

Abstract
The human adenovirus type 5 (Ad5) E1B 55-kDa protein is required for selective nuclear export of viral late mRNAs from the nucleus and concomitant inhibition of export of cellular mRNAs in HeLa cells and some other human cell lines, but its contributions(s) to replication in normal human cells is not well understood. We have therefore examined the phenotypes exhibited by viruses carrying mutations in the E1B 55-kDa protein coding sequence in normal human fibroblast (HFFs). Ad5 replicated significantly more slowly in HFFs than it does in tumor cells, a difference that is the result of delayed entry into the late phase of infection. The A143 mutation, which specifically impaired export of viral late mRNAs from the nucleus in infected HeLa cells (R. A. Gonzalez and S. J. Flint, J. Virol. 76:4507-4519, 2002), induced a more severe defect in viral mRNA export in HFFs. This observation indicates that the E1B 55-kDa protein regulates mRNA export during the late phase of infection of normal human cells. Other mutants exhibited phenotypes not observed in HeLa cells. In HFFs infected by the null mutant Hr6, synthesis of viral late mRNAs and proteins was severely impaired. Such defects in late gene expression were the result of inefficient progression into the late phase of infection, for viral DNA synthesis was 10-fold less efficient in Hr6-infected HFFs than in cells infected by Ad5. Similar, but less severe, defects in viral DNA synthesis were induced by the insertion mutation H224, which has been reported to inhibit binding of the E1B 55-kDa protein to p53 (C. C. Kao, P. R. Yew, and A. J. Berk, Virology 179:806-814, 1990).
During the late phase of human adenovirus type 5 (Ad5) infection, cellular protein synthesis is severely inhibited, while viral late proteins are made in large quantities (3, 6). Such preferential expression of viral genes is the result of posttranscriptional regulatory mechanisms: several viral gene products, including VA-RNA1 and the L4 100-kDa protein, contribute to selective translation of viral late mRNAs (see references 18, 64, and 85 for reviews), while the E1B 55-kDa and E4 Orf 6 proteins induce selective export of these mRNAs from the nucleus (reviewed in references 25, 33, and 39). In infected cells, these last two early proteins form a complex (84) that has been implicated in regulation of mRNA export (12, 19). Indeed, the E1B 55-kDa and E4 Orf 6 proteins colocalize to specific sites within infected cell nuclei, the peripheral zones of replication centers (70). Transcription of viral late genes and at least initial processing of viral pre-mRNAs take place at these same sites (4, 14, 74, 75). Mutations that prevent synthesis of the E4 Orf 6 protein or reduce binding of this to the E1B 55-kDa protein (83) result in both mislocalization of the E1B 55-kDa protein and defects in export of viral late mRNAs (39, 70). These properties indicate that E4 Orf 6 protein-dependent recruitment of the E1B protein to the specialized nuclear sites at which viral late pre-mRNAs are synthesized promotes selective export of the processed mRNAs. The observation that the accumulation of viral mRNAs at enlarged interchromatin granules, which form in infected cells as the late phase progresses, correlates with efficient late mRNA export (4, 13) provides further support for the view that efficient mRNA export is intimately coupled to the organization of infected cell nuclei. However, the molecular basis of such coupling remains unknown, nor has the cellular export pathway by which viral late mRNAs are transported from the nucleus to the cytoplasm been identified.
The E1B 55-kDa protein contains a leucine-rich nuclear export signal (NES) that is recognized by the cellular exportin 1 export receptor and mediates shuttling of the viral protein when it is synthesized either alone or in Ad5-infected cells (26, 58). It has also been reported that the E4 Orf 6 protein contains a similar NES necessary for exportin-dependent shuttling of the E1B 55-kDa-E4 Orf 6 protein complex and sufficient to allow export when fused to an heterologous protein (24). The specific inhibitor of exportin 1, leptomycin B, has been observed to inhibit shuttling of this E4 protein substantially (78). On the other hand, it has also been reported that the E4 Orf 6 NES failed to direct efficient export of a heterologous protein and that export of this E4 protein is insensitive to leptomycin B (26, 58). Regardless of whether the E4 Orf 6 protein leaves the nucleus via the exportin 1 pathway, there is clear evidence that this export receptor does not mediate export of viral late mRNAs: the inhibition of exportin 1 by leptomycin B or an NES containing peptide had no effect on synthesis of viral late proteins (15, 78) or viral late mRNA export (34).
Mutants of Ad5 null for expression of the E1B 55-kDa protein coding sequence exhibit conditional phenotypes in HeLa cells and other established lines of human cells. In the first place, replication of such mutants is temperature dependent, with significantly more-severe defects observed at 32.5 to 33°C than at 37°C and efficient growth at 38.5 to 39.5°C (41, 46, 51, 60, 86). Secondly, the yield of the mutants is increased by a factor of 7 when HeLa cells are infected in S phase compared to the yield from cells that are in G1 phase at the time of infection (40). Both temperature-dependent and cell cycle-restricted replication appear to be the result of defects in export of viral mRNAs: late mRNA export and synthesis of late proteins are substantially impaired when E1B mutant virus-infected cells are maintained at low temperatures (46, 60, 96) and when cells are infected in G1 phase (42). Furthermore, the cell cycle-dependent replication of these mutants is substantially relieved at 39°C (41). These properties imply that cellular components that are synthesized, active, or present at a sufficient concentration only under specific conditions (e.g., elevated temperature) can compensate, at least partially, for the absence of the E1B 55-kDa protein by facilitating export of viral late mRNAs.
Reproduction of mutants that cannot direct synthesis of the E1B 55-kDa protein also varies with the identity of the host cells (8, 41, 44, 46, 82, 94). In some established lines of human cells, including A549, the yield of these mutants, compared to that of the wild type, is reduced only two- to fivefold, whereas in others, including H1299 and U2OS, growth defects of several orders of magnitude have been observed. The molecular mechanisms responsible for these differences in host cell permissiveness are not fully understood (see references 21 and 45 for reviews) and appear to be cell type specific (28). However, in at least one case (H1299 cells), defects in viral late gene expression have been shown to make a major contribution to the low replication efficiency of an E1B 55-kDa protein null mutant (46).
The conditional replication and mRNA export regulation phenotypes exhibited by viruses carrying mutations in the E1B 55-kDa protein coding sequence in established lines of human cells raise the question of whether regulation of mRNA export is necessary for efficient growth of Ad5 in normal human cells. To address this issue, we have examined the phenotypes exhibited by the E1B 55-kDa protein null mutant Hr6 (51, 96) and a set of mutants carrying insertions in this gene (97, 98) in normal human foreskin fibroblasts. These normal host cells have allowed identification of defects in entry into the late phase of the infectious cycle associated with certain mutations, phenotypes that are not observed in transformed human host cells.
MATERIALS AND METHODS
Cells and viruses.
Maintenance of HeLa and 293 cells in monolayer cultures and propagation and plaque assay of Ad5 and mutant viruses carrying alterations in the E1B 55-kDa protein coding sequence were as described previously (39). Primary human foreskin fibroblasts (HFFs), the gift of C. Patterson and T. Shenk, were maintained in monolayer cultures in Dulbecco's modified Eagle's medium supplemented with 10% (vol/vol) fetal calf serum (Gibco-Invitrogen Corp.) for no more than 15 passages. Unless otherwise stated, HFFs were infected with Ad5 or E1B mutant viruses at 30 PFU/cell.
Viral yields.
To examine the kinetics of Ad5 replication, HFFs and HeLa cells at 90% confluence were infected at 0.1 or 10.0 PFU/cell, and infected cells were harvested after increasing periods of infection. Cells were washed in phosphate-buffered saline (Gibco-Invitrogen Corp) and suspended in 0.25 M Tris-HCl, pH 7.4, containing 0.15 M KCl, 5 mM KCl, 10 mM MgCl2, and 0.1% (vol/vol) dextrose. Following five cycles of freezing and thawing, cellular debris was removed by centrifugation. Concentrations of extracted virions were determined by plaque assay on HeLa cells. To compare yields of Ad5 and E1B mutant viruses, HFFs at 90% confluence were infected in parallel with 10 PFU/cell and harvested 72 h after infection. Virus particles were extracted as described above, and the concentrations of virions were determined by plaque assay on 293 cells, which synthesize the viral E1B 55-kDa protein (2).
Detection of viral DNA.
