

Abstract
ADAR2 is a double-stranded-RNA-specific adenosine deaminase involved in the editing of mammalian RNAs by the site-selective conversion of adenosine to inosine. Previous studies from our laboratory have demonstrated that ADAR2 can modify its own pre-mRNA to create a proximal 3′ splice site containing a noncanonical adenosine-inosine dinucleotide. Alternative splicing to this proximal acceptor adds 47 nucleotides to the mature ADAR2 transcript, thereby resulting in the loss of functional ADAR2 protein expression due to premature translation termination in an alternate reading frame. To examine whether the editing of ADAR2 transcripts represents a negative autoregulatory strategy to modulate ADAR2 protein expression, we have generated genetically modified mice in which the ability of ADAR2 to edit its own pre-mRNA has been selectively ablated by deletion of a critical sequence (editing site complementary sequence [ECS]) required for adenosine-to-inosine conversion. Here we demonstrate that ADAR2 autoediting and subsequent alternative splicing are abolished in homozygous ΔECS mice and that ADAR2 protein expression is increased in numerous tissues compared to wild-type animals. The observed increases in ADAR2 protein expression correlate with the extent of ADAR2 autoediting observed with wild-type tissues and correspond to increases in the editing of ADAR2 substrates, indicating that ADAR2 autoediting is a key regulator of ADAR2 protein expression and activity in vivo.
The conversion of adenosine to inosine (A to I) by RNA editing is a widespread posttranscriptional modification resulting from the hydrolytic deamination of selective adenosine residues that alters the nucleotide sequence of RNA transcripts from that encoded by genomic DNA. The majority of well-characterized A-to-I editing events involve nonsynonymous codon changes in mRNA sequences, resulting in the production of proteins with altered functional properties. In mammals, the most prominent examples of A-to-I editing have been described for transcripts encoding ionotropic glutamate receptor subunits (GluR), a voltage-gated potassium channel subunit (Kv1.1), and the 2C subtype of the serotonin receptor (5-HT2CR), which lead to the production of channels with altered electrophysiological and ion permeation properties (6, 27, 37, 38, 43, 54) and receptors with decreased G-protein coupling efficiency (5, 10, 46). A-to-I modifications have also been described for nontranslated RNA species and noncoding regions of RNA transcripts, suggesting that such RNA modifications may also affect other aspects of RNA function, including splicing, trafficking, translation efficiency, and transcript stability (1, 7, 35, 42, 45).
The enzymes responsible for the site-specific deamination of A to I in mRNA transcripts are known as adenosine deaminases that act on RNA (ADARs) (2, 3, 34, 53). For mammals, three ADAR proteins (ADAR1, ADAR2, and ADAR3) and their corresponding genes have been identified (12, 28, 36, 44). ADAR1 and ADAR2 have been shown to be ubiquitously expressed (44, 50, 59) and mediate A-to-I conversion in synthetic double-stranded RNAs (dsRNAs) and naturally occurring pre-mRNA substrates (18, 41, 49, 53). ADAR3 is expressed exclusively in the brain but has not been shown to have any catalytic activity using synthetic dsRNA or known ADAR targets (12). ADAR2, an 80-kDa nuclear protein with two dsRNA-binding domains (dsRBDs) in the amino-terminal region and an adenosine deaminase domain near the carboxyl terminus, is responsible for the majority of known codon-altering A-to-I modifications in mammals (25, 26, 61). Genetically modified mice lacking ADAR2 expression develop progressive seizures and die postnatally by day 21 due to a lack of editing (Q/R site) in transcripts encoding a subunit of the α-amino-3-hydroxy-5-methyl-isoxazole-4-propionic acid (AMPA)-subtype glutamate receptor (GluR-2) (26), demonstrating the critical role that ADAR2 plays in the normal development and function of the central nervous system (CNS).
Multiple splice variants of ADAR2 have been identified for rats, mice, and humans; yet only a subset of these RNA-processing events are conserved in all three species (22, 50, 56). One such splicing event results in the production of ADAR2 protein isoforms containing an additional 40 (human) or 10 (rat or mouse) amino acids between the second and third zinc coordination residues of the deaminase domain, generating enzymes that are approximately twice as active as those lacking the insertion (22, 50). Alternative 3′ splice site selection near the 5′ end of the coding region introduces an additional 47 nucleotides (nt) to the mature ADAR2 mRNA, generating a −1 frameshift that is predicted to produce a 9-kDa protein lacking the dsRBDs and the deaminase domain required for protein function (50, 56). Previous studies have demonstrated that this alternative splicing event is dependent upon the ability of ADAR2 to edit its own pre-mRNA, converting an adenosine residue within intron 4 (−1 site) to inosine, thereby creating a noncanonical A-I dinucleotide which serves as a novel 3′ splice acceptor (50). As with all identified ADAR substrates, editing at the −1 site of ADAR2 requires an imperfect RNA duplex formed by the base-pairing of sequences surrounding the −1 site with an inverted region referred to as the editing site complementary sequence (ECS). In the rat, the ADAR2 ECS is located approximately 1.4 kb upstream of the −1 site in intron 4 (14, 50), and this region of the intron demonstrates a high level of sequence conservation with corresponding sequences from ADAR2 genes isolated from all characterized vertebrate species (see Fig. S1 in the supplemental material) (21, 50, 56). Deletion of the ECS region resulted in the ablation of editing at the −1 site in heterologous expression systems, while deletion of intronic sequences between the ECS and the region surrounding the −1 site had little effect on the extent of A-to-I conversion (14, 50).
The ability of ADAR2 to edit and direct alternative splicing of its own pre-mRNA may represent an autoregulatory mechanism by which ADAR2 can prevent its own overexpression to avoid increased levels of editing for numerous ADAR2 substrates. In this report, we have generated genetically modified mice in which the ability of ADAR2 to edit its own transcript has been selectively ablated. Here we show that disruption of the predicted duplex required for A-to-I editing resulted in a loss of editing at the −1 site, ablation of alternative splicing, increased ADAR2 protein expression in multiple tissues, and a concomitant increase in the editing of ADAR2 target RNAs. The observed increases in ADAR2 protein expression correlated with the extent of editing-dependent alternative splicing observed with wild-type tissues, further demonstrating that autoediting represents a key regulatory step in the modulation of ADAR2 expression and the extent of A-to-I conversion for ADAR2 substrates.
MATERIALS AND METHODS
Generation of mutant mice.
