishnamachary, S. Mukherjee, M. Wittner, R. N. Kitsis, R. G. Pestell, M. P. Lisanti, C. Albanese, and H. B. Tanowitz. 2003. Activation of transcription factors (AP-1 and NF-κB) in murine chagasic myocarditis. Infect. Immun. 71:2859-2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hulit, J., T. Bash, M. Fu, F. Galbiati, C. Albanese, D. R. Sage, A. Schlegel, J. Zhurinsky, M. Shtutman, A. Ben-Ze'ev, M. P. Lisanti, and R. G. Pestell. 2000. The cyclin D1 gene is transcriptionally repressed by caveolin-1. J. Biol. Chem. 275:21203-21209. [DOI] [PubMed] [Google Scholar]
- 16.Jelicks, L. A., M. Chandra, J. Shirani, V. Shtutin, B. Tang, G. J. Christ, S. M. Factor, M. Wittner, H. Huang, L. M. Weiss, S. Mukherjee, B. Bouzahzah, S. B. Petkova, M. M. Teixeira, S. A. Douglas, M. L. Loredo, P. D'Orleans-Juste, and H. B. Tanowitz. 2002. Cardioprotective effects of phosphoramidon on myocardial structure and function in murine Chagas' disease. Int. J. Parasitol. 32:1497-1506. [DOI] [PubMed] [Google Scholar]
- 17.Kedzierski, R. M., and M. Yanagisawa. 2001. Endothelin system: the double-edged sword in health and disease. Annu. Rev. Pharmacol. Toxicol. 41:851-876. [DOI] [PubMed] [Google Scholar]
- 18.Kim, H. S., H. J. Cho, H.-J. Cho, S.-J. Park, K.-W. Park, I.-H. Chae, B.-H. Oh, Y.-B. Park, and M.-M. Lee. 2004. The essential role of p21 in radiation-induced cell cycle arrest of vascular smooth muscle cell. J. Mol. Cell. Cardiol. 37:871-880. [DOI] [PubMed] [Google Scholar]
- 19.Kitagawa, M., H. Higashi, H. K. Jung, I. Suzuki-Takahashi, M. Ikeda, K. Tamai, J. Kato, K. Segawa, E. Yoshida, S. Nishimura, and Y. Taya. 1996. The consensus motif for phosphorylation by cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. EMBO J. 15:7060-7069. [PMC free article] [PubMed] [Google Scholar]
- 20.Leon, J. S., and D. M. Engman. 2003. The significance of autoimmunity in the pathogenesis of Chagas heart disease. Front. Biosci. 8:e315-e322. [DOI] [PubMed] [Google Scholar]
- 21.Masaki, T. 1998. The discovery of endothelins. Cardiovasc. Res. 39:530-533. [DOI] [PubMed] [Google Scholar]
- 22.Montes de Oca, M., S. H. Torres, J. G. Loyo, F. Vasquez, N. Hernandez, B. Anchustegui, and J. J. Puigbo. 2004. Exercise performance and skeletal muscles in patients with advanced Chagas disease. Chest 125:1306-1324. [DOI] [PubMed] [Google Scholar]
- 23.Morris, S. A., H. B. Tanowitz, M. Wittner, and J. P. Bilezikian. 1990. Pathophysiological insights into the cardiomyopathy of Chagas' disease. Circulation 82:1900-1999. [DOI] [PubMed] [Google Scholar]
- 24.Morris, S. A., L. M. Weiss, S. M. Factor, J. P. Bilezikian, H. B. Tanowitz, and M. Wittner. 1989. Verapamil ameliorates clinical, pathologic and biochemical manifestations of experimental chagasic cardiomyopathy in mice. J. Am. Coll. Cardiol. 14:782-789. [DOI] [PubMed] [Google Scholar]
- 25.Mukherjee, S., H. Huang, S. B. Petkova, C. Albanese, R. G. Pestell, V. L. Braunstein, G. J. Christ, M. Wittner, M. P. Lisanti, J. W. Berman, L. M. Weiss, and H. B. Tanowitz. 2004. Trypanosoma cruzi infection activates extracellular signal-regulated kinase in cultured endothelial and smooth muscle cells. Infect. Immun. 72:5274-5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park, D. S., H. Lee, P. G. Frank, B. Razani, A. V. Nguyen, A. F. Parlow, R. G. Russell, J. Hulit, R. G. Pestell, and M. P. Lisantiet. 2002. Caveolin-1-deficient mice show accelerated mammary gland development during pregnancy, premature lactation, and hyperactivation of the Jak-2/STAT5a signaling cascade. Mol. Biol. Cell 13:3416-3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petkova, S. B., A. Ashton, B. Bouzahzah, H. Huang, R. G. Pestell, and H. B. Tanowitz. 2000. Cell cycle molecules and diseases of the cardiovascular system. Front. Biosci. 5:D452-D460. [DOI] [PubMed] [Google Scholar]
- 28.Petkova, S. B., H. Huang, S. M. Factor, R. G. Pestell, B. Bouzahzah, L. A. Jelicks, L. M. Weiss, S. A. Douglas, M. Wittner, and H. B. Tanowitz. 2001. The role of endothelin in the pathogenesis of Chagas' disease. Int. J. Parasitol. 31:499-511. [DOI] [PubMed] [Google Scholar]