Nuclei and cytoplasmic extracts were prepared from HFFs infected with Ad5 for increasing periods, or from cells infected in parallel with Ad5 and E1B mutant viruses, by extraction with NP-40 as described previously (32, 39). Nuclei were lysed by addition of five volumes of 0.01 M Tris-HCl, pH 7.5, containing 0.5 M NaCl and 1 mM EDTA. An equal volume of 0.01 M Tris-Cl, pH 7.5, containing 1 mM EDTA and 2% (wt/vol) sodium dodecyl sulfate (SDS), was then added, and samples were incubated with 100 μg/ml proteinase K for 30 min at 37°C. Nucleic acids were purified by phenol-CHCl3 extraction and ethanol precipitation. They were then collected by centrifugation, dissolved in 0.01 M Tris-HCl, pH 7.5, containing 0.05 M NaCl and 1 mM EDTA, and digested sequentially for 30 min at 37°C with 80 μg/ml RNase A and 50 μg/ml proteinase K. Following phenol-CHCl3 extraction and ethanol precipitation, DNA samples were digested with 200 units EcoRI for 90 min at 37°C. Equal quantities of DNA were then denatured and loaded onto nylon membranes as described previously (53). Membrane-bound DNA was hybridized to plasmids containing the viral IVa2 or E1A gene, viral L3 DNA or human ribosomal DNA labeled with [α-32P]dCTP (3,000 Ci/mmol; Perkin Elmer Life Sciences) by the random priming method (31). Signals were quantified using the PhosphorImager Storm system and ImageQuant software (Molecular Dynamics).
Steady-state concentrations of viral proteins.
The kinetics of accumulation of viral proteins were examined by immunoblotting of cytoplasmic extracts prepared as described in the previous section from equal numbers of HFFs infected with Ad5 for increasing periods. SDS-polyacrylamide gel electrophoresis, transfer of proteins to membranes, and detection of bound antibodies were done as described previously (39). The viral E1A, E1B 55-kDa, E2 72-kDa, and E4 Orf 6 proteins were detected by using monoclonal antibodies M73 (48), 2A6 (84), B6 (79), and M45 (69), respectively. To compare the concentrations of the E1B 55-kDa protein accumulating in cells infected by Ad5 and E1B mutant viruses, total cell lysates were prepared from cells infected for 20 or 40 h as described previously (39). Extracts from equal numbers of infected cells were analyzed by immunoblotting using the 2A6 monoclonal antibody (84). Viral late proteins were examined by immunoblotting of cytoplasmic extracts prepared as described in the previous section from HFFs infected in parallel with Ad5 or E1B mutant viruses for 30 h. Protein V was detected using monoclonal antibody F58 #4 (61).
Synthesis of viral and cellular proteins.
HFFs were infected in parallel with Ad5 and E1B mutant viruses or mock infected. At 29 h after infection, the medium was removed and replaced with Dulbecco's modified Eagle medium lacking methionine and cysteine (Gibco-Invitrogen Corp.) supplemented with 5% fetal bovine serum. Cells were then incubated with 100 μCi/ml 35S Easy Tag Express (175.0 Ci/mmol; Perkin Elmer Life Sciences) from 30 to 32 h after infection and harvested. Cytoplasmic and nuclear fractions were separated by NP-40 extraction as described above. The newly synthesized cytoplasmic proteins recovered from equal numbers of infected cells were examined by electrophoresis in 10% polyacrylamide-SDS gels and autoradiography of dried gels.
Immunofluorescence.
HFFs infected with Ad5 or the mutant A143 or mock infected were processed for immunofluorescence as described previously for HeLa cells (34, 39). The viral E1B 55-kDa and 72-kDa DNA-binding proteins were detected by using monoclonal antibody 2A6 immunoglobulin G labeled with AlexaFlour 488 and the B6 monoclonal antibody (80) and Cy5-labeled antimouse immunoglobulin G (Jackson Laboratories), respectively. Double-labeled samples were examined using a Zeiss LSM 510 confocal microscope with a C-Apochromat 1.2 NA water objective.
Steady-state concentrations of viral late RNA.
HFFs were infected with Ad5 or E1B mutant virus for 30 to 32 h. Nuclear and cytoplasmic fractions were separated by NP-40 extraction, and the cytoplasmic extract was digested with 100 μg/ml proteinase K as described above. RNA was then purified by phenol-CHCl3 extraction and ethanol precipitation. Precipitates were collected by centrifugation, dissolved in 0.01 M Tris-HCl, pH 7.5, containing 0.05 M NaCl and 0.01 M MgCl2, and incubated with 50 units DNase I (RNase free; Promega) for 30 min at 37°C. Samples were then digested with proteinase K as described and RNA purified by organic extraction and ethanol precipitation. Nuclear pellets were resuspended in 0.01 M Tris-HCl, pH 7.5, containing 0.5 M NaCl and 0.01 M MgCl2 and vortexed extensively. Following nuclear lysis, samples were diluted 1 to 2 with water and digested sequentially with 100 units DNase I and 100 μg/ml proteinase K, phenol-CHCl3 extracted, and ethanol precipitated. Nucleic acids were collected by centrifugation, dissolved in 0.01 M Tris-HCl, pH 7.5, containing 0.05 M NaCl and 0.01 M MgCl2, and subjected to a second cycle of DNase I and proteinase K digestion under the same conditions.
Viral late mRNAs were detected and quantified by using two-step real time reverse transcription-PCR (RT-PCR). Reverse transcription was performed manually under the conditions described previously (17, 56). Reaction mixtures contained cytoplasmic or nuclear RNA isolated from equal numbers of cells infected by Ad5 or E1B mutant viruses, 4.5 pmol of the reverse primer of a human β-actin amplicon (Applied Biosystems), and 15 pmol oligonucleotide primers specific for processed L2 penton or L5 fiber mRNAs. These last two primers spanned the splice junctions between the ML tripartite leader sequence and the mRNA bodies. Both primers were complementary to 11 bases upstream of the splice sites, and the penton and fiber primers were complementary to 12 and 14 bases, respectively, downstream of the splice junctions. cDNAs were then quantified by multiplex, real-time PCR using the AB1 Prism 7700 sequence detection system and TaqMan (Applied Biosystems) chemistry. The probes of an amplicon within the major late (ML) tripartite sequence, which was designed using Primer Express software (Applied Biosystems), and the human β-actin amplicon were labeled with VIC and 6-carboxyfluorescein, respectively. The ML amplicon comprised a forward primer corresponding to positions 69 to 90 of the tripartite leader sequence, a probe corresponding to positions 94 to 114, and a reverse primer complementary to positions 135 to 150. Relative cDNA, and hence mRNA, concentrations were determined by the standard curve method.
RESULTS
The kinetics of Ad5 replication in normal human fibroblasts.
The vast majority of studies of the productive cycle of human subgroup C adenoviruses, such as Ad5, have used established human cell lines as hosts. Such immortal cell lines are transformed and indeed are often derived from human tumors (e.g., HeLa cells, the most common laboratory host for Ad5). They are therefore abnormal (and no doubt variable) in both genotype and many aspects of growth and metabolism. In contrast, the human foreskin fibroblasts used in these experiments were diploid cells that exhibited such characteristic properties of normal primary cells as contact inhibition and senescence after a finite number of passages in culture (data not shown). As the infectious cycle of Ad5 in these normal cells had not been characterized previously, we initially examined the kinetics of production of virions, viral DNA synthesis, and viral early and late gene expression.
We first compared the time courses of production of infectious virus particles in HeLa cells and HFFs. Typical results obtained when cells were infected at low multiplicity, 0.1 PFU/cell, are shown in Fig. 1. The replication of Ad5 proceeded considerably more slowly in HFFs than in HeLa cells. Production of progeny virions in HeLa cells began between 18 and 24 h after infection and was complete by 36 h. By contrast, progeny virions were not detected until 36 h after infection of HFFs, and their concentration increased at a slower rate than that observed with HeLa cells until 72 h after infection (Fig. 1). Increasing the multiplicity of infection to 10 PFU/cell did not alter the time at which virions were first detected in either cell type (data not shown).
FIG. 1.

The kinetics of Ad5 replication in HeLa cells and HFFs. Following infection at 0.1 PFU/cell, cells were harvested at regular intervals, and yields of infectious particles were determined by duplicate plaque assays of multiple dilutions of each sample. p.i., postinfection.