The targeting vector was generated from two 129S6/SvEvTac mouse genomic bacterial artificial chromosome clones (Genome Systems, Inc.) containing the ADAR2 gene. A short-arm fragment extending from the 3′ end of intron 2 to a region upstream of the ECS element in intron 4 (−4,053 to −1,575 base pairs relative to the −1 editing site) was floxed and inserted between XhoI and BamHI sites, replacing the first LoxP site within sites in the LoxP-Neo-TK vector (a generous gift from Mark Magnuson, Vanderbilt University), where Neo is the neomycin resistance cassette and TK is thymidine kinase. A 7.4-kb long arm harboring the remainder of intron 4, downstream from the ECS element through intron 6 (−1,369 to +6,061 relative to the −1 editing site), was inserted between SalI and NotI sites in the LoxP-Neo-TK vector, and a 195-bp ECS containing an intronic fragment (−1,517 to −1,322 bp relative to the −1 site) was inserted between the two adjacent LoxP sites using BamHI digestion. The thymidine kinase gene was excised from the targeting vector by EcoRI digestion to eliminate the previously described toxicity of thymidine kinase to the male germ line (8, 17). The ClaI-linearized targeting construct was electroporated into 129S6/SvEvTac-derived embryonic stem cells. The integration of the targeting vector and the fidelity of homologous recombination were confirmed by Southern blotting analysis using 5′ and 3′ probes outside the region of homology, and the correctly targeted embryonic stem cells were microinjected into C57BL/6J blastocysts. High-percentage chimeras were mated with 129S6/SvEvTac animals to obtain heterozygous mice bearing the targeted allele (3lox [see Fig. 1A ]), which were then mated with EIIA-Cre transgenic mice developed on a C57BL/6J background (a generous gift from Richard Breyer, Vanderbilt University) to delete the region between the loxP sites early in embryogenesis (40, 62). Progeny from this mating were analyzed by PCR and direct DNA sequence analysis for either loss of the phosphoglycerate kinase (PGK)-neomycin cassette alone (lox-ECS-lox) or recombination of the entire region containing the selectable marker and a 209-bp region of ADAR2 containing the ECS (ΔECS). Mutant mice bearing the ΔECS alleles were mated with wild-type C57BL/6J animals, and subsequent progeny were assessed for the presence of the modified ADAR2 allele and loss of the Cre transgene by segregation.
FIG. 1.

Targeting strategy for selective ablation of ADAR2 autoediting. (A) Schematic diagram of a portion of the mouse ADAR2 gene, extending from exon 3 through exon 6, before and after targeted gene modification. The positions of the positive selectable marker (PGK-neo), the ECS, and the alternative 3′ splice site generated by ADAR2 autoediting (*) are indicated. The positions of primer pairs used for genotype analysis (→), introduced loxP recombination sites for selective deletion of the PGK-neo cassette and the ECS region (▹), and DNA sequences outside the region of targeting vector homology (---) are shown. (B) Analysis of mouse genotype by PCR amplification of mouse tail genomic DNA. The migration positions for PCR amplicons corresponding to the wild-type (+/+; 343 bp) and ΔECS (−/−; 172 bp) alleles are indicated.
Genotype analysis.
The wild-type and targeted (3lox [see Fig. 1A]) alleles were distinguished using a PCR-based strategy with sense (5′-TGTGAGCAGATGTAGCGTGTGAT-3′) and antisense (5′-AATTTTCATTTATAACATCCTC-3′) primers, flanking the upstream loxP site to generate PCR amplicons of 140 and 174 bp for wild-type and targeted alleles, respectively. To distinguish between the wild-type and ΔECS alleles, PCR was performed using sense (5′-TGTGAGCAGATGTAGCGTGTGAT-3′) and antisense (5′-GCGGGCAGGG TTAATGTGATGA-3′) primers flanking the ECS region to generate 343- and 172-bp amplicons for the wild-type and ΔECS alleles, respectively.
RNA characterization.
Male ΔECS mice and wild-type littermates were sacrificed at 4 weeks of age, organs were immediately frozen in liquid nitrogen, and RNA was isolated and purified using a Versagene RNA tissue kit and DNase kit (Gentra Systems) according to the manufacturer's instructions. To quantify relative expression of ADAR2 mRNA splice variants in RNAs isolated from tissues in wild-type and ΔECS mice, first-strand cDNA was synthesized using avian myeloblastosis virus reverse transcriptase (Promega) and then amplified using PCR with a 6-carboxyfluorescein-labeled sense primer (5′-CGCTTGCTATTTTAGTGCTGCGG-3′) and a nonlabeled antisense primer (5′-GCGGTTTTCTTTAACATCAGTGC-3′). Amplicons corresponding to alternatively spliced ADAR2 variants were resolved by 2.5% agarose gel electrophoresis and quantified by phosphorimager analysis (Amersham Biosciences).
To determine the extents of site-selective editing for different ADAR substrates, first-strand cDNA was synthesized and amplified using gene-specific primers and assessed using a modified primer extension analysis (50); the primers and mixtures of deoxy- and dideoxynucleotides used in each primer extension assay are listed in Table S1 in the supplemental material. The extension products were resolved by denaturing polyacrylamide gel electrophoresis and quantified using phosphorimager analysis.
To quantify levels of ADAR1 and ADAR2 mRNA expression, first-strand cDNA was synthesized as described above and subjected to TaqMan real-time PCR analysis (Applied Biosystems). All primers and probes used for the real-time PCRs were products of Assay-On-Demand from Applied Biosystems (ADAR1, assay ID Mm00508001_m1; ADAR2, assay ID Mm00504621_m1). Eukaryotic 18S rRNA (product no. 4319413E; Applied Biosystems) was included in each multiplex PCR as an internal control. Real-time PCR and subsequent analysis were performed with an ABI Prism 7900HT sequence detection system (SDS v2.1; Applied Biosystems). Quantitation of target gene expression in all samples was normalized to 18S rRNA expression by the equation CT(target) − CT(18S) =ΔCT, where CT is the threshold cycle number. The mean ΔCT value of samples from each tissue for all wild-type animals was determined and used as a reference point for the samples from the same tissues for ΔECS mice. Differences between wild-type and ΔECS mice, including individual variation, were calculated by the equation ΔCT(individual ΔECS) − ΔCT(mean wild-type) = ΔΔCT. Changes in target gene expression (n-fold) in each sample were calculated by 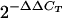 , from which the means and standard errors of the means (SEM) were derived.
