

Abstract
The preferential use of older antimicrobial agents is, in general, sound public health policy and is meant to maintain susceptibility to newer agents. In the case of fluoroquinolones, however, this strategy is flawed and may actually hasten the spread of Streptococcus pneumoniae strains resistant to newer members of the class. In a mouse thigh infection model, we were unable to isolate clones of pneumococci resistant to the newer fluoroquinolone levofloxacin at 2 × or 4 × the baseline MIC. An initial exposure in vivo to the older agent, ciprofloxacin, allowed straightforward selection of clones resistant to levofloxacin in a subsequent experiment. The original ciprofloxacin exposure generated clones without changes in the parC/E and gyrA/B quinolone target sites almost exclusively but did allow overexpression of a reserpine-responsive pump. While this caused only minimal change in the levofloxacin MIC (0.6 mg/liter to 0.8 mg/liter), it allowed a major change in the mutational frequency to resistance for levofloxacin (<1/108.5 to approximately 1/104.5), which allowed levofloxacin-resistant clones to be isolated in a subsequent in vivo experiment. The reason underlying ciprofloxacin's propensity to select for pump-overexpressed clones is likely related to its hydrophilicity. To preserve the susceptibility of Streptococcus pneumoniae to newer members of the class of quinolones, use of ciprofloxacin for community-acquired respiratory infections should be minimized.
The pathogen Streptococcus pneumoniae is a frequent cause of mortality and morbidity from community-acquired infections. It remains the most common cause of meningitis. However, the majority of the mortality and morbidity it causes is as the pathogen most frequently responsible for serious community-acquired pneumonia.
Beta-lactam antibiotics have been a mainstay of therapy for these indications. Unfortunately, the pneumococcus has become much less susceptible to this class of agents over the last decade (23). During this time, the bacterium has also become less susceptible to many other agents in the physician's therapeutic armamentarium that have traditionally been effective. The fraction of organisms tested that show full resistance to the macrolides and azalides (erythromycin, clarithromycin, and azithromycin) has shown a marked increase (23). Trimethoprim-sulfamethoxazole has also been shown to have markedly decreased activity (23).
Over the past 5 to 10 years, the fluoroquinolone antimicrobials have been used extensively for the treatment of pneumococcal pneumonia. These antimicrobials have been among the agents of choice recommended for the treatment of pneumonia by several expert committees, including the American Thoracic Society (19) and the Infectious Disease Society of America (3). However, concurrent with increased use of the fluoroquinolones, several surveillance studies of pneumococcal susceptibility have shown a small percentage of isolates demonstrating resistance and, troublingly, reports of failures of therapy with these agents (5, 12).
Chen and colleagues demonstrated that the rate of resistance increased in direct proportion to the use of these agents (5). We have previously demonstrated that it is possible to dose fluoroquinolones to suppress the amplification of resistant clones of Pseudomonas aeruginosa (13). Given that levofloxacin is the most used fluoroquinolone for the treatment of pneumococcal pneumonia, we wished to evaluate the antibacterial effects of this agent at multiple different drug exposures and to ascertain the pharmacodynamic variable most closely linked to the ability to kill Streptococcus pneumoniae in the mouse thigh infection model. We also wished to examine the impact of levofloxacin therapy on the emergence of resistance in Streptococcus pneumoniae in an in vivo model of infection. In addition, as the widely used fluoroquinolone ciprofloxacin has lost patent protection, we wished to examine its impact on the emergence of resistance in Streptococcus pneumoniae. Finally, we felt it was important to ascertain the mechanism of resistance in resistant clones recovered from the in vivo experiments and to replicate these findings in vitro.
MATERIALS AND METHODS
Microorganisms.
A strain of Streptococcus pneumoniae isolated from an Albany Medical Center Hospital patient (AMC-058), and an isogenic pair of the readily transformable, nonencapsulated laboratory strains R6 and R6N were studied. Subsequently, daughter isolates which expressed different resistance mechanisms were recovered from mouse thigh infection experiments. Strains RC2 (efflux pump overexpressed) and RC4 (efflux pump overexpressed and with a Ser79Tyr mutation in parC) were also subsequently studied. The strains were stored at −70°C in skim milk.
Antimicrobial agents.
Levofloxacin, ciprofloxacin, and sparfloxacin were used as test compounds. Levofloxacin powder (assay no., 85632; lot no., DW136; potency, 97.2%), ciprofloxacin (lot, 5GFC; potency, 84.3%), and sparfloxacin (potency, 99.5%) were supplied by R. W. Johnson Pharmaceutical Research and Development, Bayer Corporation, and Rhone-Poulenc Rorer (now Sanofi-Aventis) Pharmaceuticals, respectively. Stock solutions of the drugs at 1 mg/ml in saline were prepared, aliquoted, and stored at −70°C. Prior to each MIC experiment, an aliquot of the drug was thawed and diluted to the desired concentrations with Mueller-Hinton broth supplemented with lysed horse blood. Drugs were prepared fresh for all in vivo studies.
Susceptibility studies.
MIC (defined as the lowest drug concentration that resulted in no visible growth after 24 h of incubation at 35°C in 5% CO2) and minimum bactericidal concentration (MBC) (99.9% density reduction compared to the growth control) studies were conducted using standard CLSI (formerly NCCLS) broth macrodilution testing methods (18). MIC and MBC determinations were conducted in 5% nonheparinized lysed horse blood. Serial twofold dilutions of drugs were used to determine the geometric MICs, followed by a narrower range of drug concentrations to determine the arithmetic MICs. MBCs were determined by quantitative culturing of broth macrodilution tube material from MIC determination studies. MIC and MBC studies were conducted three times each by two independent technicians.
Animals.
Female 24- to 26-g outbred ICR/Swiss mice were used in all in vivo studies (Taconic Farms, NY). They received food and water ad libitum. All animal experimentation procedures were approved by and conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee of Albany Medical College, Albany, N.Y.
Murine thigh infection model.
A mouse thigh infection model, pioneered by Eagle et al. (9) and greatly expanded upon by Craig (6, 7), was adapted to examine the relationship between fluoroquinolone exposure and reduction in the density of bacteria in the thigh muscles of mice. These experiments were performed with normal (nonneutropenic) mice.
Single-dose pharmacokinetics studies of infected mice.