At the latest time point examined following low-multiplicity infection, the yield of infectious virus particles per cell was an order of magnitude lower in HFFs than in HeLa cells (Table 1). Infection at 10 PFU/cell did not change this ratio (Table 1). This difference in yield per cell could indicate that production of virions is significantly less efficient in HFFs than in HeLa cells or that a multiplicity of infection sufficient for infection of all HeLa cells (10 PFU/cell) failed to result in infection of all normal fibroblasts. To distinguish between the possibilities, we first used indirect immunofluorescence to measure the percentage of HFFs that produced the viral hexon protein as a function of multiplicity of infection. While a multiplicity of 50 PFU/cell resulted in infection of all cells, only some 80% stained positive for hexon when 25 PFU/cell was used (data not shown). This observation indicates that HFFs are significantly less susceptible to Ad5 infection than are HeLa cells. Consequently, a multiplicity of 30 PFU/cell was used in all subsequent experiments. Furthermore, measurement of virus yield when all cells were infected indicated that each normal fibroblast supports production of a smaller number of virions than does a HeLa cell (Table 1).
TABLE 1.
Comparison of yields of Ad5 in HeLa cells and HFFs
| Multiplicitya of infection | Yield, PFU/cellb
|
Yield ratioc | |
|---|---|---|---|
| HFF | HeLa | ||
| 0.1 | 1.7 | 14 | 0.12 |
| 10.0 | 36.7 | 322.2 | 0.11 |
| 50.0 | 125.0 | 319.4 | 0.39 |
Based on titer determined by plaque assay with HeLa cells.
Yields were determined by duplicate plaque assays with HeLa cells. For clarity, standard deviations (± 15%) are not shown.
Ratio of yield for HFFs to that for HeLa cells.
Entry into the late phase of infection is delayed in Ad5-infected HFFs.
To investigate the reason for the slow Ad5 replication in HFFs compared to that in HeLa cells, we first examined the kinetics of viral DNA synthesis. HFFs infected with 30 PFU/cell Ad5 were harvested at regular intervals and DNA purified as described in Materials and Methods. DNA recovered from an equal numbers of cells was loaded onto nylon membranes and hybridized to 32P-labeled viral E1A DNA as described in Materials and Methods. Following quantification of signals, membranes were stripped and hybridized to 32P-labeled human ribosomal DNA to provide an internal control. As shown in Fig. 2A, a large increase in the concentration of viral DNA was first detected between 20 and 24 h after infection, and synthesis of viral DNA ceased by 36 to 40 h after infection. The length of the period during which the concentration of intranuclear viral DNA increased is very similar to that observed with HeLa cells (66). Furthermore, cytoplasmic fiber mRNA reached its maximal concentration some 8 to 10 h after the onset of viral DNA synthesis in HFFs (data not shown), indicating that the kinetics of viral late gene expression are also similar in HFFs and HeLa cells. By contrast, the onset of viral DNA replication occurs much earlier in HeLa cells, between 6 and 8 h after infection (66). We therefore conclude that the early phase of Ad5 infection is extended by some fourfold in normal human fibroblasts compared to that in transformed HeLa cells.
FIG. 2.

The kinetics of synthesis of viral DNA and early proteins in Ad5-infected HFFs. A. The concentrations of viral DNA present in HFFs infected with 30 PFU/cell Ad5 for the periods indicated were determined by blotting and hybridization to viral DNA as described in Materials and Methods. Signals were quantified and corrected using rRNA genes as an internal control and are expressed relative to the maximal quantity of viral DNA detected. The values shown represent means for two independent experiments. B. Total cell extracts prepared from HFFs infected with 30 PFU/cell Ad5 for the periods indicated were assayed for the presence of the E1A and E1B 55-kDa proteins by using immunoblotting as described in Materials and Methods. p.i., postinfection.
We next examined the accumulation of viral early proteins in Ad5-infected HFFs by using immunoblotting of soluble extracts prepared from cells harvested after increasing periods of infection. The immediate-early E1A proteins are the first to be synthesized and are required for efficient expression of viral early genes (see references 35, 87, and 88). Although these proteins are present from 2 to 4 h after infection of HeLa cells, they were only just detectable at 14 h after infection of HFFs (Fig. 2B). The various forms of the E1A proteins (48) then increased in concentration until 20 h after infection. Consistent with the requirement of E1A proteins for efficient transcription of viral early genes, the E1B 55-kDa protein was not made at 14 h after infection and was present at a low concentration by 20 h (Fig. 2B). This protein attained its maximal concentration by 32 h after infection, well into the late phase of infection (Fig. 2A), as is also the case with established lines of human cells (9, 83, 90, 92). The kinetics of accumulation of the E2 72-kDa DNA-binding protein and the E4 Orf 6 protein were similar to those of the E1B 55-kDa protein (data not shown). These results indicate that expression of the viral E1A gene is much less efficient in normal human fibroblasts than in HeLa cells. Because efficient expression of E2 coding sequences for the viral replication proteins requires both the 289R and 243R E1A proteins (see references 35 and 67 for reviews), this property makes a major contribution to the extended-early-phase characteristic of Ad5 infection of HFFs (Fig. 2A).
We also examined the intracellular locations of the E1B 55-kDa and E4 Orf 6 proteins as the infectious cycle progressed by immunofluorescence using monoclonal antibodies 2A6 and M45, respectively. The intracellular localization of both proteins closely resembled the patterns previously described for Ad5-infected HeLa cells (39, 70), albeit with delayed kinetics of entry into the nucleus (data not shown).
Effects of mutations in the E1B 55-kDa protein coding sequence on Ad5 replication in HFFs.
To assess the role of the E1B 55-kDa protein in regulation of mRNA export in normal cells infected by Ad5, we examined the phenotypes displayed by mutant viruses carrying insertions in the E1B protein coding sequence. These mutants, A143, H224, and H354 (Fig. 3A), which are a subset of a series that has been characterized in some detail (37, 55, 83, 97), were chosen on the basis of the properties exhibited by the altered E1B 55-kDa proteins in HeLa cells. We first compared the steady-state concentrations of the E1B 55-kDa protein in HFFs infected with Ad5 or the mutant viruses by using immunoblotting of total cell extracts prepared 20 or 40 h after infection. As illustrated in Fig. 3B, none of the mutations resulted in reduced accumulation, compared to the wild-type level, of the altered E1B proteins either early or late in infection. On the other hand, all of the mutations impaired viral replication in HFFs (Table 2), although the defect induced by the H354 mutation was modest.
FIG. 3.

Effects of insertion mutations on accumulation of the E1B 55-kDa protein. A. The 496-residue Ad5 E1B 55-kDa protein is represented by the rectangle at the top, on which are shown the positions of the leucine-rich export signal (NES), an RNP RNA-binding motif (RNP), and a putative C2H2 zinc finger (Zn finger). The sites at which the protein is modified are indicated above the protein. The sites of the four-amino-acid insertions carried by the mutants used in these experiments are indicated below the protein. The effects of these mutations on binding of the E1B 55-kDa protein to other proteins (34, 55, 83) and on viral late mRNA export in HeLa cells (39) are summarized below the insertion sites, where - and - indicate inhibition of the activity or function listed at the left. B. HFFs were infected for 20 or 40 h with 30 PFU/cell Ad5 (lanes 1 and 6), A143 (lanes 2 and 7), H224 (lanes 3 and 8), or H354 (lanes 4 and 9) or mock infected (lanes 5 and 10). The E1B 55-kDa protein was examined by immunoblotting, as described in Materials and Methods.
TABLE 2.
Replication of E1B mutant viruses in HFFs
| Virus | Relative yielda
|
|
|---|---|---|
| Expt 1 | Expt 2 | |
| Ad5 | 1.0 | 1.0 |
| A143 | 3.3 × 10−3 | 7.6 × 10−3 |
| H224 | 1.6 × 10−2 | 7.3 × 10−3 |
| H354 | 1.4 × 10−1 | 6.9 × 10−1 |
| Hr 6 | 3.5 × 10−3 | 1.5 × 10−3 |
Yields, determined by plaque assay with 293 cells, are shown relative to the values determined for Ad5.
The synthesis of viral late proteins in HFFs infected with these mutants was then examined. The E1B 55-kDa protein null virus Hr6 (51) was included in this and subsequent analyses. Cells infected by the mutants or Ad5, as well as mock-infected cells, were labeled with [35S]methionine and [35S]cysteine for 2 h during the late phase of infection, as described in Materials and Methods. Newly synthesized cytoplasmic proteins were analyzed by SDS-polyacrylamide gel electrophoresis and autoradiography. In Ad5-infected HFFs, efficient synthesis of viral late protein was accompanied by severe inhibition of cellular protein synthesis (Fig. 4A, compare lanes 1 and 2). Production of viral late protein was also readily evident in H354-infected cells, although it was somewhat less efficient, and cellular protein synthesis was partially inhibited (Fig. 4A, compare lanes 1, 2, and 5). By contrast, reduction in labeling of cellular proteins was not observed in cells infected by any of the other mutants (Fig. 4A, lanes 1, 3, 4, and 6). Furthermore, reduced quantities of viral proteins (for example, hexon polypeptide II and pVII) were synthesized in A143-infected HFFs but could not be detected readily in cells infected by H224 or Hr6 (Fig. 4A, compare lanes 2, 3, 4, and 6). To circumvent the difficulty in detecting small quantities of newly synthesized viral proteins when cellular proteins were also labeled, we used immunoblotting to compare the steady-state concentrations of protein V 30 h after infection of HFFs with Ad5 and the two mutants. As illustrated in Fig. 4B, this late protein was indeed produced in cells infected by H224 or Hr6 but at concentrations some 50-fold lower than those observed with Ad5-infected HFFs.