, from which the means and standard errors of the means (SEM) were derived.
For quantification of expression levels for ADAR2 mRNA isoforms, RNase protection analysis was performed, essentially as described previously (19), using an [α-32P]UTP-labeled antisense probe corresponding to a region from −88 to +314 relative to the start codon of the mouse ADAR2 cDNA (GenBank accession no. AF403109). A 111-nt antisense RNA probe for cyclophilin (nt 34 to 144; GenBank accession no. M19533) was used as an internal control for RNA loading and normalization of expression levels. The protected fragments were resolved by denaturing polyacrylamide gel electrophoresis and quantified by phosphorimager analysis.
Quantitative Western blotting analysis.
Expression levels of ADAR2 and β-actinin tissue homogenates from wild-type and ΔECS mice were quantified by Western blotting analysis using an anti-ADAR2 polyclonal antibody (1:250 [vol/vol]) (50, 52) and an anti-β-actin monoclonal antibody (1:500 [vol/vol], sc-1616; Santa Cruz Biotechnology), respectively, followed by an Alexa Fluor 680 anti-goat secondary antibody (1:10,000 [vol/vol], A21084; Molecular Probes). The secondary antibody was detected directly using an Odyssey infrared imaging system in accordance with the manufacturer's instructions (Li-Cor Biosciences). The linear range of detection for the assay was defined by quantifying serial dilutions of tissue homogenates.
Statistical analysis.
Student's t test was performed using GraphPadPRISM (GraphPad Software, Inc.). Values are reported as means ± SEM, and P values of <0.05 were considered significant.
RESULTS
Generation of ΔECS mice.
To examine whether the ability of ADAR2 to edit its own pre-mRNA represents a negative autoregulatory mechanism to modulate ADAR2 protein expression, we generated genetically modified mice in which ADAR2 autoediting was selectively ablated. Previous analysis of the cis-active elements necessary for ADAR2 autoediting revealed the requirement for an extended RNA duplex formed by base-pairing interactions between the region surrounding the proximal 3′ splice site in intron 4 (−1 site) and an ECS element located more centrally in the intron (see Fig. S1 in the supplemental material) (14, 50, 56). To selectively eliminate ADAR2 autoediting, we deleted the ECS region to disrupt the conserved duplex structure (Fig. 1A), similarly to previous studies in which editing of the Q/R site in GluR-2 pre-mRNAs was disrupted by deletion of the intronic ECS (9). Since previous studies demonstrated that ablation of ADAR2 expression resulted in early-onset seizures and postnatal lethality (26), we chose to circumvent the possibility of a lethal phenotype by generating mutant mouse strains in which the ECS region could be conditionally deleted by taking advantage of the P1 bacteriophage Cre-loxP system (62). Mutant mice that were heterozygous for the modified ADAR2 allele (3-lox [Fig. 1A]) were mated to transgenic animals expressing Cre recombinase under the control of the EIIA promoter (40) to delete the DNA sequences between the loxP sites early in embryogenesis. Progeny from this mating lost either the PGK-neomycin cassette alone (lox-ECS-lox [Fig. 1A]) or the region of ADAR2 gene containing the ECS (lox-neo-lox) or both (ΔECS [Fig. 1A]). Heterozygous ΔECS mice were interbred, and progeny demonstrated the expected Mendelian frequency (34 wild type [+/+], 67 heterozygous mutants [+/−], and 44 homozygous mutants [−/−]) (χ2 = 2.2; P > 0.25), as assessed using a PCR-based assay (Fig. 1B); no obvious phenotypic alterations were observed with animals homozygous for the ΔECS allele.
Ablation of ADAR2 pre-mRNA editing and alternative splicing in ΔECS mice.
To confirm that deletion of the ECS region resulted in the loss of ADAR2 autoediting, we took advantage of a primer extension-based assay (50) to quantify the editing efficiency of ADAR2 pre-mRNA transcripts isolated from multiple tissues in wild-type and homozygous ΔECS mice. The extents of ADAR2 pre-mRNA editing (−1 site) varied widely in tissues from wild-type animals, with the greatest and least levels of editing observed in lung (32%) and testis (8%), respectively. By contrast, only background levels of editing were observed with tissues from ΔECS mice (Fig. 2A), thereby demonstrating that the 195-bp ECS region was critical for A-to-I conversion in vivo.
FIG. 2.

Ablation of ADAR2 autoediting and alternative splicing in ΔECS mice. (A) Extent of editing at the −1 site in ADAR2 pre-mRNA isolated from different tissues of wild-type (open columns) and ΔECS (filled columns) mice was determined by primer extension analysis. Values were obtained from tissue isolated from three animals per genotype (mean ± SEM). (B) Schematic diagram of four possible ADAR2 mRNA isoforms resulting from alternative splicing of exon 4 and inclusion or exclusion of 47-nt cassette (+47 and −47, respectively) at the 5′ boundary of exon 5. (C) Quantitative analysis of ADAR2 alternative splicing. The positions of RT-PCR amplicons corresponding to the four alternatively spliced ADAR2 transcripts described for panel B are indicated. The percentages of total ADAR2 transcripts containing the 47-nt cassette but not exon 4 (3/5 +47) are shown (mean ± SEM; n = 3).
Since the generation of an A-I dinucleotide to form a functional 3′ splice site in intron 4 requires ADAR2 autoediting, it was predicted that abrogation of this RNA-processing event would result in sole use of the distal splice acceptor immediately preceding exon 5 to produce ADAR2 mRNAs that lack the 47-nt cassette and encode a full-length, functional protein product. However, in addition to alternative 3′ splice site selection in this region of the ADAR2 gene, recent studies have identified an additional exon (exon 4) that occurs 174 bp downstream of exon 3 within the ADAR2 gene (56). While inclusion of this 73-bp exon is predicted to result in a +1 frameshift, the extent of exon 4 inclusion in ADAR2 transcripts isolated from mouse brain was very low (56). As a result of these alternative splicing events, up to four ADAR2 splice variants can be generated in the region between exons 3 and 5 (Fig. 2B). Among these four mRNA isoforms, only the isoform lacking both exon 4 and the 47-nt cassette (designated 3/5 −47) can encode a full-length ADAR2 protein. To quantify the relative abundance of ADAR2 splice variants, we developed a quantitative reverse transcription (RT)-PCR-based strategy using a 6-carboxyfluorescein-labeled sense PCR primer. The quantitative accuracy of this method was confirmed using a series of templates with mixtures of plasmids containing or lacking the 47-nt cassette at fixed ratios (see Fig. S2 in the supplemental material). Consistent with previous reports (56), mRNA isoforms containing exon 4 represented a small percentage of total ADAR2 transcripts in all tissues (Fig. 2C), while use of the proximal A-I splice site roughly paralleled the tissue-specific pattern of −1 site editing in wild-type animals (Fig. 2A and C). In RNA isolated from various tissues of ΔECS mice however, ADAR2 mRNA isoforms containing the 47-nt cassette were not observed, demonstrating that deletion of the ECS region concomitantly ablated ADAR2 autoediting and the ability to use the proximal splice site in intron 4. The single exception to this observation was the presence of a PCR amplicon generated from testis RNA that comigrated with the 3/5 +47 isoform, representing less than 2% of the ADAR2 mRNA transcripts.