Dose range studies were conducted to determine the pharmacokinetic parameters for levofloxacin when administered intraperitoneally (i.p.) as a single dose. Normal mice were inoculated intramuscularly (1 × 107.5 CFU/thigh) in both posterior thigh muscles. Two hours later, groups of mice were injected i.p. with 25, 50, 150, 200, 250, and 350 mg/kg (body weight) of levofloxacin in 0.2 ml of saline. Three animals from each group were sacrificed at 0.25, 0.5, 1, 2, 3, 4, and 6 h after drug administration. Blood was collected by cardiac puncture and allowed to clot on ice. The serum was separated from the clot by centrifugation and stored at −70°C. The concentration of levofloxacin in each serum sample was determined using the well diffusion microbiological assay and/or the high-performance liquid chromatography (HPLC) assay. The microbiological assay was performed early in the set of studies while the HPLC assay was being validated. The assays were cross-validated.
Drug concentration quantitation: microbiological assay.
Levofloxacin concentrations were measured in mouse serum from pharmacokinetic studies using a microbiological assay. Escherichia coli KL16 was used as the assay organism. Pour plates containing 105 CFU/ml of organism in heart infusion agar were prepared. Five-millimeter wells were made in the agar. Twenty microliters of sample or standards was pipetted into the wells. The plates were incubated overnight for 24 h at 35°C in an ambient air incubator. The diameters of zones of inhibition for samples and standards were measured to the nearest 0.1 mm with a Vernier caliper. Drug concentrations in the samples were calculated using the data from the curves derived from the drug standards.
Levofloxacin HPLC assay.
The concentration of levofloxacin in each serum sample was also determined using a reverse-phase isocratic levofloxacin HPLC assay adapted from Wong et al. (27). Levofloxacin was obtained as the hemihydrate powder (assay no., 85632; lot no., DW136; potency, 97.2%) from the R. W. Johnson Pharmaceutical Research Institute (Raritan, NJ). Ciprofloxacin, the internal standard, was obtained as the hydrochloride salt from Sigma (St. Louis, MO). HPLC analysis was performed using a Hewlett-Packard series 1100 system (Hewlett-Packard, Waldbronn, Germany). The detector wavelength was set at 330 nm. Quantitation of levofloxacin concentrations in plasma samples was based on linear regression analysis of peak area ratios of standard levofloxacin concentrations. Separation was accomplished on a Hewlett-Packard Eclipse XDB-C18 (5 μm; 250- by 4.6-mm internal diameter) column (Agilent Technologies, Palo Alto, CA) maintained at 35°C. The isocratic mobile phase was composed of copper(II) sulfate pentahydrate (5 mM) and l-isoleucine (10 mM) in methanol (87.5:12.5 [vol/vol]) and was extracted with 4 ml of dichloromethane and reconstituted in 100 μl of HPLC mobile phase. The assay was linear over a range of 0.01 to 10 mg/liter. Within-day coefficients of variation were less than 9%, and the between-day coefficients of variation were less than 13%. The limit of quantitation was 0.01 mg/liter.
Ciprofloxacin HPLC assay.
The concentration of ciprofloxacin in mouse serum was determined using a modification of the methodology described by Wright et al. (28). One-hundred-microliter aliquots of mouse serum were mixed with 200 μl of ice-cold acetonitrile and centrifuged for 5 min at 13,000 × g. Two hundred microliters of supernatant fluid was withdrawn and mixed with 200 μl of 0.02 M sodium phosphate containing 0.2% triethylamine and 0.2% sodium dodecyl sulfate, adjusted to pH 3 with phosphoric acid. Ciprofloxacin standards were prepared in mouse serum and treated exactly as described for the experimental samples. Analysis was performed using an Agilent 1100 series HPLC system equipped with a model 1046A fluorescence detector. The mobile phase was a 60:40 mixture of the above-described buffer and acetonitrile, delivered at a rate of 1.5 ml/min. The stationary phase consisted of an Alltech Adsorbosphere HS C18 column (150 × 4.6 mm). Ciprofloxacin was detected fluorometrically, using excitation and emission wavelengths of 275 and 450, respectively. The assay response was linear over a concentration range of 0.1 to 20 μg/ml.
Quinolone dose range studies: establishing the stasis, ED20 (exposure to drug mediating 20% of the maximal effect), ED50, and ED80 doses.
Preliminary thigh infection experiments were performed to establish bacterial inocula (5.0 to 8.5 log10 CFU/thigh) for the clinical S. pneumoniae isolate AMC-058 that resulted in reproducible infections over a 24-hour period for the test microorganism. The bacterial suspension was prepared from bacteria in log-phase growth and was washed by centrifugation prior to dilution in sterile saline. Then, 0.1 ml of the bacterial suspension was injected into each posterior thigh muscle. The concentration of the bacterial inoculum was confirmed by quantitative culture.
The mice were separated into groups, each consisting of five or six animals. Two hours after inoculation of the bacteria, the mice were given one i.p. injection of a predetermined dose of drug. The following doses of drug were evaluated: 0, 10, 20, 35, 50, 65, 100, and 200 mg/kg. Twenty-four hours later the mice were humanely sacrificed by CO2 asphyxiation. Both posterior thigh muscles from each mouse were aseptically collected in 0.9% saline, homogenized, serially diluted 1:9 in 0.9% saline, and plated for quantitation on drug-free (total-population enumeration) and drug-containing (drug-resistant-subpopulation enumeration) agar plates. An additional four infected mice were sacrificed just before therapy initiation to determine the bacterial density in the thigh muscles at the start of treatment. The density of bacteria in each posterior thigh muscle was calculated by enumeration of growing colonies on heart infusion agar plates. The densities of bacteria in both thighs of each mouse were averaged and considered one specimen for statistical analysis. The results from this study were evaluated using an inhibitory sigmoid Emax (maximal observed kill) dose-response model. The data were subsequently used to determine the doses that resulted in no net change in bacterial density at the infection site and 20%, 50%, and 80% of maximum drug activity (stasis, ED20, ED50, and ED80). These doses are important because they coincide with the steepest part of the Emax dose-response curve. This means that larger and therefore more readily identifiable changes in microbiological effects are associated with these drug doses. These values were important for the delineation of the effect of the schedule on microbiological activity (see below). Additional dose-response studies were performed in order to fully delineate the sigmoid Emax dose-response relationship.