FIG. 4.

Synthesis of viral late proteins in HFFs infected by E1B mutant viruses. A. HFFs infected with 30 PFU/cell of the viruses indicated at the top or mock infected (M) were labeled with [35S]methionine plus [35S]cysteine during the late phase of infection, and cytoplasmic proteins were examined by SDS-polyacrylamide gel electrophoresis and autoradiography. The positions to which prestained molecular mass markers migrated are listed at the left, and those of prominent viral late proteins are indicated at the right. B. The steady-state concentrations attained by protein V 30 h after infection of HFFs with the viruses indicated at the top or in mock-infected cells (M) were compared by immunoblotting as described in Materials and Methods.
Previous studies have established that mutations in the E1B 55-kDa protein coding sequence that result in impaired export of viral late mRNAs from the nucleus induce severe defects in synthesis of viral late proteins (40, 41, 42, 46, 73, 96). Quantitatively, these defects are significantly greater than can be accounted for by the reduced quantities of viral late mRNAs entering the cytoplasm. However, it is not clear whether the E1B 55-kDa protein facilitates late mRNA translation directly or indirectly as a result of efficient export of viral mRNAs encoding proteins necessary for efficient ML transcription (IVa2 protein [62, 71, 93]) or late mRNA translation (L4 100-kDa protein 49, 85). As impaired export of viral late mRNAs from the nucleus to the cytoplasm could account for the defects in synthesis of viral late proteins illustrated in Fig. 4, we compared this process in HFFs infected by Ad5 or the mutant viruses. Measurement of the concentrations of processed fiber mRNA in the cytoplasmic fractions of the Ad5-infected HFFs used for analysis of the kinetics of viral DNA synthesis (Fig. 2A) established that this late mRNA attained its maximal concentration between 28 and 32 h after infection (data not shown). Cells infected with 30 PFU/cell Ad5 or the mutants were therefore harvested 30 to 32 h after infection, and cytoplasmic and nuclear RNAs were purified from them as described in Materials and Methods. The relative concentrations of processed L2 penton and L5 fiber mRNAs were then determined by using quantitative RT-PCR. Reverse transcription was performed manually with primers complementary to the splice junctions between the coding exons of these mRNAs and the ML tripartite leader sequence. Relative cDNA, and hence mRNA, concentrations were then determined by using real-time PCR to amplify a segment of the ML tripartite sequence common to all ML mRNAs. A human β-actin mRNA sequence was reverse transcribed and amplified in the same reactions to provide an internal control. No amplification of either the viral or the cellular sequence was observed when reverse transcriptase was omitted from primer extension reactions (data not shown), confirming the specificity of the detection method for RNA. The results obtained for fiber mRNA production and export in several independent experiments are summarized in Fig. 5.
FIG. 5.
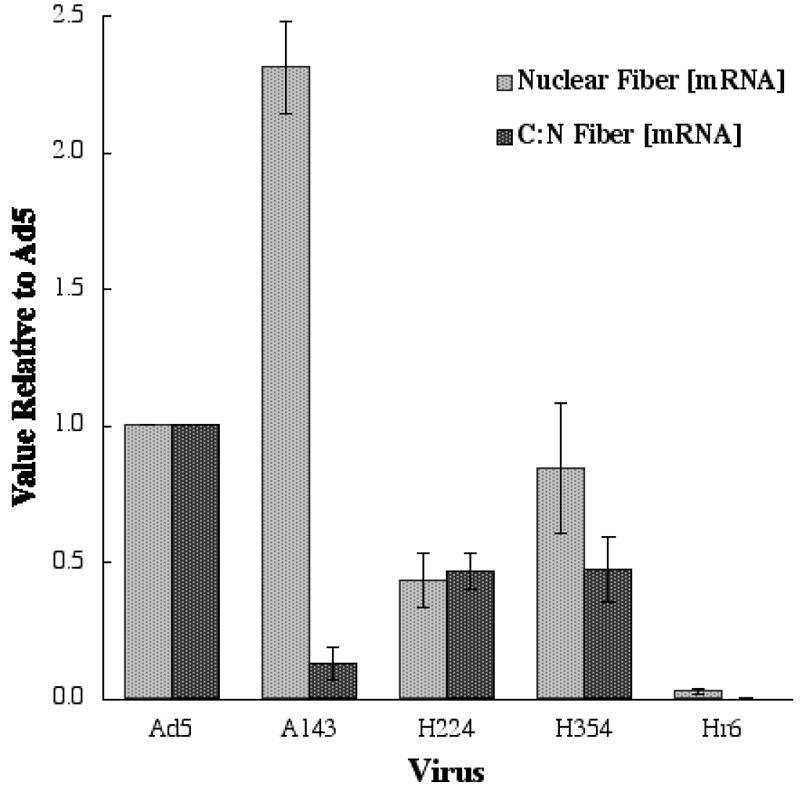
Effects of E1B mutations on the production and export of L2 fiber mRNA in HFFs. The concentrations of mature fiber mRNA present in nuclear and cytoplasmic fractions of HFFs infected for 30 to 32 h with 30 PFU/cell of the viruses indicated were determined by real-time RT-PCR as described in Materials and Methods. The nuclear concentrations and cytoplasmic/nuclear (C:N) concentration ratios, a measure of export efficiency, are expressed relative to the values observed for Ad5-infected cells. The values represent the means for three independent experiments.
We have previously reported that the A143 mutation, which results in impaired interaction of the E1B 55-kDa protein with the E4 Orf 6 protein (83), induces a specific defect in export of viral late mRNAs and disrupts the association of the E1B protein with the peripheral zones of viral replication centers in infected HeLa cells (39). The same mRNA export defect was observed in HFFs, although the effects of the mutation were more dramatic: export of fiber mRNA was reduced by a factor of 5, with a concomitant 2.5-fold increase in nuclear mRNA concentration (Fig. 5). It is likely that increased intranuclear turnover of fiber mRNA molecules that are not exported accounts for the difference between the degree of export inhibition and the increase in nuclear concentration of the mRNA. We also examined the effects of the A143 mutation on the location of the E1B 55-kDa protein by using immunofluorescence, as described in the Materials and Methods section. In Ad5-infected HFF nuclei, this viral protein was concentrated around viral replication centers, which were identified by the presence of the 72-kDa single-stranded DNA-binding protein (Fig. 6e to i). Some colocalization of the two viral proteins was observed (Fig. 6, panels h and i), but much less than that seen in Ad5-infected HeLa cells (39, 70). Rather, the E1B 55-kDa protein more typically was present in a reticular network that surrounded and connected viral replication centers (Fig. 6, panels h and i). This difference seems likely to reflect the larger size and greater flatness of infected HFF nuclei compared to infected HeLa cell nuclei and hence increased spatial resolution in the former. Regardless, the A143 mutation severely reduced the association of the E1B 55-kDa protein with viral replication centers in HFF nuclei (Fig. 6, panels j to n); in A143-infected cells, the protein was seen diffusely distributed throughout the nucleus and in speck-like structures well separated from viral replication centers.
FIG. 6.

Effects of the A143 mutation on the intracellular location of the E1B 55-kDa protein. HFFs infected with 30 PFU/cell of Ad5 or A143 or mock infected, as indicated, were processed for immunofluorescence 30 h after infection, and the locations of the viral E1B 55-kDa and E2 72-kDa proteins were examined as described in Materials and Methods. Panels i and n show merged images of Ad5- and A143-infected cells, respectively, at higher magnification. White arrows show colocalization of the two viral proteins.