ADAR2 protein expression in ΔECS mice.
The ablation of ADAR2 autoediting and proximal splice acceptor use in ΔECS mice resulted in the predominant production of an ADAR2 mRNA isoform (3/5 −47) that encodes full-length, functional ADAR2 protein. By contrast, expression of the 3/5 +47 isoform in wild-type mice, representing a significant portion of the total ADAR2 transcripts in most tissues, is predicted to generate a truncated, inactive ADAR2 protein resulting from the premature translation termination. We therefore anticipated that the ablation of ADAR2 autoediting would lead to elevated ADAR2 protein expression in ΔECS mice. Homogenates prepared from multiple tissues of wild-type and ΔECS animals were assessed for ADAR2 protein levels by quantitative Western blotting analysis using antisera directed against ADAR2 and β-actin. In the lung, where the greatest level of ADAR2 autoediting and 47-nt inclusion was observed with wild-type animals (Fig. 2C), a 3.7-fold increase in ADAR2 protein expression was identified with ΔECS mice (Fig. 3). Similarly, in the brain and heart, where the +47 isoform represented >50% of the total ADAR2 mRNA, 2.4- and 1.7-fold increases in ADAR2 protein were exhibited, respectively. In tissues such as the liver and testis, where ADAR2 autoediting and proximal splice site use occurred at a low frequency in wild-type animals, the ADAR2 protein expression in ΔECS mice was not significantly different from that of control animals (Fig. 3).
FIG. 3.

Quantitative Western blotting analysis of ADAR2 protein levels in tissues isolated from wild-type (WT) and ΔECS mice. A representative Western blot using 20 μg of total protein from each tissue is presented; the expected migration positions for ADAR2 and β-actin are indicated. The increase of ADAR2 protein expression in ΔECS versus wild-type tissues was quantified after normalization to the internal β-actin control (mean ± SEM; n = 5; **, P < 0.01; *, P < 0.05).
Characterization of ADAR2 mRNA expression in ΔECS mice.
To confirm that the elevated ADAR2 protein levels in ΔECS mice resulted from alterations in 3′ splice site selection rather than an increase in the steady-state levels of ADAR2 transcripts, an RNase protection assay was employed to quantify isoform-specific ADAR2 mRNA expression in tissues from control and mutant animals. Due to the complexity of alternative splicing events between exons 3 and 5, generating as many as four mRNA isoforms, the RNase protection probe was designed to generate a 481-nt fragment from the 3/5 −47 mRNA, whereas partial protection by all other mRNA isoforms and unspliced pre-mRNA gave rise to a 332-nt protected species (Fig. 4A). We were unable to develop a probe that specifically recognized the mRNA isoform containing the 47-nt cassette, due to the presence of multiple editing sites at positions 10, 23, and 24 (relative to the −1 site) in this region (14). Results from this analysis demonstrated the expected increase in the relative level of 3/5 −47 ADAR2 mRNA, when normalized to a constitutively expressed cyclophilin RNA control, in the brains and lungs of ΔECS mice. A trend toward an increase in 3/5 −47 mRNA levels was also observed for RNA from the hearts of mutant mice; however, the difference did not reach statistical significance with the current sample size. Consistent with the lower levels of autoediting observed with wild-type animals (Fig. 2A), no significant increase in the 3/5 −47 isoform was seen with RNAs isolated from the liver and the testis in ΔECS mice (Fig. 4B). To further confirm that deletion of the ECS region did not result in alterations in the steady-state level of ADAR2 transcripts, a real-time RT-PCR-based strategy using a TaqMan probe spanning exons 9 and 10 was performed to quantify total ADAR2 mRNAs in multiple tissues from wild-type and ΔECS mice. Results from this analysis revealed that despite the increase in ADAR2 protein expression in ΔECS animals (Fig. 3), there was no significant change in the total level of ADAR2 mRNAs in any tissue examined, except for the brain, where there was a 30% decrease in ADAR2 mRNA (Fig. 4C). This decrease in the steady-state level of ADAR2 transcripts could result from a tissue-specific instability of the modified ADAR2 pre-mRNA, alterations in splicing efficiency, or a compensatory transcriptional alteration to counteract elevated ADAR2 protein levels.
FIG. 4.

Characterization of ADAR2 mRNA isoform expression. (A) Schematic diagram indicating the structures and sizes of predicted RNase protection products resulting from ADAR2 pre-mRNAs or ADAR2 mRNA isoforms containing or lacking the 47-nt cassette or exon 4. (B) RNase protection analysis of total RNA (10 μg) from tissues of wild-type (WT) and ΔECS mice. The predicted migration positions for protected RNA fragments are indicated. The increase (n-fold) in ADAR2 RNA isoforms lacking both the 47-nt cassette and exon 4 (3/5 −47) in ΔECS versus wild-type tissues was quantified after normalization to the internal cyclophilin control (mean ± SEM; n = 6 for brain, lung, and heart; n = 3 for liver and testis; **, P < 0.01; *, P < 0.05.) (C) Quantification of relative ADAR2 mRNA expression between wild-type (open columns) and ΔECS (filled columns) mice by use of real-time RT-PCR analysis (mean ± SEM; n = 6 for brain, lung, and heart; n = 3 for liver and testis; **P < 0.01; *P < 0.05).
Alterations in editing of ADAR substrates in ΔECS mice.