Ciprofloxacin was also evaluated in this system at exposures producing 40/1 and 80/1 area under the curve (AUC))/MIC ratios (doses of 25 mg/kg every 12 h [q12h] and 50 mg/kg q12h, respectively). The doses were chosen on the basis of previous data on ciprofloxacin pharmacokinetics and pharmacodynamics (15), where it was demonstrated that the AUC/MIC ratio was the pharmacodynamically linked variable.
Effect of dose scheduling on bacterial densities in infected tissue.
Simultaneous dose fractionation and dose range studies were performed with AMC-058 in order to determine the pharmacodynamic parameter (AUC/MIC ratio, maximum concentration of drug in serum/MIC ratio, or the time > MIC) that best predicted the microbiological outcome. It is important to perform these studies simultaneously in order to eliminate the impact of interday variability due to (i) bacterial-inoculum preparation and viability, (ii) limits in treatment drug concentration reproducibility, and (iii) the “drift” in the ED20, ED50, and ED80 that may occur because of the effect of factors i and ii on the study results. The doses selected for the dose fractionation study are those that, based on the first dose range study, were predicted to fall on the steep portion of the sigmoid Emax dose-response curve, since these doses result in maximum differences in the microbiological outcome. Differences in efficacy may be difficult to observe if dosages associated with minimal changes in microbiological outcome (those doses on the flat parts of the exposure-response curve) are used.
Infected mice were treated with each selected total dose (corresponding to the ED20, ED50, and ED80) as either a single injection, two equally divided doses of half the total daily dose given 12 h apart, or four equally divided doses of one-quarter the total daily dose given 6 h apart. The no-treatment (ED0) group was the control group and received saline. Power analysis was conducted on the original dose-response data to determine the minimum number of animals needed in each group to have a 90% probability of identifying a 0.7 log10 difference between treatment groups.
Microbiological-outcome results from groups that received the same total dose on different schedules were compared with each other to determine the pharmacodynamic parameter most closely linked to microbiological activity. If the peak/MIC ratio was most closely linked to the outcome, the once-daily dosing group would have the best bacterial-cell kill. If the AUC/MIC ratio was most important, all dosing groups would be equivalent. If time > MIC was most closely linked, the most fractionated dosing schedule (every 6 h) would provide the best results. The results were tested for differences by analysis of variance.
Streptococcus pneumoniae-infected thigh homogenates were plated on 3% lysed horse blood agar plates containing 2 × and 4 × MIC/ml drug, as well as on drug-free blood agar plates. These concentrations were chosen because a mutation in the target site for fluoroquinolones most commonly causes a ≥4-fold increase from the baseline MIC. In contrast, efflux pump overexpression usually causes changes of slightly less than this value.
PCR amplification and DNA sequence determination and analysis of changes in the parC/E and gyrA/B genes of clones recovered from resistance plates.
For amplification of the gyrA, parC, parE, and gyrB open reading frame regions, oligonucleotide primers were designed from the published S. pneumoniae sequences of these genes (2, 16, 17, 20, 21). One microgram of chromosomal DNA, a 1 μM concentration of each synthetic oligonucleotide primer, a 200 μM concentration of deoxynucleoside triphosphate, and 5 mM MgCl2 in the reaction buffer recommended by the manufacturer were employed. Amplification was achieved with an initial cycle of 5 min of denaturation at 95°C, followed by 15 min of annealing at 55°C (7 min before and 8 min after the addition of the enzyme), and 6 min of polymerase extension at 72°C. Then, 20 cycles of 1 min of denaturation at 95°C, 2 min of annealing at 55°C, and 2.5 min of polymerase extension at 72°C, with a final 20 min of extension at 72°C with subsequent slow cooling at 4°C, were carried out. Oligonucleotides SP-gyrA-1f (5′-CGTCGCATTCTCTACGGAATGAATGAATT-3′) and SP-gyrA-1r (5′-AGTTGCTCCATTAACCAAAAGGTTTGGAAA-3′) were used for the amplification of gyrA. Oligonucleotides SP-parC-3f (5′-TGGGTTGAAGCCGGTTC-3′) and SP-parC-3r (5′-TGCTGGCAAGACCGTTGG-3′) and oligonucleotides SP-gyrB-2f (5′-TTCTCCGATTTCCTCATG-3′) and SP-gyrB-2r (5′-AGAAGGGTACGAATGTGG-3′) were used for the amplification of parC and gyrB, respectively. Amplification of the parE region was done with oligonucleotides SP-parE-4f (5′-AAGGCGCGTGATGAGAGC-3′) and SP-parE-4r (5′-TCTGCTCCAACACCCGCA-3′). All sequences shown in this study were determined with both DNA strands. DNA and protein sequence comparisons were done with Intelligenetics PC Gene 6.0 software.
In vitro selection of resistant clones of Streptococcus pneumoniae.
Plates containing 2 × and 4 × the baseline MIC of ciprofloxacin were inoculated with 108 CFU of S. pneumoniae strain AMC-058. From this, the mutation frequencies at 2 × MIC and 4 × MIC were calculated.
Uptake and efflux of EtBr by S. pneumoniae isolates.
Measurement of the levels of ethidium bromide (EtBr) accumulation and efflux in Streptococcus pneumoniae test strains (AMC-058, RC2, R6, and R6N) was based on previously described methods (11). Bacterial suspensions with a standardized initial density of 107 CFU/ml (determined turbidometrically) were prepared in minimum growth uptake buffer (NaCl, 100 mM; KCl, 7 mM; NH4Cl, 50 mM; Na2HPO4, 0.4 mM; Tris base, 52 mM; glucose, 0.2%; adjusted to pH 7.5 with HCl). They were then exposed to 2 μg/ml EtBr. The increase in fluorescence as EtBr entered the cells was recorded fluorometrically with a spectrofluorometer at 30°C (excitation and emission wavelengths [λ] were set at 530 nm and 630 nm, respectively).
EtBr efflux was determined from bacterial suspensions first exposed to EtBr (2 μg/ml) for 20 min at 37°C in the presence of reserpine (10 μg/ml) in order to maximize the loading of EtBr into the bacteria. The cells were pelleted by centrifugation and resuspended in fresh minimal growth medium (without reserpine). EtBr efflux from bacterial cells was measured as a decrease in fluorescence. Reserpine inhibition of EtBr loss was determined in experiments where reserpine (10 μg/ml) was also added to the minimal growth medium.