The only other mutation associated with a specific (albeit modest) defect in late mRNA export was H354. In cells infected by this mutant, the nuclear concentration of fiber mRNA was not significantly altered, but export efficiency was reduced to some 50% of the value observed in Ad5-infected HFFs (Fig. 6). By contrast, production and export of the viral mRNA were decreased to closely similar extents in H224-infected cells (Fig. 5). Since it is not known whether the rate of viral late mRNA export is a function of the nuclear concentration of the export substrates, we can draw no conclusion about the effect of the H224 mutation on viral mRNA export. In HeLa cells, Hr6 and other E1B 55-kDa protein null mutants are defective for selective export of viral late mRNAs but exhibit neither other primary phenotypes nor perturbations in progression through the early phase of infection (60, 72, 73, 96). By contrast, only very small quantities of fiber (or penton) mRNA were detected in the nuclei of Hr6-infected HFFs, and the cytoplasmic concentrations were too low to allow measurement of export efficiency (Fig. 5).
As these data suggested that entry into the late phase of infection was blocked or delayed by the Hr6 mutation, viral DNA synthesis was examined in HFFs infected by Ad5 or the mutant viruses. Total nuclear DNA was isolated 30 h after infection and examined by blotting, as described in Materials and Methods. The raw data from one such experiment are shown in Fig. 7A. Quantification of this same experiment using rRNA genes as a control and of the signals observed in three independent infections are summarized in Fig. 7B (gray and black bars, respectively). These data clearly indicate that viral DNA synthesis was significantly impaired in cells infected by H224 or Hr6 compared to HFFs infected by Ad5 or the other mutant viruses. This difference was not the result of delayed entry into the late phase of infection, since incubation of infected cells until 40 h after infection did not increase the relative concentrations of viral DNA accumulating in cells infected by either H224 or Hr6 (data not shown).
FIG. 7.

Effects of E1B mutations on viral DNA synthesis in HFFs. Viral DNA was purified from HFFs infected for 30 h with the viruses indicated, loaded onto nylon membranes, and hybridized to labeled Ad5 DNA sequences as described in Materials and Methods. In the example shown in panel A, hybridization was to L3 viral DNA. Quantification of several such blots using rRNA genes as the internal control is shown by the gray bars in panel B. The variance shown for Ad5 was obtained by arbitrarily setting one of the signals as 1.0 and calculating the others relative to it. The black bars summarize the results obtained from three separate infections.
DISCUSSION
The low susceptibility of human foreskin fibroblasts to Ad5 infection (Table 1) is consistent with the results of previous studies of infection of human fibroblasts from various tissues (22, 50, 82). In the case of adult lung fibroblasts, this property has been attributed to the absence from the cell surface of the major receptor for Ad5, CAR (50). Whether the foreskin fibroblasts used in these experiments also lack CAR has not been established, but the high concentration of virions required to achieve efficient infection would be consistent with entry via one or more lower-affinity receptors, such as major histocompatibility complex class I proteins (7), heparan sulfate (20), or αvβ3 or αvβ5 integrins, which appear to mediate entry in the absence of the viral fiber-CAR interaction (50). At a multiplicity at which essentially all HFFs were infected, Ad5 replicated considerably more slowly than it does in transformed HeLa cells (Fig. 1). Similar kinetics of virion production and/or viral DNA synthesis have been observed with normal human lung cells (68, 89). In HFFs, it is the early phase that is extended, primarily because the E1A immediate-early proteins are not produced in appreciable quantities until 14 h after infection (Fig. 2). This property could be a consequence of inefficient transcription of the E1A gene, for example, a result of repression by Kruppel-related proteins (36), or of slow rates of one or more steps preceding the onset of viral gene expression, such as uncoating or movement of the viral genome to and into the nucleus.
Considerable effort is being devoted to the development of replicating, oncolytic derivatives of human adenoviruses for the treatment of cancer (reviewed in references 23, 30, and 54). Nevertheless, all but a few studies of adenovirus replication and the functions of viral gene products have employed as hosts established lines of transformed human cells. Indeed, HeLa cells, which were derived from a cervical carcinoma (38), have been the most commonly used host. Such transformed cell lines are genetically abnormal (often aneuploid) and have lost many mechanisms that restrain growth and proliferation of normal cells. Consequently, it remains unclear whether the molecular actions of viral gene products that allow efficient replication in, for example, HeLa cells are sufficient or function in the identical manner when the virus infects normal human cells. This question is particularly acute in the case of regulation of mRNA export by the E1B 55-kDa protein (see the introduction). The properties of the A143 mutant reported here establish that the E1B 55-kDa protein regulates viral late mRNA export in normal human fibroblasts, as it does in HeLa cells, and is required for maximally efficient replication. Furthermore, the A143 mutation also disrupted association of the E1B protein with the peripheral zones of viral replication centers in infected HFF nuclei (Fig. 6), as previously observed with HeLa cells (39). The close similarities of the phenotypes exhibited by A143 in HeLa cells and HFFs are consistent with the conclusion that the E1B 55-kDa protein promotes selective export of viral late mRNAs by the same mechanism(s) in transformed HeLa cells and normal foreskin fibroblasts. While these studies were in progress, O'Shea and colleagues reported that the Ad5 E1B 55-kDa protein is required for efficient export of viral late mRNAs in primary small airway epithelial cells (68). The results reported here extend this conclusion to a second, distinct type of normal human cell. Furthermore, they have revealed a previously undescribed requirement for the E1B 55-kDa protein during the early phase of infection.
The insertion mutant H224 and the null mutant Hr6 exhibited defects in HFFs that have not been observed when HeLa cells are used as hosts (39, 96, 97). The H224 mutation, which has been reported to perturb the interaction of the E1B 55-kDa protein with both the viral E4 Orf 6 protein (83) and the cellular p53 proteins (55) (Fig. 3A), impaired both production of viral late mRNA (Fig. 5) and replication of the viral genome (Fig. 7). Similar but more-severe defects were observed in HFFs infected by Hr6; in the absence of the E1B 55-kDa protein, only very small quantities of viral late mRNA and proteins were made (Fig. 4 and 5), and viral DNA synthesis was reduced by a factor of 5 to 10 (Fig. 7).
Binding of the E1B 55-kDa protein to the N terminus of the cellular p53 protein blocks activation of p53-dependent transcription and, in in vitro reactions, converts p53 from a transcriptional activator to a repressor (55, 63, 98). In conjunction with the E4 Orf 6 protein, the E1B protein also induces degradation of p53, via formation of a complex that contains these and several additional components, including a cullin E3 ubiquitin ligase (43, 47, 76, 81, 90). These activities of the E1B protein have been postulated to facilitate adenovirus replication by blocking p53-dependent apoptosis induced by viral E1A proteins (reviewed in references 11 and 77). Our results indicate that disruption of the interaction of the E1B and p53 proteins correlates with inefficient viral DNA synthesis in HFFs; the H224 and Hr6 mutations disrupt this interaction, whereas the A143 insertion alters neither the efficiency of viral DNA synthesis nor association with p53 (55). It is therefore possible that failure to counter activation and/or accumulation of p53 accounts for inefficient progression through the early phase of infection in HFFs infected by H224 or Hr6 (Fig. 7). However, the E1B 55-kDa protein also interacts with the E4 Orf3 protein during the early phase of infection, and these two proteins colocalize to nuclear, PML protein-containing structures, termed PML bodies or nuclear domain 10s, that are reorganized early in infection (16, 27, 57, 59). Furthermore, the cellular double-stranded-DNA break repair complex that contains the Mre11, Rad50, and Nbs1 proteins (the MRN complex) is relocalized to similar structures by the E4 Orf3 protein (29). Inactivation of this complex prevents concatamerization of the viral genome (10, 91, 95) and is necessary to allow replication of the linear viral DNA genome (29). The defects in viral DNA synthesis observed in HFFs infected by H224 and Hr6 could therefore also be, at least in part, a consequence of impaired interaction of the E1B 55-kDa protein with the E4 Orf3 protein early in infection. Experiments are in progress to define in more detail the effects of these mutations on early events in the infectious cycle.
In stark contrast to the properties of Hr6 described here, it has been reported that OXNYX015 (dl1520), which is also a mutant null for production of the E1B 55-kDa protein (5), exhibits no defects in viral DNA synthesis in small airway epithelial cells (68). Furthermore, compared to the wild-type yield, the yield of this mutant was reduced only some 20-fold (68), compared to the 500- to 1,000-fold decrease observed in Hr6-infected HFFs (Table 2). These differences could be consequences of the nature of the cells used as hosts. Indeed, variations in the contributions of the E1B 55-kDa protein to Ad5 replication with the type of primary cell used as a host have been observed (68, 82). It is also possible that the proliferative state of primary cells at the time of infection modulates the requirement for the E1B 55-kDa protein: in our experiments, HFFs were infected prior to becoming contact inhibited and supplied with fresh growth factors immediately after virus adsorption, whereas O'Shea and colleagues employed contact-inhibited cells with no addition of growth factors (68). This explanation implies that one or more functions of the E1B 55-kDa protein are important during the early phase, when infected cells are not only subject to the mitogenic actions of the viral E1A proteins (65, 67) but also receive external signals that induce proliferation. In this context, it is noteworthy that replication of ONYX015 (dl1520) is more severely impaired in proliferating than in contact-inhibited human mammary epithelial cells (68). We are currently evaluating the effects of primary host cell type and proliferation status on early steps in the replication of null mutant viruses.