ADAR2 has been shown to be responsible for the site-selective editing of numerous mammalian RNAs (6, 10, 46, 50, 60). To examine whether the increase in ADAR2 protein expression observed with ΔECS mice resulted in editing changes for ADAR2 substrates, we quantified the extent of editing for known ADAR2 targets, including the D site of 5-HT2CR transcripts, the R/G site of the GluR-2 pre-mRNA, and the I/V site of the Kv1.1 mRNA (6, 25, 27, 60, 61). Since expression of most identified ADAR2 substrates is restricted to the nervous system (26), we determined the editing efficiencies of ADAR2 targets in RNA isolated from whole brains of wild-type and ΔECS mice by using a modified primer extension analysis (10). While the level of editing at the D site for 5-HT2CR transcripts (47.8%) isolated from wild-type mice was comparable to that found by previous studies (61), a 15% increase in editing was seen with ΔECS animals (Table 1). Similarly, editing for GluR-2 (R/G site) and Kv1.1 (I/V site) was increased by 11% and 10%, respectively, in ΔECS animals (Table 1). To determine whether the changes in editing were limited to ADAR2-selective sites, we also examined the editing efficiencies of two known ADAR1 sites, the +60 site in the GluR-2 pre-mRNA and the A site in 5-HT2CR transcripts (10, 25, 26, 60). The extent of editing at the A site in 5-HT2CR transcripts decreased from 74% in wild-type animals to 64% in ΔECS mice, yet editing at the +60 site was not significantly changed. ADAR1 mRNA expression in the brains of ΔECS mice was not different from that of wild-type animals, as determined by real-time RT-PCR analysis (see Fig. S3 in the supplemental material), suggesting that the decreased editing observed to occur at the A site did not result from a decrease in ADAR1 expression. Recent studies have demonstrated that ADAR2 may compete with ADAR1 binding to the duplex in 5-HT2CR transcripts (12), thus providing a direct molecular mechanism by which increased ADAR2 expression can affect ADAR1-selective editing.
TABLE 1.
Site-specific RNA editing of ADAR substrates
| Transcript | Editing site | Tissue | % Editing (mean ± SEM)
|
|
|---|---|---|---|---|
| Wild type | ΔECSa | |||
| 5-HT2cR | D | Brain | 47.8 ± 2.5 | 63.0 ± 4.7** |
| GluR-2 | R/G | Brain | 68.6 ± 1.8 | 79.8 ± 0.9** |
| Kv1.1 | I/V | Brain | 37.0 ± 3.1 | 46.7 ± 2.6* |
| 5-HT2cR | A | Brain | 74.3 ± 1.2 | 64.4 ± 0.7*** |
| GluR-2 | +60 | Brain | 61.8 ± 1.3 | 60.2 ± 1.7 |
*, P < 0.05; **, P < 0.01; ***, P < 0.001.
Despite significant alterations in the editing of ADAR substrates in ΔECS mice (Table 1), no overt phenotypic alterations were observed with mutant mice from birth through adulthood. Since any expected changes in ΔECS mice are dependent upon the repertoire of ADAR targets affected by changes in ADAR2 expression and the majority of characterized editing events are limited to neurotransmitter receptors in the CNS, we assessed alterations in animal behavior by using a gross examination based upon the neurological screen developed by Irwin (29). This standardized method provides a behavioral and functional profile of wild-type and mutant mice by observational assessment of potential defects in gait or posture, motor control and coordination, changes in excitability and aggression, salivation, lacrimation, piloerection, defecation, muscle tone, and body temperature. Results from this neurological screen identified no significant alterations between ΔECS and wild-type mice (data not shown).
DISCUSSION
Unlike the static changes imparted by genetic polymorphisms, the conversion of A to I by RNA editing represents a dynamic cellular strategy to increase the spatiotemporal diversity of gene expression in response to extracellular cues (23, 24, 63, 65). Selective ablation of ADAR2 or ADAR1 in mutant mice can give rise to pathological conditions ranging from postnatal seizures to embryonic lethality (25, 26, 58, 60, 61), and alterations in A-to-I conversion have been associated with a number of CNS disorders, including depression, schizophrenia, seizure susceptibility, and amyotrophic lateral sclerosis (24, 30, 32, 39, 47, 57, 58). While numerous studies have identified novel ADAR targets in the human and mouse transcriptomes (1, 7, 13, 35, 42, 45), little is known about the molecular mechanisms regulating ADAR expression under basal conditions and in response to a variety of physiologic stimuli. Previous studies have demonstrated that ADAR2 may be regulated by nucleolar sequestration (15, 52) and alternative splicing (22, 50). One such splicing event is dependent upon the ability of ADAR2 to edit its own pre-mRNA, thereby promoting the insertion of a 47-nt cassette near the 5′ end of the open reading frame to abort translation prematurely (50). To determine whether ADAR2 autoediting and subsequent alternative splicing represent important regulatory mechanisms for modulating ADAR2 protein expression and activity, we generated mutant mice in which the ability of ADAR2 to edit its own transcript was selectively ablated.
To eliminate the ability of ADAR2 to generate a proximal 3′ splice site preceding exon 5, we disrupted the predicted RNA duplex required for A-to-I conversion using Cre-mediated recombination to remove the evolutionarily conserved ECS region in intron 4 (14, 50, 56). In the current study, deletion of the ECS region in mice efficiently eliminated editing at the −1 site (Fig. 2A) and inclusion of the 47-nt cassette in mature ADAR2 transcripts (Fig. 2C), further confirming that the ECS element is essential for −1 site editing and subsequent alternative splicing in vivo. Ablation of −1 site editing promoted the use of the distal 3′ splice site immediately preceding exon 5 (Fig. 2C) to allow exclusion of the 47-nt cassette and increase the relative expression of the ADAR2 mRNA isoform (3/5 −47) that encodes the full-length, functional protein (Fig. 4B). Accordingly, increased ADAR2 protein expression in multiple tissues (Fig. 3) and a concomitant increase in the editing of ADAR2 substrates were observed with ΔECS mice (Table 1), strongly supporting the hypothesis that ADAR2 autoediting serves as a negative-feedback mechanism for modulating ADAR2 protein expression.