Pharmacokinetic modeling.
Changes in drug concentration in the sera of infected mice over time were analyzed by the NPEM2 approach (26) with one- and two-compartment open models with first-order elimination from the central compartment and a first-order input. The most appropriate models for fitting pharmacokinetic data were determined using model selection criteria based on a modified form of Akaike's information criterion (29). The inverse of the standard deviation of measured mean concentrations was used for weighting the pharmacokinetic data. Population pharmacokinetic parameters, including drug clearance from the serum compartment (liters/h), the volume of the central compartment (liters), the first-order absorption rate constant (h−1), and the first-order transfer constants between the central and peripheral compartments (h−1), were estimated.
Statistical analysis.
The relationship between the exposure to levofloxacin and the bacterial density in the thigh muscles of infected mice was evaluated by an inhibitory sigmoid Emax dose-response model using the identification module of the ADAPT II package of programs of D'Argenio and Schumitzky (8). Experimental data were weighted by the inverse of the observation variance. The significance of differences between bacterial densities in groups that received the same total dose of levofloxacin in one, two, or four divided doses were evaluated by comparison of group means and their associated variances. A difference was considered statistically significant at a P value of <0.05. All statistical tests were determined using the software program STATISTICA (StatSoft Inc., Tulsa, OK).
RESULTS
Levofloxacin MIC and MBC determinations for S. pneumoniae.
The geometric and arithmetic dilution series MICs and geometric dilution series MBCs for the S. pneumoniae strain (AMC-058) used in these studies were 0.5 to 1.0 mg/liter, 0.6 mg/liter, and 1.0 mg/liter, respectively, in cation-adjusted Mueller-Hinton broth and 5% unheparinized lysed horse blood. MIC and MBC determinations were performed in quadruplicate (in duplicate by two independent testers) at least three times.
Levofloxacin single-dose pharmacokinetics in infected mice.
Mice were infected with S. pneumoniae and separated into different treatment groups, each consisting of four or more animals. Two hours after bacterial inoculation, mice in each group were treated with single incremental doses from 0 to 350 mg/kg of levofloxacin (i.p.). Levofloxacin single-dose dose range pharmacokinetics studies were performed twice. The calculated clearance and serum half-life of levofloxacin in infected mice were 1.91 liters/h/kg and 1.5 h, respectively. The intercompartmental transfer rate constants between the central blood compartment and the peripheral compartment from population PK analysis were 1.3 h−1 and 0.86 h−1. The absorption rate constant was 35 h−1. Levofloxacin displayed linear pharmacokinetics over the range of doses tested.
Single-dose dose-response studies in a murine thigh infection model of S. pneumoniae.
Mice infected with known bacterial densities of AMC-058 were treated with a range of different doses of levofloxacin. An inhibitory sigmoid Emax dose-response model was used to determine the exposure-therapeutic-response relationship between the administered dose of drug and the number of surviving bacteria. Figure 1 demonstrates the dose-response relationships obtained for S. pneumoniae. The general dose-response relationship of bacterial-density (log10 CFU) changes relative to drug exposure is given by the following relationship:
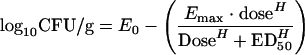 |
where, E0 is the bacterial density found in the untreated animals at the time of sacrifice, Emax is the maximal observed kill, H is the relative steepness of the dose-effect relationship, and ED50 is the exposure measure at which half-maximal kill (effect) is observed. The “stasis dose” was defined as the dose of drug that resulted in no net change in bacterial density (log10 CFU/thigh) at experiment termination from the starting bacterial burden (determined at therapy initiation). A dose range experiment was performed (Fig. 1A). Inhibitory sigmoid Emax modeling showed that the AUC/MIC ratio resulting in half the maximal response was 51.2. AUC/MIC and free-drug AUC/MIC ratios generating stasis (return to baseline in CFU/ml at the infection site), as well as 1.0, 2.0, and 3.0 log10 CFU/ml declines from stasis, were 15, 34, 59, and 111 and 11, 24, 41, and 78, respectively.
FIG. 1.

(A) Dose-response relationship between levofloxacin and changes in S. pneumoniae (strain AMC-058) density in thigh muscles (mean ± 1 standard deviation) following ∼108-CFU/thigh inoculation. In this evaluation, all doses were given on a once-daily basis. In neither experiment (A and B) were any drug-resistant mutants recovered by direct plating on levofloxacin-contaning plates. (B) Dose fractionation experiments were performed (QD refers to the whole dose given once; BID refers to half the dose given every 12 h twice; QID refers to one-quarter of the dose given every 6 h). The data are displayed with the AUC/MIC ratio as the independent variable. The peak/MIC ratio and time > MIC are also evaluated as independent variables, but AUC/MIC displayed the best fit of the model to the data. The inoculum was 6.5 log10 CFU/thigh. When tested by analysis of variance, no difference was seen between q24h (the whole dose once), q12h (a half dose q12h), and q6h (a quarter dose q6h) administration of the same total daily dose.
Figure 1B demonstrates that the AUC/MIC ratio is the pharmacodynamically linked variable for Streptococcus pneumoniae AMC-058 for this fluoroquinolone. Doses were fractionated (the whole dose once, a half dose q12h × 2, and a quarter dose q6h × 4) at doses producing circa 20%, 50%, and 80% of the maximal effect. No significant differences (P > 0.05) in effect were seen based on the schedule of administration, indicating that the AUC/MIC ratio was the pharmacodynamically linked variable.
In vivo isolation of levofloxacin-resistant S. pneumoniae mutants.
Attempts in both experiments shown in Fig. 1A and B to isolate levofloxacin-resistant S. pneumoniae mutants of AMC-058 in vivo failed at any exposure.
In vivo isolation of ciprofloxacin-resistant S. pneumoniae mutants.
Thigh homogenates from animals treated with ciprofloxacin were plated on plates containing ciprofloxacin. Unlike with levofloxacin, ciprofloxacin-resistant S. pneumoniae mutants from the in vivo experiments were isolated on plates containing 2 × and 4 × MIC following treatment with a ciprofloxacin exposure equal to an AUC/MIC ratio of 80/1 (50 mg/kg q12h).
Phenotypic characterization of isolated in vivo ciprofloxacin-resistant S. pneumoniae mutants.