It has been proposed that the induction of selective export of viral late mRNAs by the E1B 55-kDa protein is the major determinant of selective replication of E1B 55-kDa protein null mutants of Ad5, such as ONYX015, in tumor cells (68). The implication of this hypothesis, that viral late mRNAs and cellular mRNAs encoding proteins important for proliferation or tumorigenesis are exported by the same mechanism, has yet to be examined. Nevertheless, the notion that the efficiency of viral late mRNA export determines tumor cell selectivity appears overly simplistic. Regulation of mRNA export during adenovirus infection and the roles of the E1B 55-kDa and E4 Orf 6 proteins in this process were first identified for HeLa (6, 19, 73, 96), a tumor-derived cell line. More importantly, in these tumor cells at 37°C, the reductions in viral late mRNA export efficiency observed in the absence of the E1B 55-kDa protein (39, 52, 73, 96) are similar to or greater in magnitude than that measured in ONYX015-infected small airway epithelial cells (68). Rather, the data available from these and many previous studies of the basis of selective replication of E1B 55-kDa-null mutants (21, 68) suggest that multiple functions of the E1B 55-kDa protein can be important for Ad5 replication. Furthermore, the relative contributions of mRNA export regulation by, and other functions of, the E1B protein appear to depend on the repertoire of cellular genes expressed in both tumor and normal cells and the outcome of a complex interplay between this viral protein and multiple cellular components.
Acknowledgments
We thank Catherine Patterson and Tom Shenk for human foreskin fibroblasts, Norma Caputo and Ellen Brindle-Clark for assistance with preparation of the manuscript, and Humayra Ali and Nishal Mohan for their critical comments.
This work was supported by a grant from the National Institute of Allergy and Infectious Disease, National Institutes of Health.
REFERENCES
- 1.Reference deleted.
- 2.Aiello, L., R. Guilfoyle, K. Huebner, and R. Weinmann. 1979. Adenovirus 5 DNA sequences present and RNA sequences transcribed in transformed human embryo kidney cells (HEK-Ad-5 or 293). Virology 94:460-469. [DOI] [PubMed] [Google Scholar]
- 3.Anderson, C. W., P. R. Baum, and R. F. Gesteland. 1973. Processing of adenovirus 2-induced proteins. J. Virol. 12:241-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aspegren, A., C. Rabino, and E. Bridge. 1998. Organization of splicing factors in adenovirus-infected cells reflects changes in gene expression during the early to late phase transition. Exp. Cell Res. 245:203-213. [DOI] [PubMed] [Google Scholar]
- 5.Barker, D. D., and A. J. Berk. 1987. Adenovirus proteins from both E1B reading frames are required for transformation of rodent cells by viral infection and DNA transfection. Virology 156:107-121. [DOI] [PubMed] [Google Scholar]
- 6.Beltz, G. A., and S. J. Flint. 1979. Inhibition of HeLa cell protein synthesis during adenovirus infection: restriction of cellular messenger RNA sequences to the nucleus. J. Mol. Biol. 131:353-373. [DOI] [PubMed] [Google Scholar]
- 7.Bergelson, J. M., J. A. Cunningham, G. Droguett, E. A. Kurt-Jones, A. Krithivas, J. S. Hong, M. S. Horwitz, R. L. Crowell, and R. W. Finberg. 1997. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 275:1320-1323. [DOI] [PubMed] [Google Scholar]
- 8.Bischoff, J. R., D. H. Kirn, A. Williams, C. Heise, S. Horn, M. Muna, L. Ng, J. A. Nye, A. Sampson-Johannes, A. Fattaey, and F. McCormick. 1996. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 274:373-376. [DOI] [PubMed] [Google Scholar]
- 9.Boivin, D., M. R. Morrison, R. C. Marcellus, E. Querido, and P. E. Branton. 1999. Analysis of synthesis, stability, phosphorylation, and interacting polypeptides of the 34-kilodalton product of open reading frame 6 of the early region 4 protein of human adenovirus type 5. J. Virol. 73:1245-1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boyer, T. G., M. E. Martin, E. Lees, R. P. Ricciardi, and A. J. Berk. 1999. Mammalian Srb/Mediator complex is targeted by adenovirus E1A protein. Nature 399:276-279. [DOI] [PubMed] [Google Scholar]
- 11.Braithwaite, A. W., and A. Russell. 2001. Induction of cell death by adenoviruses. Apoptosis 6:359-370. [DOI] [PubMed] [Google Scholar]
- 12.Bridge, E., and G. Ketner. 1990. Interaction of adenoviral E4 and E1b products in late gene expression. Virology 174:345-353. [DOI] [PubMed] [Google Scholar]
- 12a.Bridge, E., and U. Pettersson. 1996. Nuclear organization of adenovirus RNA biogenesis. Exp. Cell Res. 229:233-239. [DOI] [PubMed] [Google Scholar]
- 13.Bridge, E., K. U. Riedel, B. M. Johansson, and U. Pettersson. 1996. Spliced exons of adenovirus late RNAs colocalize with snRNP in a specific nuclear domain. J. Cell Biol. 135:303-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bridge, E., D. X. Xia, M. Carmo-Fonesca, B. Cardinali, A. I. Lamond, and U. Pettersson. 1995. Dynamic organization of splicing factors in adenovirus-infected cells. J. Virol. 69:281-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carter, C. C., R. Izadpanah, and E. Bridge. 2003. Evaluating the role of CRM1-mediated export for adenovirus gene expression. Virology 315:224-233. [DOI] [PubMed] [Google Scholar]
- 16.Carvalho, T., J. S. Seeler, K. Ohman, P. Jordan, U. Pettersson, G. Akusjärvi, M. Carmo-Fonseca, and A. Dejean. 1995. Targeting of adenovirus E1A and E4-ORF3 proteins to nuclear matrix-associated PML bodies. J. Cell Biol. 131:45-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen, H., R. Vinnakota, and S. J. Flint. 1994. Intragenic activating and repressing elements control transcription from the adenovirus IVa2 initiator. Mol. Cell. Biol. 14:676-685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cuesta, R., Q. Xi, and R. J. Schneider. 2001. Preferential translation of adenovirus mRNAs in infected cells. Cold Spring Harb. Symp. Quant. Biol. 66:259-267. [DOI] [PubMed] [Google Scholar]
- 19.Cutt, J. R., T. Shenk, and P. Hearing. 1987. Analysis of adenovirus early region 4-encoded polypeptides synthesized in productively infected cells. J. Virol. 61:543-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dechecchi, M. C., A. Tamanini, A. Bonizzato, and G. Cabrini. 2000. Heparan sulfate glycosaminoglycans are involved in adenovirus type 5 and 2-host cell interactions. Virology 268:382-390. [DOI] [PubMed] [Google Scholar]
- 21.Dix, B. R., S. J. Edwards, and A. W. Braithwaite. 2001. Does the antitumor adenovirus ONYX-015/dl1520 selectively target cells defective in the p53 pathway? J. Virol. 75:5443-5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dix, B. R., S. J. O'Carroll, C. J. Myers, S. J. Edwards, and A. W. Braithwaite. 2000. Efficient induction of cell death by adenoviruses requires binding of E1B55k and p53. Cancer Res. 60:2666-2672. [PubMed] [Google Scholar]
- 23.Dobbelstein, M. 2004. Replicating adenoviruses in cancer therapy. Curr. Top. Microbiol. Immunol. 273:291-334. [DOI] [PubMed] [Google Scholar]
- 24.Dobbelstein, M., J. Roth, W. T. Kimberly, A. J. Levine, and T. Shenk. 1997. Nuclear export of the E1B 55-kDa and E4 34-kDa adenoviral oncoproteins mediated by a rev-like signal sequence. EMBO J. 16:4276-4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dobner, T., and J. Kzhyshkowska. 2001. Nuclear export of adenovirus RNA. Curr. Top. Microbiol. Immunol. 259:25-34. [DOI] [PubMed] [Google Scholar]
- 26.Dosch, T., F. Horn, G. Schneider, F. Kratzer, T. Dobner, J. Hauber, and R. H. Stauber. 2001. The adenovirus type 5 E1B-55k oncoprotein actively shuttles in virus-infected cells, whereas transport of E4Orf6 is mediated by a CRM1-independent mechanism. J. Virol. 75:5677-5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doucas, V., A. M. Ishov, A. Romo, H. Juguilon, M. D. Weitzman, R. M. Evans, and G. G. Maul. 1996. Adenovirus replication is coupled with the dynamic properties of the PML nuclear structure. Genes Dev. 10:196-207. [DOI] [PubMed] [Google Scholar]