- 29.Petkova, S. B., H. B. Tanowitz, H. I. Magazine, S. M. Factor, J. Chan, R. G. Pestell, B. Bouzahzah, S. A. Douglas, V. Shtutin, S. A. Morris, E. Tsang, L. M. Weiss, G. J. Christ, M. Wittner, and H. Huang. 2000. Myocardial expression of endothelin-1 in murine Trypanosoma cruzi infection. Cardiovasc. Pathol. 9:257-265. [DOI] [PubMed] [Google Scholar]
- 30.Rossi, M. A. 1990. Microvascular changes as a cause of chronic cardiomyopathy in Chagas' disease. Am. Heart J. 120:233-236. [DOI] [PubMed] [Google Scholar]
- 31.Rossi, M. A., and S. G. Ramos. 1996. Coronary microvascular abnormalities in Chagas' disease. Am. Heart J. 132:207-210. [DOI] [PubMed] [Google Scholar]
- 32.Rowin, K. S., H. B. Tanowitz, M. Wittner, H. T. Nyguen, and B. Nadal-Ginard. 1983. Inhibition of muscle differentiation by Trypanosoma cruzi. Proc. Natl. Acad. Sci. USA 80:6390-6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sherr, C. J., and J. M. Roberts. 1999. CDK inhibitors: positive and negative regulators of G1 phase progression. Genes Dev. 13:1501-1512. [DOI] [PubMed] [Google Scholar]
- 34.Tanowitz, H. B., E. R. Burns, A. K. Sinha, N. N. Kahn, S. A. Morris, S. M. Factor, V. B. Hatcher, J. P. Bilezikian, and M. Wittner. 1990. Enhanced platelet adherence and aggregation in Chagas' disease: a potential pathogenic mechanism for cardiomyopathy. Am. J. Trop. Med. Hyg. 43:274-281. [DOI] [PubMed] [Google Scholar]
- 35.Tanowitz, H. B., J. P. Gumprecht, D. Spurr, T. M. Calderon, M. C. Ventura, C. Raventos-Suarez, S. M. Factor, V. Hatcher, M. Wittner, and J. W. Berman. 1992. Cytokine gene expression of endothelial cells infected with Trypanosoma cruzi. J. Infect. Dis. 166:598-603. [DOI] [PubMed] [Google Scholar]
- 36.Tanowitz, H. B., H. Huang, L. A. Jelicks, M. Chandra, M. L. Loredo, L. M. Weiss, S. M. Factor, V. Shtutin, S. Mukherjee, R. N. Kitsis, G. J. Christ, M. Wittner, J. Shirani, Y. Y. Kisanuki, and M. Yanagisawa. 2005. Role of endothelin-1 in the pathogenesis of chronic chagasic heart disease. Infect. Immun. 73:2496-2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanowitz, H. B., D. K. Kaul, B. Chen, S. A. Morris, S. M. Factor, L. M. Weiss, and M. Wittner. 1996. Compromised microcirculation in acute Trypanosoma cruzi infection. J. Parasitol. 82:124-130. [PubMed] [Google Scholar]
- 38.Tanowitz, H. B., L. V. Kirchhoff, D. Simon, S. A. Morris, L. M. Weiss, and M. Wittner. 1992. Chagas' disease. Clin. Microbiol. Rev. 4:400-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanowitz, H. B., S. A. Morris, S. M. Factor, L. M. Weiss, and M. Wittner. 1992. Parasitic disease of the heart I: acute and chronic Chagas' disease. Cardiovasc. Pathol. 1:7-15. [DOI] [PubMed] [Google Scholar]
- 40.Tanowitz, H. B., M. Wittner, B. Chen, H. Huang, L. M. Weiss, G. J. Christ, V. L. Braunstein, J. P. Bilezikian, and S. A. Morris. 1996. Effects of verapamil on acute Chagas' disease. J. Parasitol. 82:814-819. [PubMed] [Google Scholar]
- 41.Tarleton, R. L. 2001. Parasite persistence in the aetiology of Chagas disease. Int. J. Parasitol. 31:550-554. [DOI] [PubMed] [Google Scholar]
- 42.Torres, F. W., H. Acqualella, J. A. Condado, R. Dinsmore, and I. F. Palacios. 1995. Coronary reactivity is abnormal with Chagas' disease. Am. Heart J. 129:995-1001. [DOI] [PubMed] [Google Scholar]
- 43.Torres, S. H., H. J. Finol, M. Montes de Oca, F. Vasquez, J. J. Puigbo, and J. G. Loyo. 2004. Capillary damage in skeletal muscle in advanced Chagas' disease patients. Parasitol. Res. 93:364-368. [DOI] [PubMed] [Google Scholar]
- 44.Wittner, M., G. J. Christ, H. Huang, L. M. Weiss, V. B. Hatcher, S. A. Morris, G. A. Orr, J. W. Berman, G. A. Zeballos, S. A. Douglas, and H. B. Tanowitz. 1995. Trypanosoma cruzi induces endothelin release from endothelial cells. J. Infect. Dis. 171:493-497. [DOI] [PubMed] [Google Scholar]
- 45.Zabalgoittia, M., J., Ventura, J. Lozano, L. Anderson, K. Carey, G. Hubbard, J. Williams, and J. Vandeberg. 2004. Myocardial contrast echocardiography in assessing microcirculation in baboons with Chagas disease. Microcirculation 11:271-278. [DOI] [PubMed] [Google Scholar]
- 46.Zhao, W., and G. J. Christ. 1995. Endothelin-1 as a putative modulator of erectile dyfunction. II. Calcium mobilization in cultured human corporal smooth muscle cells. J. Urol. 154:1571-1577. [PubMed] [Google Scholar]