Mutations underlying quinolone resistance can be inferred by profiling MICs of parent strains and resistant isolates to different quinolones, each with different octanol-water partition coefficients (log10 P) and which preferentially bind to different topoisomerase targets. Thus, changes in the quinolone MICs for the parent strains versus mutant strains give qualitative information on the mutations that underlie the resistance phenotype based on the hydrophilicity of the quinolones and differences in the test compound target (gyrA or parC) affinity. Efflux pump-mediated resistance is further characterized by the effect of the plant alkaloid reserpine, which is an inhibitor of multidrug efflux pumps, on the MIC. An exposure to 10 μg/ml of reserpine was employed.
Ciprofloxacin (a hydrophilic quinolone that preferentially binds to ParC in S. pneumoniae), levofloxacin (a hydrophobic quinolone that preferentially binds to S. pneumoniae ParC), and sparfloxacin (which is more hydrophobic than levofloxacin and preferentially binds to S. pneumoniae GyrA) were used to compare the susceptibilities of the parent S. pneumoniae AMC-058 strain and RC2 (a mutant derived from an in vivo experiment, isolated from a 2 × MIC ciprofloxacin-containing plate) (Table 1). A reserpine-reversible decrease in the susceptibility of the RC2 and RC4 (a mutant isolated from an in vivo experiment from a 4 × MIC ciprofloxacin-containing plate) strains to ciprofloxacin was observed, while levofloxacin and sparfloxacin susceptibilities were unchanged for S. pneumoniae AMC-058, RC2, and RC4. These MICs were determined using a geometric (twofold) dilution series.
TABLE 1.
Phenotypic evaluation of mutations underlying RC2 and RC4, single-passage ciprofloxacin-resistant mutants
| Druga | log10P | MIC (mg/liter)
|
||
|---|---|---|---|---|
| AMC-058 | RC2 | RC4b | ||
| Ciprofloxacin + reserpine | 1.25 | 0.6/0.6 | 3.5/1.0 | >32/8 |
| Levofloxacin + reserpine | 0.48 | 0.6/0.6 | 0.8/0.6 | 2/1 |
| Sparfloxacin + reserpine | 0.36 | 0.2/0.1 | 0.2/0.2 | 0.5/0.5 |
10 mg/liter reserpine was added to the fluoroquinolone MIC test medium.
Geometric dilution MICs were determined once in duplicate.
PCR amplification and DNA sequencing.
The entire open reading frames of all targets (gyrA/B and parC/E) for the parent strain (AMC-058), as well as RC2 and RC4, were sequenced in order to determine if point mutations were involved in resistance development. Sequence analysis of gyrA, parC, gyrB, and parE showed no difference between the parent strain, AMC-058, and the daughter strain, RC2. These results, coupled with the decrease in the ciprofloxacin MIC in the presence of reserpine (Table 1), imply that RC2 is a first-stage ciprofloxacin-resistant mutant with no topoisomerase mutations whose resistance phenotype is efflux pump mediated.
A mutation in RC4 was found in parC Ser79Tyr. This strain's MIC was also decreased with exposure to ciprofloxacin and reserpine, indicating a combination of pump overexpression and the identified target mutation.
Uptake and efflux of EtBr by S. pneumoniae whole cells.
Experiments on EtBr uptake into and efflux from whole S. pneumoniae cells were designed to differentiate efflux pump activities in the parent strain, AMC-058, and the daughter strain, RC2. S. pneumoniae laboratory strains, R6 (the parent strain) and R6N (an isogenic derivative of R6 that was transformed with DNA from a laboratory isolate exposed to norfloxacin that subsequently overexpressed pmrA, an efflux pump present in S. pneumoniae), were also characterized and served as controls for the efflux experiments. Experiments were conducted comparing EtBr influx and efflux in isogenic pairs of the mother-daughter pairs AMC-058-RC2 and R6-R6N. EtBr efflux in R6N was more rapid than in R6 (data not shown). This result was expected, because R6N is an overexpresser of pmrA, an efflux pump that can be inhibited by reserpine (4). The well-defined characteristics of strains R6 and R6N made them suitable controls in efflux studies for the in vivo clinical-isolate parent, AMC-058, and the daughter strain, RC2. EtBr efflux studies of S. pneumoniae AMC-058 and RC2 cells demonstrated that RC2 showed more rapid efflux of ethidium bromide than AMC-058 (Fig. 2). The addition of reserpine decreased the rate of EtBr efflux in both AMC-058 and AMC-058-RC2. These data suggest that RC2 possesses constitutive overexpression of an efflux pump relative to the basal expression seen in AMC-058.
FIG. 2.

Comparison of efflux rates of EtBr from the parent strain (AMC-058) and its isogenic efflux-mediated-resistance daughter (RC2). The change in fluorescence is normalized to the initial observed levels for direct comparison of initial EtBr efflux rates. The time scale is in seconds. The error bars indicate standard deviations.
In vitro repetition of the in vivo experiment.
To obtain in vitro data supporting our animal studies showing that the initial selection of low-level resistance with ciprofloxacin was due to overexpression of an efflux pump rather than a topoisomerase mutation, we exposed AMC-058 to concentrations of ciprofloxacin at 2 × and 4 × the MIC. The mutation frequencies at 2 × the MIC were 1/104.71 ± 0.42 (n = 5). At 4 × the MIC, the mutation frequency for ciproflocacin was 1/105.69 ± 0.53 (n = 4). Among 21 clones showing ciprofloxacin MICs of ≥4 mg/liter, 12 clones were selected at random for further testing. All isolates showed a >4-fold fall in MIC with exposure to reserpine. In 11/12, no changes were seen in the quinolone resistance-determining region for gyrA/B or parC/E. In one clone, a parC Ser79Tyr substitution was found, along with reserpine responsiveness, exactly mimicking the in vivo experiment for ciprofloxacin selection.
Treatment of AMC-058 and RC2 infections with ciprofloxacin and levofloxacin.
Following complete phenotypic characterization, S. pneumoniae AMC-058 and RC2 were used as the infection inocula in another mouse thigh study in which levofloxacin and ciprofloxacin treatments were conducted in parallel for both strains. Treatment was with pharmacodynamically equivalent regimens of ciprofloxacin or levofloxacin. Mice were exposed to quinolone AUC/MIC ratios of 0, 40/1, and 80/1. The results from these studies are shown in Fig. 3 and 4. Total-population and resistant-population densities for AMC-058 (Fig. 3) and RC2 (Fig. 4) are presented for both ciprofloxacin and levofloxacin. When no resistant mutants were isolated, the data point was plotted at the limit of detection.