In the simplest model, where ADAR2 autoediting represents the rate-limiting step for determining alternative splice site selection, the equation Y = 100/(100 − X) can define the relationship between the extent of tissue-specific proximal splice site use in wild-type tissues (X) and the expected increase in ADAR2 protein expression (Y) in ΔECS mice (Fig. 5). For example, only a 1.05-fold increase in ADAR2 protein expression would be predicted for the testis of ΔECS animals, since the +47 RNA isoforms represent only 5% of the total ADAR2 mRNA transcripts in this tissue from wild-type mice [1/(1 − 0.05) = 1.05] (Fig. 2C), whereas a 4.8-fold increase in ADAR2 protein levels would be anticipated for lung samples from mutant mice [1/(1 − 0.79) = 4.8]. A comparison of the relationship between ADAR2 protein expression in ΔECS mice and proximal 3′ splice site use in wild-type tissues revealed a good correlation between predicted and observed values (Fig. 5), confirming that ADAR2 autoediting is a key regulator for modulating ADAR2 protein expression in vivo.
FIG. 5.

ADAR2 autoediting is a key regulator of ADAR2 protein levels. Correlation between ADAR2 autoediting levels in isolated tissues from wild-type mice (from Fig. 3) and relative increases in ADAR2 protein expression from the corresponding tissues in ΔECS animals (from Fig. 4) was determined by nonlinear regression with the equation Y = A/(B − X) (A = 70; B = 98). The best-fit lines defined by the observed [Y = 70/(98−X)] and predicted [Y = 100/(100 − X)] data are indicated with solid and dashed lines, respectively.
The possibility that the increased ADAR2 expression in ΔECS mice resulted from alterations in the steady-state level of ADAR2 transcripts, via changes in RNA stability or transcription, were ruled out by real-time PCR analysis; no change in the level of ADAR2-derived RNAs was observed for most tissues, with the exception of the brain, where the level of mature ADAR2 mRNA was decreased by 30% compared to that of control animals (Fig. 4C). Decreased levels of GluR-2 mRNA have been reported previously for mice with a deletion of the ECS element required for GluR-2 editing (Q/R site), due to nuclear accumulation of incompletely processed primary GluR-2 transcripts (9). A similar decrease in GluR-2 mRNA expression was also observed with ADAR2-null mice, where a low level of GluR-2 editing (Q/R site) was observed (26), indicating that RNA editing may be a prerequisite for efficient splicing and processing of GluR-2 pre-mRNAs (26). RNase protection analysis of ADAR2 RNA isoforms from wild-type mouse brain indicated that a majority of the transcripts (75%) represented a combination of RNAs containing the 47-nt cassette, exon 4, or intron 4 (Fig. 4A and B). Since quantitative RT-PCR analysis further indicated that the major ADAR2 mRNA isoform contained the 47-nt cassette (74%) and isoforms containing exon 4 represented only 3% of the total ADAR2 mRNA species, these data suggest that only a small percentage of the total ADAR2-derived RNAs are represented by unprocessed precursors in wild-type animals. By contrast, analysis of ΔECS mice revealed no detectable RNA isoforms with the 47-nt cassette and only 2.5% of the transcripts containing exon 4 (Fig. 2C), indicating that the 332-nt protected species observed from RNase protection analysis of ΔECS mice (Fig. 4B) (22% of total ADAR2 RNAs) represented almost exclusively ADAR2 pre-mRNA. As with mutant mice in which GluR-2 (Q/R site) editing was eliminated, the ablation of ADAR2 autoediting results in an accumulation of unprocessed RNA precursors that could explain the decreased level of mature ADAR2 transcripts in RNA isolated from ΔECS mouse brain (Fig. 4C).
Consistent with the increased expression of ADAR2 protein, the extent of editing at ADAR2-selective sites was elevated in the brains of ΔECS mice, whereas a significant decrease in editing at an ADAR1-preferred site also was observed (5-HT2CR A site) (Table 1). Previous in vitro studies have indicated that ADAR1 and ADAR2 can affect the site selectivity of one another, presumably due to sequence-independent competition for binding dsRNA (12). Further support for this hypothesis was observed with mice lacking ADAR1 expression, where editing at the ADAR2-selective D site was increased (25). Molecular recognition of dsRNA is a key event for numerous biological pathways, including the trafficking, editing, and maturation of cellular RNA, the interferon-mediated antiviral response, and RNA interference (11, 16, 64). A lack of sequence-specific binding for members of the dsRBD-containing family (20, 51) suggests that ADAR2 may bind to a wide variety of dsRNA substrates, thus competing with the functions of other dsRNA-binding proteins within the cell.
Comparisons of ADAR2 genes between multiple vertebrate species (zebra fish, pufferfish, chicken, rat, mouse, dog, chimpanzee, and humans) have revealed >90% intronic sequence conservation in the predicted region of the inverted repeats (see Fig. S1 in the supplemental material), with a majority of the nucleotide differences clustered in predicted bulge regions within the duplex (14, 50, 56). Furthermore, alternative 3′ splice site selection has been shown to be conserved among these species (56), suggesting that ADAR2 autoediting and alternative splicing represent an important biological process to modulate ADAR2 protein expression in vertebrates. Recent studies of the Drosophila melanogaster ADAR gene (dADAR) have demonstrated that the dADAR enzyme is capable of editing its own mRNA to generate a protein isoform with a serine-to-glycine substitution close to the dADAR active site (33). This single amino acid alteration appears to restrict dADAR function, since the glycine-containing isoform encoded by the edited dADAR mRNA is less active than the genomically encoded protein product. Ubiquitous or muscle-specific expression of a dADAR transgene that cannot be modified by A-to-I conversion resulted in increased editing of a voltage-gated calcium channel transcript (Ca-alpha 1D) and subsequent lethality (33), suggesting that the ability of dADAR to edit its own mRNA is critical for Drosophila viability. In an example of what may represent convergent evolution, dADAR and ADAR2 have developed distinct autoediting strategies to modulate ADAR activity. Explanations for the difference in phenotypic consequences between Drosophila mutants and ΔECS mice include the level of heterologous-promoter-driven dADAR transgene expression in flies compared to increased ADAR2 expression in ΔECS mice, as well as the potential for compensatory alterations in vertebrates that could serve to circumvent the adverse phenotypic consequences resulting from increased ADAR2 activity.