- 28.Edwards, S. J., B. R. Dix, C. J. Myers, D. Dobson-Le, L. Huschtscha, M. Hibma, J. Royds, and A. W. Braithwaite. 2002. Evidence that replication of the antitumor adenovirus ONYX-015 is not controlled by the p53 and p14 (ARF) tumor suppressor genes. J. Virol. 76:12483-12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Evans, J. D., and P. Hearing. 2005. Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. J. Virol. 79:6207-6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Everts, B., and H. van der Poel. 2005. Replication-selective oncolytic viruses in the treatment of cancer. Cancer Gene Ther. 12:141-161. [DOI] [PubMed] [Google Scholar]
- 31.Feinberg, A. P., and B. Vogelstein. 1983. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal. Biochem. 137:266-267. [DOI] [PubMed] [Google Scholar]
- 32.Flint, S. J., P. H. Gallimore, and P. A. Sharp. 1975. Comparison of viral RNA sequences in adenovirus 2-transformed and lytically infected cells. J. Mol. Biol. 96:47-68. [DOI] [PubMed] [Google Scholar]
- 33.Flint, S. J., and R. A. Gonzalez. 2003. Regulation of mRNA production by the adenoviral E1B 55kDa and E4 Orf6 proteins. Curr. Top. Microbiol. Immunol. 272:287-330. [DOI] [PubMed] [Google Scholar]
- 34.Flint, S. J., W. Huang, J. Goodhouse, and S. Kyin. 2005. A peptide inhibitor of exportin1 blocks shuttling of the adenoviral E1B 55 kDa protein but not export of viral late mRNAs. Virology 337:7-17. [DOI] [PubMed] [Google Scholar]
- 35.Flint, S. J., and T. Shenk. 1989. Adenovirus E1A protein: paradigm viral transactivator. Annu. Rev. Genet. 23:141-161. [DOI] [PubMed] [Google Scholar]
- 36.Fognani, C., G. Della Valle, and L. E. Babiss. 1993. Repression of adenovirus E1A enhancer activity by a novel zinc finger-containing DNA-binding protein related to the GLI-Kruppel protein. EMBO J. 12:4985-4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gabler, S., H. Schutt, P. Groitl, H. Wolf, T. Shenk, and T. Dobner. 1998. E1B 55-kilodalton-associated protein: a cellular protein with RNA-binding activity implicated in nucleocytoplasmic transport of adenovirus and cellular mRNAs. J. Virol. 72:7960-7971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gey, G. O., F. B. Bang, and M. K. Gey. 1954. Responses of a variety of normal and malignant cells to continuous cultivation, and some practical applications of these responses to problems in the biology of disease. Ann. N. Y. Acad. Sci. 58:976-999. [DOI] [PubMed] [Google Scholar]
- 39.Gonzalez, R. A., and S. J. Flint. 2002. Effects of mutations in the adenoviral E1B 55-kDa protein coding sequence on viral late mRNA metabolism. J. Virol. 76:4507-4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goodrum, F. D., and D. A. Ornelles. 1997. The early region 1B 55-kilodalton oncoprotein of adenovirus relieves growth restrictions imposed on viral replication by the cell cycle. J. Virol. 71:548-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goodrum, F. D., and D. A. Ornelles. 1998. p53 status does not determine outcome of E1B 55-kilodalton mutant adenovirus lytic infection. J. Virol. 72:9479-9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goodrum, F. D., and D. A. Ornelles. 1999. Roles for the E4 orf6, orf3, and E1B 55-kilodalton proteins in cell cycle-independent adenovirus replication. J. Virol. 73:7474-7488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grand, R. J., M. L. Grant, and P. H. Gallimore. 1994. Enhanced expression of p53 in human cells infected with mutant adenoviruses. Virology 203:229-240. [DOI] [PubMed] [Google Scholar]
- 44.Hall, A. R., B. R. Dix, S. J. O'Carroll, and A. W. Braithwaite. 1998. p53-dependent cell death/apoptosis is required for a productive adenovirus infection. Nat. Med. 4:1068-1072. [DOI] [PubMed] [Google Scholar]
- 45.Hann, B., and A. Balmain. 2003. Replication of an E1B 55-kilodalton protein-deficient adenovirus (ONYX-015) is restored by gain-of-function rather than loss-of-function p53 mutants. J. Virol. 77:11588-11595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harada, J. N., and A. J. Berk. 1999. p53-Independent and -dependent requirements for E1B-55K in adenovirus type 5 replication. J. Virol. 73:5333-5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harada, J. N., A. Shevchenko, D. C. Pallas, and A. J. Berk. 2002. Analysis of the adenovirus E1B-55K-anchored proteome reveals its link to ubiquitination machinery. J. Virol. 76:9194-9206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harlow, E., B. Franza, Jr., and C. Schley. 1985. Monoclonal antibodies specific for adenovirus early region 1A proteins: extensive heterogeneity in early region 1A products. J. Virol. 55:533-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hayes, B. W., G. C. Telling, M. M. Myat, J. F. Williams, and S. J. Flint. 1990. The adenovirus L4 100-kilodalton protein is necessary for efficient translation of viral late mRNA species. J. Virol. 64:2732-2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hidaka, C., E. Milano, P. L. Leopold, J. M. Bergelson, N. R. Hackett, R. W. Finberg, T. J. Wickham, I. Kovesdi, P. Roelvink, and R. G. Crystal. 1999. CAR-dependent and CAR-independent pathways of adenovirus vector-mediated gene transfer and expression in human fibroblasts. J. Clin. Investig. 103:579-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ho, Y. S., R. Galos, and J. F. Williams. 1982. Isolation of type 5 adenovirus mutants with a cold-sensitive phenotype: genetic evidence of an adenovirus transformation maintenance function. Virology 122:109-124. [DOI] [PubMed] [Google Scholar]
- 52.Huang, W., J. Kiefer, D. Whalen, and S. Flint. 2003. DNA synthesis-dependent relief of transcription from the adenoviral type 2 IVa2 promoter by a cellular protein. Virology 314:394-402. [DOI] [PubMed] [Google Scholar]
- 53.Iftode, C., and S. J. Flint. 2004. Viral synthesis-dependent titration of a cellular repressor activates transcription of the human adenovirus type 2 IVa2 Gene Pro. Natl. Acad. Sci. USA 101:17831-17836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kanerva, A., and A. Hemminki. 2005. Adenoviruses for treatment of cancer. Ann. Med. 37:33-43. [DOI] [PubMed] [Google Scholar]
- 55.Kao, C. C., P. R. Yew, and A. J. Berk. 1990. Domains required for in vitro association between the cellular p53 and the adenovirus 2 E1B 55K proteins. Virology 179:806-814. [DOI] [PubMed] [Google Scholar]
- 56.Kasai, Y., H. Chen, and S. J. Flint. 1992. Anatomy of an unusual RNA polymerase II promoter containing a downstream TATA element. Mol. Cell. Biol. 12:2884-2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Konig, C., J. Roth, and M. Dobbelstein. 1999. Adenovirus type 5 E4orf3 protein relieves p53 inhibition by E1B-55-kilodalton protein. J. Virol. 73:2253-2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kratzer, F., O. Rosorius, P. Heger, N. Hirschmann, T. Dobner, J. Hauber, and R. H. Stauber. 2000. The adenovirus type 5 E1B-55K oncoprotein is a highly active shuttle protein and shuttling is independent of E4orf6, p53 and Mdm2. Oncogene 19:850-857. [DOI] [PubMed] [Google Scholar]
- 59.Leppard, K. N., and R. D. Everett. 1999. The adenovirus type 5 E1b 55K and E4 Orf3 proteins associate in infected cells and affect ND10 components. J. Gen. Virol. 80:997-1008. [DOI] [PubMed] [Google Scholar]
- 60.Leppard, K. N., and T. Shenk. 1989. The adenovirus E1B 55 kd protein influences mRNA transport via an intranuclear effect on RNA metabolism. EMBO J. 8:2329-2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lunt, R., M. E. Vayda, M. Young, and S. J. Flint. 1988. Isolation and characterization of monoclonal antibodies against the adenovirus core proteins. Virology 164:275-279. [DOI] [PubMed] [Google Scholar]
- 62.Maderious, A., and S. Chen-Kiang. 1984. Pausing and premature termination of human RNA polymerase II during transcription of adenovirus in vivo and in vitro. Proc. Natl. Acad. Sci. USA 81:5931-5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martin, M. E., and A. J. Berk. 1998. Adenovirus E1B 55K represses p53 activation in vitro. J. Virol. 72:3146-3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mathews, M. B., and T. E. Shenk. 1991. Adenovirus virus-associated RNA and translation control. J. Virol. 65:5657-5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moran, E. 1993. DNA tumor virus transforming proteins and the cell cycle. Curr. Opin. Genet. Dev. 3:63-70. [DOI] [PubMed] [Google Scholar]
- 66.Nayak, D. P. 1977. The molecular biology of animal viruses. M. Dekker, New York, N.Y.