FIG. 3.

In vivo outcome study results for S. pneumoniae AMC-058 with pharmacodynamically equivalent exposures to ciprofloxacin (CIP) and levofloxacin (LVX). Total bacterial population densities (black) enumerated on drug-free plates and drug-resistant-subpopulation (CIPr and LVXr) densities (gray) isolated on 3 × MIC-containing plates are shown for ciprofloxacin (A) and levofloxacin (B). Levofloxacin-exposed organisms were plated on levofloxacin-containing plates, and ciprofloxacin-exposed organisms were plated on ciprofloxacin-containing plates. In panel B, no clones resistant to levofloxacin were isolated; this is displayed by plotting at the detection limit. The error bars indicate standard deviations.
FIG. 4.

In vivo outcome study results for S. pneumoniae RC2 exposed to pharmacodynamically equivalent exposures of ciprofloxacin (CIP) and levofloxacin (LVX). Total bacterial population densities (black) enumerated on drug-free plates and drug-resistant-subpopulation (CIPr and LVXr) densities (gray) isolated on 3 × MIC-containing plates are shown for ciprofloxacin (A) and levofloxacin (B). Levofloxacin-exposed organisms were plated on levofloxacin-containing plates, and ciprofloxacin-exposed organisms were plated on ciprofloxacin-containing plates. The error bars indicate standard deviations.
Exposure of strain AMC-058 to ciprofloxacin resulted in ciprofloxacin resistance selection at both drug exposures (Fig. 3). However, levofloxacin treatment was not permissive for emergence of resistance to levofloxacin at either exposure. Use of ciprofloxacin-containing plates for selection did allow selection under these circumstances. In contrast, exposure of RC2 to ciprofloxacin or levofloxacin (Fig. 4) resulted in the selection of both ciprofloxacin- and levofloxacin-resistant mutants upon treatment with either drug at both drug exposures.
DISCUSSION
In the initial evaluation, we demonstrated that the AUC/MIC ratio is the pharmacodynamically linked variable for fluoroquinolones (specifically, for levofloxacin) and Streptococcus pneumoniae, as had also been demonstrated by Scaglione et al. (24). This linkage has been shown for this drug class and the pneumococci by others (1). This allowed us to pursue a strategy of administering a large range of doses on a once-daily basis and to evaluate the effect on the total population, as well as to identify an exposure that might suppress the amplification of a resistant mutant subpopulation.
After extensive effort, we were unable to isolate any mutants in mice that were resistant to levofloxacin at the level of twice the baseline MIC, irrespective of the exposure employed. A report by Lacy and colleagues (14) based on an in vitro pharmacodynamic model system was consistent with this finding. In that report, organisms that were treated with suboptimal levofloxacin regimens did not have altered MICs, whereas strains with increased ciprofloxacin MICs were rapidly isolated with the ciprofloxacin control regimen. However, given the report by Chen et al. (5), we were puzzled when we were unable to isolate organisms with elevated MICs to levofloxacin, especially as the total organism population burden approximated 8.5 log10 CFU/g at the primary infection site at the initiation of therapy. It should be appreciated that, on average, there are approximately 6 g of mouse thigh. As we were employing both mouse thighs as one statistical entity, the total population burden of Streptococcus pneumoniae exceeded 9.5 log10 CFU.
Because of our inability to isolate resistant mutants with levofloxacin, we decided to evaluate ciprofloxacin in this in vivo infection model. While levofloxacin did not allow selection of resistant mutants even with AUC/MIC ratios as low as 13 (free-drug AUC/MIC ratio = 9), we were readily able to select mutants in vivo with ciprofloxacin. Indeed, the frequency with which this occurred was approximately 1/3,000 to 1/10,000 colonies.
We could isolate many clones that would grow on plates containing 2 × the baseline ciprofloxacin MIC. We were also able to isolate a single clone on a plate that contained ciprofloxacin at 4 × the baseline MIC. When we examined the mechanisms mediating resistance, we found that the isolate tested from plates containing 2 × the baseline MIC of ciprofloxacin contained no mutations in parC/E or gyrA/B. There was a drop, however, in the MIC when it was determined in the presence of reserpine, a known inhibitor of pneumococcal efflux pumps. The single clone that was isolated from the plate containing 4 × the baseline ciprofloxacin MIC concentration also responded to reserpine with a fall in the MIC (Table 1), suggesting that this isolate had two resistance mechanisms, the pump overexpression and Ser79Tyr of parC. This substitution is a well-known mutation conferring some fluoroquinolone resistance, particularly for drugs that bind preferentially to ParC/E.
To provide further data for the presence of pump overexpression, we examined how the parent strain (AMC-058) and a mutant (RC2) performed when challenged with ethidium bromide, a known substrate for the pmrA pump and homologous efflux pumps in Staphylococcus aureus and Bacillus subtilis. As can be seen in Fig. 2, the mutant strain pumps this molecule more efficiently than the parent strain. The combination of ethidium bromide pumping and MIC decrement in the presence of reserpine, coupled with the inability to identify any changes in any of the targets of quinolone binding, provides strong evidence that the mutants isolated under in vivo pressure on plates containing twice the baseline ciprofloxacin MIC represent, in the main, pure pump overexpression.
This finding may also explain the difference noted between ciprofloxacin and levofloxacin, in that we could not select these mutants with levofloxacin, but it was straightforward to do so with ciprofloxacin. Others have demonstrated that ciprofloxacin is better pumped than levofloxacin in Staphylococcus aureus (25), likely on the basis of its hydrophilicity (while this was demonstrated with norA, this pump is in the same major facilitator superfamily as pmrA and is the homologue of pmrA in staphylococci). Table 1 indicates that the log10 P for ciprofloxacin is 1.25, while that of levofloxacin is 0.48. Because of the logarithmic transformation, this indicates that ciprofloxacin preferentially partitions into the water phase 5.9-fold more than levofloxacin, perhaps providing the explanation for ciprofloxacin's increased propensity to allow the emergence of resistance, particularly in the absence of target site mutations.