It is commonly believed that the process of negative feedback provides a noise reduction mechanism for biological systems and can serve to minimize the effects of fluctuations on downstream processes (4, 31, 48, 55). A simple model for ADAR2 autoregulation would suggest that the extent of A-to-I conversion at the −1 site is dictated by ADAR2 activity that exceeds a critical threshold value in the cell (Fig. 6). Analysis of ADAR2 autoediting in wild-type mice, however, revealed a broad range of editing efficiency at the −1 site in multiple tissues (Fig. 2). The tissue-specific threshold for ADAR2 autoediting could be determined by any number of cellular processes affecting ADAR2 activity, including the level of ADAR2 protein or the expression of other ADAR2 targets which could inhibit the ability of ADAR2 to modify its own pre-mRNA. In this regard, autoediting could serve as a sensor to modulate editing activity in response to changing levels of ADAR2 targets, thereby maintaining steady-state levels of A-to-I conversion.
FIG. 6.

Model of ADAR2 autoregulation. A schematic diagram of the biosynthetic processes involved in the production of ADAR2 is presented, showing a region of the ADAR2 pre-mRNA between exon 3 and 5, the predicted RNA duplex required for A-to-I editing, and the position of the −1 site (*). The RNA processing pathway lacking −1 site editing, leading to the production of full-length (711-amino-acid [aa]) ADAR2b is indicated with dashed arrows; the functional domains in ADAR2 (nuclear localization signal [NLS], dsRBD, and adenosine deaminase domain) are indicated. The biosynthetic pathway involving A-to-I conversion at the −1 site (*), leading to the production of a predicted 9-kDa (83-aa) protein, is indicated with solid arrows; the cross-hatched box represents the amino acid sequences encoded by an altered reading frame resulting from proximal 3′ splice acceptor use and inclusion of an additional 47 nt in the mature ADAR2 transcript.
Supplementary Material
Acknowledgments
We thank Jim Patton and members of the Emeson laboratory for critical reading of the manuscript.
This work was supported by a grant to R.B.E. from the National Institutes of Health (NS33323).
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Athanasiadis, A., A. Rich, and S. Maas. 2004. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2:e391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bass, B. L. 2002. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 71:817-846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bass, B. L., K. Nishikura, W. Keller, P. H. Seeburg, R. B. Emeson, M. A. O'Connell, C. E. Samuel, and A. Herbert. 1997. A standardized nomenclature for adenosine deaminases that act on RNA. RNA 3:947-949. [PMC free article] [PubMed] [Google Scholar]
- 4.Becskei, A., and L. Serrano. 2000. Engineering stability in gene networks by autoregulation. Nature 405:590-593. [DOI] [PubMed] [Google Scholar]
- 5.Berg, K. A., J. D. Cropper, C. M. Niswender, E. Sanders-Bush, R. B. Emeson, and W. P. Clarke. 2001. RNA-editing of the 5-HT2C receptor alters agonist-receptor-effector coupling specificity. Br. J. Pharmacol. 134:386-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhalla, T., J. J. Rosenthal, M. Holmgren, and R. Reenan. 2004. Control of human potassium channel inactivation by editing of a small mRNA hairpin. Nat. Struct. Mol. Biol. 11:950-956. [DOI] [PubMed] [Google Scholar]
- 7.Blow, M., P. A. Futreal, R. Wooster, and M. R. Stratton. 2004. A survey of RNA editing in human brain. Genome Res. 14:2379-2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braun, R. E., D. Lo, C. A. Pinkert, G. Widera, R. A. Flavell, R. D. Palmiter, and R. L. Brinster. 1990. Infertility in male transgenic mice: disruption of sperm development by HSV-tk expression in postmeiotic germ cells. Biol. Reprod. 43:684-693. [DOI] [PubMed] [Google Scholar]
- 9.Brusa, R., F. Zimmermann, D. S. Koh, D. Feldmeyer, P. Gass, P. H. Seeburg, and R. Sprengel. 1995. Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science 270:1677-1680. [DOI] [PubMed] [Google Scholar]
- 10.Burns, C. M., H. Chu, S. M. Rueter, L. K. Hutchinson, H. Canton, E. Sanders-Bush, and R. B. Emeson. 1997. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature 387:303-308. [DOI] [PubMed] [Google Scholar]
- 11.Carlson, C. B., O. M. Stephens, and P. A. Beal. 2003. Recognition of double-stranded RNA by proteins and small molecules. Biopolymers 70:86-102. [DOI] [PubMed] [Google Scholar]
- 12.Chen, C. X., D. S. Cho, Q. Wang, F. Lai, K. C. Carter, and K. Nishikura. 2000. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA 6:755-767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clutterbuck, D. R., A. Leroy, M. A. O'Connell, and C. A. Semple. 2005. A bioinformatic screen for novel A-I RNA editing sites reveals recoding editing in BC10. Bioinformatics 21:2590-2595. [DOI] [PubMed] [Google Scholar]
- 14.Dawson, T. R., C. L. Sansam, and R. B. Emeson. 2004. Structure and sequence determinants required for the RNA editing of ADAR2 substrates. J. Biol. Chem. 279:4941-4951. [DOI] [PubMed] [Google Scholar]
- 15.Desterro, J. M., L. P. Keegan, M. Lafarga, M. T. Berciano, M. O'Connell, and M. Carmo-Fonseca. 2003. Dynamic association of RNA-editing enzymes with the nucleolus. J. Cell Sci. 116:1805-1818. [DOI] [PubMed] [Google Scholar]
- 16.Doyle, M., and M. F. Jantsch. 2002. New and old roles of the double-stranded RNA-binding domain. J. Struct. Biol. 140:147-153. [DOI] [PubMed] [Google Scholar]
- 17.Ellison, A. R., H. Wallace, R. al-Shawi, and J. O. Bishop. 1995. Different transmission rates of herpesvirus thymidine kinase reporter transgenes from founder male parents and male parents of subsequent generations. Mol. Reprod. Dev. 41:425-434. [DOI] [PubMed] [Google Scholar]
- 18.Emeson, R., and M. Singh. 2001. Adenosine-to-inosine RNA editing: substrates and consequences, p. 109-138. In B. Bass (ed.), RNA editing. Oxford University Press, Oxford, United Kingdom.