- 67.Nevins, J. R. 1992. E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science 258:424-429. [DOI] [PubMed] [Google Scholar]
- 68.O'Shea, C., L. Johnson, B. Bagus, S. Choi, C. Nicholas, A. Shen, L. Boyle, K. Pandey, C. Soria, J. Kunich, Y. Shen, G. Habets, D. Ginzinger, and F. McCormick. 2004. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell 6:611-623. [DOI] [PubMed] [Google Scholar]
- 69.Obert, S., R. J. O'Connor, S. Schmid, and P. Hearing. 1994. The adenovirus E4-6/7 protein transactivates the E2 promoter by inducing dimerization of a heteromeric E2F complex. Mol. Cell. Biol. 14:1333-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ornelles, D., and T. Shenk. 1991. Location of the adenovirus early region 1B 55 kilodalton protein during lytic infection: association with nuclear viral inclusions requires the early region 4 34-kilodalton protein. J. Virol. 65:424-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pardo-Mateos, A., and C. S. Young. 2004. A 40 kDa isoform of the type 5 adenovirus IVa2 protein is sufficient for virus viability. Virology 324:151-164. [DOI] [PubMed] [Google Scholar]
- 72.Pilder, S., J. Logan, and T. E. Shenk. 1984. Deletion of the gene encoding the adenovirus 5 early region 1b 21,000-molecular weight polypeptide leads to degradation of viral and host cell DNA. J. Virol. 52:664-671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pilder, S., M. Moore, J. Logan, and T. Shenk. 1986. The adenovirus E1B-55K transforming polypeptide modulates transport or cytoplasmic stabilization of viral and host cell mRNAs. Mol. Cell. Biol. 6:470-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pombo, A., J. Ferreira, E. Bridge, and M. Carmo-Fonseca. 1994. Adenovirus replication and transcription sites are spatially separated in the nucleus of infected cells. EMBO J. 13:5075-5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Puvion-Dutilleul, F., R. Roussev, and E. Puvion. 1992. Distribution of viral RNA molecules during the adenovirus type 5 infectious cycle in HeLa cells. J. Struct. Biol. 108:209-220. [DOI] [PubMed] [Google Scholar]
- 76.Querido, E., R. C. Marcellus, A. Lai, R. Charbonneau, J. G. Teodoro, G. Ketner, and P. E. Branton. 1997. Regulation of p53 levels by the E1B 55-kilodalton protein and E4orf6 in adenovirus-infected cells. J. Virol. 71:3788-3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Querido, E., M. R. Morisson, H. Chu-Pham-Dang, S. W. Thirlwell, D. Boivin, and P. E. Branton. 2001. Identification of three functions of the adenovirus e4orf6 protein that mediate p53 degradation by the E4orf6-E1B55K complex. J. Virol. 75:699-709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rabino, C., A. Aspegren, K. Corbin-Lickfett, and E. Bridge. 2000. Adenovirus late gene expression does not require a Rev-like nuclear RNA export pathway. J. Virol. 74:6684-6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reich, N. C., and A. J. Levine. 1984. Growth regulation of a cellular tumor antigen, p53, in non-transformed cells. Nature 308:199-201. [DOI] [PubMed] [Google Scholar]
- 80.Reich, N. C., P. Sarnow, E. Duprey, and A. J. Levine. 1983. Monoclonal antibodies which recognise native and denatured forms of the adenovirus DNA-binding protein. Virology 128:480-484. [DOI] [PubMed] [Google Scholar]
- 81.Ridgway, P. J., A. R. Hall, C. J. Myers, and A. W. Braithwaite. 1997. p53/E1b58kDa complex regulates adenovirus replication. Virology 237:404-413. [DOI] [PubMed] [Google Scholar]
- 82.Rothmann, T., A. Hengstermann, N. J. Whitaker, M. Scheffner, and H. zur Hausen. 1998. Replication of ONYX-015, a potential anticancer adenovirus, is independent of p53 status in tumor cells. J. Virol. 72:9470-9478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rubenwolf, S., H. Schutt, M. Nevels, H. Wolf, and T. Dobner. 1997. Structural analysis of the adenovirus type 5 E1B 55-kilodalton-E4orf6 protein complex. J. Virol. 71:1115-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sarnow, P., P. Hearing, C. W. Anderson, D. N. Halbert, T. Shenk, and A. J. Levine. 1984. Adenovirus early region 1B 58,000 dalton tumor antigen is physically associated with an early region 4 25,000-dalton protein in productively infected cells. J. Virol. 49:692-700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schneider, R. J., and I. Mohr. 2003. Translation initiation and viral tricks. Trends Biochem. Sci. 28:130-136. [DOI] [PubMed] [Google Scholar]
- 86.Shen, Y., G. Kitzes, J. A. Nye, A. Fattaey, and T. Hermiston. 2001. Analyses of single-amino-acid substitution mutants of adenovirus type 5 E1B-55K protein. J. Virol. 75:4297-4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shenk, T. 1996. Adenoviridae and their replication, p. 2111-2148. In B. Fields, P. Howley, and D. Knipe (ed.), Fields virology. Raven Press, New York, N.Y.
- 88.Shenk, T. 2002. Might a vanguard of mRNAs prepare cells for the arrival of herpes simplex virus? Proc. Natl. Acad. Sci. USA 99:8465-8466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Spindler, K. R., C. Y. Eng, and A. J. Berk. 1985. An adenovirus early region 1A protein is required for maximal viral DNA replication in growth-arrested human cells. J. Virol. 53:742-750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Steegenga, W. T., N. Riteco, A. G. Jochemsen, F. J. Fallaux, and J. L. Bos. 1998. The large E1B protein together with the E4orf6 protein target p53 for active degradation in adenovirus infected cells. Oncogene 16:349-357. [DOI] [PubMed] [Google Scholar]
- 91.Stracker, T. H., C. T. Carson, and M. D. Weitzman. 2002. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 418:348-352. [DOI] [PubMed] [Google Scholar]
- 92.Takayesu, D., J. G. Teodoro, S. G. Whalen, and P. E. Branton. 1994. Characterization of the 55K adenovirus type 5 E1B product and related proteins. J. Gen. Virol. 75:789-798. [DOI] [PubMed] [Google Scholar]
- 93.Tribouley, C., P. Lutz, A. Staub, and C. Kedinger. 1994. The product of the adenovirus intermediate gene IVa2 is a transcription activator of the major late promoter. J. Virol. 68:4450-4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Turnell, A. S., R. J. Grand, and P. H. Gallimore. 1999. The replicative capacities of large E1B-null group A and group C adenoviruses are independent of host cell p53 status. J. Virol. 73:2074-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weiden, M. D., and H. S. Ginsberg. 1994. Deletion of the E4 region of the genome produces adenovirus DNA concatemers. Proc. Natl. Acad. Sci. USA 91:153-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Williams, J., B. D. Karger, Y. S. Ho, C. L. Castiglia, T. Mann, and S. J. Flint. 1986. The adenovirus E1B 495R protein plays a role in regulating the transport and stability of the viral late messages. Cancer Cells 4:275-284. [Google Scholar]
- 97.Yew, P. R., C. C. Kao, and A. J. Berk. 1990. Dissection of functional domains in the adenovirus 2 early 1B 55k polypeptide by suppressor-linker-insertional mutagenesis. Virol. 179:795-805. [DOI] [PubMed] [Google Scholar]
- 98.Yew, P. R., X. Liu, and A. J. Berk. 1994. Adenovirus E1B oncoprotein tethers a transcriptional repression domain to p53. Genes Dev. 8:190-202. [DOI] [PubMed] [Google Scholar]