Importantly, this also indicates that pumps play a central role in the pathway to emergence of resistance to fluoroquinolones for Streptococcus pneumoniae. No target site mutations were identified in the tested resistant clone isolated from the 2 × MIC plate from the in vivo experiment. This was also the finding in 11/12 instances for the clones randomly chosen from the in vitro selection experiment. The single clone with a change in the quinolone resistance-determining region had the Ser79Tyr change seen in the single clone isolated on the 4 × MIC plate from the in vivo experiment. All the strains tested had a decline in the ciprofloxacin MIC with the addition of reserpine.
This may indicate that there is interaction between pump overexpression and the probability of obtaining a target mutation. For ciprofloxacin, the MIC rise allows more rounds of replication per unit time for strains that are pump overexpressed. Alternatively, the generation of a target mutation may have occurred randomly. Arguing against the latter hypothesis is the fact that the total population burden (circa 109 CFU) did not exceed the product of the frequencies with which each is estimated to occur (circa 1/105 for pump overexpression and an estimated 1/108 to 1/109 for a target mutation). An alternative explanation is that error-prone replication mechanisms were responsible for the target site mutations.
These findings have public health importance. When we took the RC2 mutant and placed it back in the infection model, we were able to isolate resistant mutants with both levofloxacin and ciprofloxacin therapies (Fig. 4). This implies that with pump overexpression, the minor shift in the MIC (0.6 to 0.8 mg/liter) (Table 1) may be sufficient to allow generation of a target mutation. It should be noted that we were unable to isolate a single mutant on many plates with a population burden of approximately 108 to 109 colonies of AMC-058 (the wild-type isolate) when levofloxacin was the selective pressure. When the RC2 strain was employed (a pump-overexpressed strain), we were able to isolate >100 colonies on levofloxacin-containing plates (Fig. 4), even when obtained from mice not receiving drugs (circa 1 mutant/106). Under levofloxacin pressures of AUC/MIC ratios of 40 and 80, there were resistant mutants isolated at the rate of approximately 1/104.5 to 1/103.5. This is likely due to the amplification of the preexisting resistant subpopulation, as we have described previously in this model with Pseudomonas aeruginosa (13).
Ciprofloxacin has lost patent protection, and generic versions are available. There exists the possibility that admission of this drug to the formulary (because of price) in a managed care setting for community-acquired respiratory tract infections will speed the loss of susceptibility of Streptococcus pneumoniae to the whole class of fluoroquinolones. Other data from our laboratory have demonstrated that another, newer agent from this class (gemifloxacin) also did not allow selection of mutants (22). Consequently, ciprofloxacin appears to occupy a special place in allowing emergence of resistance to fluoroquinolones among pneumococci and may explain the findings of the Chen et al. study (5), as >95% of fluoroquinolone use across Canada during the time of strain collection for the Chen et al. study was ciprofloxacin.
Traditionally, from a public health perspective, practitioners have been urged not to employ newer drugs, but rather to use older, cheaper agents for the therapy of community-acquired infections. Indeed, this recommendation makes sense, as Garcia-Rey et al. (10) have shown an association between the emergence of resistance to macrolides and the use of newer members of the class that was not seen with erythromycin usage. In the case of beta-lactams, the United Kingdom and the Scandinavian countries were among the areas that did not widely adopt oral cephalosporins over penicillins for use in community-acquired infections. These areas maintain high rates of beta-lactam susceptibility, even today.
Here, however, we seem to have the inverse. An early member of the class (ciprofloxacin) seems to have a special place in selecting pump-overexpressed mutants, probably because of its hydrophilicity. Once these efflux pumps are overexpressed, the mutants are able to become resistant to newer members of the class. Use of newer agents of this class (levofloxacin, gemifloxacin, and likely moxifloxacin, although we did not study the last agent in the mouse thigh model) did not allow selection of resistance, even at very suboptimal drug exposures (a free-drug AUC/MIC of about 9). It would seem prudent for ciprofloxacin use to be minimized for patients with community-acquired respiratory tract infections. It clearly has a place for patients with serious infections by gram-negative bacteria. Other, newer members of this class (e.g., moxifloxacin, gatifloxacin, gemifloxacin, and levofloxacin) should be used preferentially for community-acquired respiratory tract infections. Further, prudence would also dictate that the use of this class of agents be minimized for less severe infections, such as run-of-the-mill flare ups of chronic obstructive pulmonary disease. By so doing, we may help preserve the utility of this class of agents for pneumococcal pneumonia.
Acknowledgments
This work was supported in part by AI 055821 and in part by Ortho-McNeil Pharmaceutical.
REFERENCES
- 1.Andes, D., and W. A. Craig. 2002. Pharmacodynamics of the new fluoroquinolone gatifloxacin in murine thigh and lung infection models. Antimicrob. Agents Chemother. 46:1665-1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balas, D., E. Férnandez-Moreira, and A. G. de la Campa. 1998. Molecular characterization of the gene encoding the DNA gyrase A subunit of Streptococcus pneumoniae. J. Bacteriol. 180:2854-2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartlett, J. G., R. F. Breiman, L. A., Mandell, and T. M. File, Jr. 1998. Community-acquired pneumonia in adults: guidelines for management. Clin. Infect. Dis. 26:811-838. [DOI] [PubMed] [Google Scholar]
- 4.Brenwald, N. P., M. J. Gill, and R. Wise. 1998. Prevalence of a putative efflux mechanism among fluoroquinolone-resistant clinical isolates of Streptococcus pneumoniae. Antimicrob. Agents Chemother. 42:2032-2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen, D. K., A. McGeer, J. C. de Azavedo, and D. E Low. 1999. Decreased susceptibility of Streptococcus pneumoniae to fluoroquinolones in Canada. N. Engl. J. Med. 341:233-239. [DOI] [PubMed] [Google Scholar]
- 6.Craig, W. A. 1998. Pharmacokinetic/pharmacodynamic parameters: rationale for antibiotic dosing of mice and men. Clin. Infect. Dis. 26:1-10. [DOI] [PubMed] [Google Scholar]
- 7.Craig, W. A. 2001. Does the dose matter? Clin. Infect. Dis. 33:S233-S237. [DOI] [PubMed] [Google Scholar]
- 8.D'Argenio, D. Z., and A. Schumitzky. 1997. ADAPT II. A program for simulation, identification, and optimal experimental design. University of Southern California, Los Angeles.
- 9.Eagle, H., R. Fleischman, and M. Levy. 1953. Continuous versus discontinuous therapy with penicillin: the effect of interval between injections on therapeutic efficacy. N. Engl. J. Med. 248:481-488. [DOI] [PubMed] [Google Scholar]
- 10.Garcia-Rey C., L. Aguilar, F. Baquero, J. Casal, and R. Dal-Re. 2002. Importance of local variations in antibiotic consumption and geographical differences of erythromycin and penicillin resistance in Streptococcus pneumoniae. J. Clin. Microbiol. 40:159-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gill, M. J., N. P. Brenwald, and R. Wise. 1999. Identification of an efflux pump gene, pmrA, associated with fluoroquinolone resistance in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 43:187-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldstein, E. J., and S. M. Garabedian-Ruffalo. 2002. Widespread use of fluoroquinolones versus emerging resistance in pneumococci. Clin. Infect. Dis. 35:1505-1511. [DOI] [PubMed] [Google Scholar]
- 13.Jumbe, N., A. Louie, R. Leary, W. Liu, M. R. Deziel, V. H Tam, R. Bacchawat, C. Freeman, J. B. Kahn, K. Bush, M. N. Dudley, M. H. Miller, and G. L. Drusano. 2003. Application of a mathematical model to prevent amplification of antibiotic-resistant bacterial populations during therapy. J. Clin. Investig. 112:275-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lacy, M. K., W. Lu, X. Xu, P. R. Tessier, D. P. Nicolau, R. Quintiliani, and C. H. Nightingale. 1999. Pharmacodynamic comparisons of levofloxacin, ciprofloxacin and ampicillin against Streptococcus pneumoniae in an in vitro model of infection. Antimicrob. Agents Chemother. 43:672-677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leggett, J. E., S. Ebert, B. Fantin, and W. A Craig. 1990. Comparative dose-effect relations at several dosing intervals for beta-lactam, aminoglycoside and quinolone antibiotics against gram-negative bacilli in murine thigh-infection and pneumonitis models. Scand. J. Infect. Dis. 74:179-184. [PubMed] [Google Scholar]
- 16.Muñoz, R., M. Bustamante, and A. G. de la Campa. 1995. Ser-127-to-Leu substitution in the DNA gyrase B subunit of Streptococcus pneumoniae is implicated in novobiocin resistance. J. Bacteriol. 177:4166-4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muñoz, R., and A. G. de la Campa. 1996. ParC subunit of DNA topoisomerase IV of Streptococcus pneumoniae is a primary target of fluoroquinolones and cooperates with DNA gyrase A subunit in forming resistance phenotype. Antimicrob. Agents Chemother. 40:2252-2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.NCCLS. 1997. NCCLS publication M7-A4. NCCLS, Wayne, PA.
- 19.Niederman, M. S., L. A. Mandell, A. Anzueto, J. B. Bass, W. A. Broughton, C. D. Campbell, N. Dean, T. File, M. J. Fine, P. A. Gross, F. Martinez, T. J. Marrie, J. F. Plouffe, J. Ramirez, G. A. Sarosi, A. Torres, R. Wilson, and V. L. Yu. 2001. Guidelines for the management of adults with community-acquired pneumonia. Am. J. Respir. Crit. Care Med. 163:1730-1754. [DOI] [PubMed] [Google Scholar]
- 20.Pan, X.-S., and L. M. Fisher. 1996. Cloning and characterization of the parC and parE genes of Streptococcus pneumoniae encoding DNA topoisomerase IV: role in fluoroquinolone resistance. J. Bacteriol. 178:4060-4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pan, X.-S., J. Ambler, S. Mehtar, and M. Fisher. 1996. Involvement of topoisomerase IV and DNA gyrase as ciprofloxacin targets in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 40:2321-2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Puthisigamani, M., G. L. Drusano, W. Liu, V. H. Tam, M. R. Deziel, and A. Louie. Gemifloxacin pharmacokinetic and pharmacodynamic studies using isogenic Streptococcus pneumoniae wild-type +/− efflux pumps +/− a parC mutation in a murine thigh infection model, abstr. 471. Prog. Abstr. Infect. Dis. Soc. Am. 39th Annu. Meet. Infectious Disease Society of America, San Francisco, Calif.
- 23.Sahm, D. F, J. A. Karlowsky, L. J. Kelly, I. A. Critchley, M. E. Jones, C. Thornsberry, Y. Mauriz, and J. Kahn. 2001. Need for annual surveillance of antimicrobial resistance in Streptococcus peumoniae in the United States: 2 year longitudinal analysis. Antimicrob. Agents Chemother. 45:1037-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scaglione, F., J. W. Mouton, R. Mattina, and F. Fraschini. 2003. Pharmacodynamics of levofloxacin and ciprofloxacin in a murine pneumonia model: peak concentration/MIC versus area under the curve/MIC ratios. Antimicrob. Agents Chemother. 47:2749-2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmitz, F.-J., A. C. Fluit, M. Luckefahr, B. Engler, B. Hoffmann, J. Verhoef, H.-P. Heinz, U. Hadding, and M. E. Jones. 1998. The effect of reserpine, an inhibitor of multidrug efflux pumps on the in vitro activities of ciprofloxacin, sparfloxacin and moxifloxacin against clinical isolates of Staphylococcus aureus. J. Antimicrob. Chemother. 42:807-810. [DOI] [PubMed] [Google Scholar]
- 26.Schumitzky, A., R. Jelliffe, and M. van Guilder. 1994. NPEM: a program for pharmacokinetic population analysis. Clin. Pharmacol. Ther. 55:163. [Google Scholar]
- 27.Wong, F. A., S. J. Juzwin, and S. C. Flor. 1997. Rapid stereospecific high-performance liquid chromatographic determination of levofloxacin in human plasma and urine. J. Pharm. Biomed. Anal. 15:765-771. [DOI] [PubMed] [Google Scholar]
- 28.Wright, D. H., V. K. Herman, F. N. Konstantinides, and J. C. Rotschafer. 1998. Determination of quinolone antibiotics in growth media by reversed-phase high-performance liquid chromatography. J. Chromatogr. B 709:97-104. [DOI] [PubMed] [Google Scholar]
- 29.Yamaoko, K., T. Nakagawa, and T. Uno. 1978. Application of Akaike's information criterion in the evaluation of linear pharmacokinetic equations. J. Pharmacokinet. Biopharm. 6:165-175. [DOI] [PubMed] [Google Scholar]