- 19.Emeson, R. B., F. Hedjran, J. M. Yeakley, J. W. Guise, and M. G. Rosenfeld. 1989. Alternative production of calcitonin and CGRP mRNA is regulated at the calcitonin-specific splice acceptor. Nature 341:76-80. [DOI] [PubMed] [Google Scholar]
- 20.Fierro-Monti, I., and M. B. Mathews. 2000. Proteins binding to duplexed RNA: one motif, multiple functions. Trends Biochem. Sci. 25:241-246. [DOI] [PubMed] [Google Scholar]
- 21.Frazer, K. A., L. Pachter, A. Poliakov, E. M. Rubin, and I. Dubchak. 2004. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32:W273-W279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gerber, A., M. A. O'Connell, and W. Keller. 1997. Two forms of human double-stranded RNA-specific editase 1 (hRED1) generated by the insertion of an Alu cassette. RNA 3:453-463. [PMC free article] [PubMed] [Google Scholar]
- 23.Gurevich, I., M. T. Englander, M. Adlersberg, N. B. Siegal, and C. Schmauss. 2002. Modulation of serotonin 2C receptor editing by sustained changes in serotonergic neurotransmission. J. Neurosci. 22:10529-10532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gurevich, I., H. Tamir, V. Arango, A. J. Dwork, J. J. Mann, and C. Schmauss. 2002. Altered editing of serotonin 2C receptor pre-mRNA in the prefrontal cortex of depressed suicide victims. Neuron 34:349-356. [DOI] [PubMed] [Google Scholar]
- 25.Hartner, J. C., C. Schmittwolf, A. Kispert, A. M. Muller, M. Higuchi, and P. H. Seeburg. 2004. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J. Biol. Chem. 279:4894-4902. [DOI] [PubMed] [Google Scholar]
- 26.Higuchi, M., S. Maas, F. N. Single, J. Hartner, A. Rozov, N. Burnashev, D. Feldmeyer, R. Sprengel, and P. H. Seeburg. 2000. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 406:78-81. [DOI] [PubMed] [Google Scholar]
- 27.Hoopengardner, B., T. Bhalla, C. Staber, and R. Reenan. 2003. Nervous system targets of RNA editing identified by comparative genomics. Science 301:832-836. [DOI] [PubMed] [Google Scholar]
- 28.Hough, R. F., and B. L. Bass. 1994. Purification of the Xenopus laevis double-stranded RNA adenosine deaminase. J. Biol. Chem. 269:9933-9939. [PubMed] [Google Scholar]
- 29.Irwin, S. 1968. Comprehensive observational assessment. Ia. A systematic, quantitative procedure for assessing the behavioral and physiologic state of the mouse. Psychopharmacologia 13:222-257. [DOI] [PubMed] [Google Scholar]
- 30.Iwamoto, K., and T. Kato. 2003. RNA editing of serotonin 2C receptor in human postmortem brains of major mental disorders. Neurosci. Lett. 346:169-172. [DOI] [PubMed] [Google Scholar]
- 31.Kaern, M., T. C. Elston, W. J. Blake, and J. J. Collins. 2005. Stochasticity in gene expression: from theories to phenotypes. Nat. Rev. Genet. 6:451-464. [DOI] [PubMed] [Google Scholar]
- 32.Kawahara, Y., K. Ito, H. Sun, H. Aizawa, I. Kanazawa, and S. Kwak. 2004. Glutamate receptors: RNA editing and death of motor neurons. Nature 427:801. [DOI] [PubMed] [Google Scholar]
- 33.Keegan, L. P., J. Brindle, A. Gallo, A. Leroy, R. A. Reenan, and M. A. O'Connell. 2005. Tuning of RNA editing by ADAR is required in Drosophila. EMBO J. 24:2183-2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keegan, L. P., A. Leroy, D. Sproul, and M. A. O'Connell. 2004. Adenosine deaminases acting on RNA (ADARs): RNA-editing enzymes. Genome Biol. 5:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim, D. D. Y., T. T. Y. Kim, T. Walsh, Y. Kobayashi, T. C. Matise, S. Buyske, and A. Gabriel. 2004. Widespread RNA editing of embedded Alu elements in the human transcriptome. Genome Res. 14:1719-1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim, U., Y. Wang, T. Sanford, Y. Zeng, and K. Nishikura. 1994. Molecular cloning of cDNA for double-stranded RNA adenosine deaminase, a candidate enzyme for nuclear RNA editing. Proc. Natl. Acad. Sci. USA 91:11457-11461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kohler, M., N. Burnashev, B. Sakmann, and P. H. Seeburg. 1993. Determinants of Ca2+ permeability in both TM1 and TM2 of high affinity kainate receptor channels: diversity by RNA editing. Neuron 10:491-500. [DOI] [PubMed] [Google Scholar]
- 38.Krampfl, K., F. Schlesinger, A. Zörner, M. Kappler, R. Dengler, and J. Bufler. 2002. Control of kinetic properties of GluR2 flop AMPA-type channels: impact of R/G nuclear editing. Eur. J. Neurosci. 15:51-62. [DOI] [PubMed] [Google Scholar]
- 39.Kwak, S., and Y. Kawahara. 2005. Deficient RNA editing of GluR2 and neuronal death in amyotropic lateral sclerosis. J. Mol. Med. 83:110-120. [DOI] [PubMed] [Google Scholar]
- 40.Lakso, M., J. G. Pichel, J. R. Gorman, B. Sauer, Y. Okamoto, E. Lee, F. W. Alt, and H. Westphal. 1996. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc. Natl. Acad. Sci. USA 93:5860-5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lehmann, K. A., and B. L. Bass. 1999. The importance of internal loops within RNA substrates of ADAR1. J. Mol. Biol. 291:1-13. [DOI] [PubMed] [Google Scholar]
- 42.Levanon, E. Y., E. Eisenberg, R. Yelin, S. Nemzer, M. Hallegger, R. Shemesh, Z. Y. Fligelman, A. Shoshan, S. R. Pollock, D. Sztybel, M. Olshansky, G. Rechavi, and M. F. Jantsch. 2004. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 22:1001-1005. [DOI] [PubMed] [Google Scholar]
- 43.Lomeli, H., J. Mosbacher, T. Melcher, T. Hoger, J. R. Geiger, T. Kuner, H. Monyer, M. Higuchi, A. Bach, and P. H. Seeburg. 1994. Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science 266:1709-1713. [DOI] [PubMed] [Google Scholar]
- 44.Melcher, T., S. Maas, A. Herb, R. Sprengel, P. H. Seeburg, and M. Higuchi. 1996. A mammalian RNA editing enzyme. Nature 379:460-464. [DOI] [PubMed] [Google Scholar]
- 45.Morse, D. P., P. J. Aruscavage, and B. L. Bass. 2002. RNA hairpins in noncoding regions of human brain and Caenorhabditis elegans mRNA are edited by adenosine deaminases that act on RNA. Proc. Natl. Acad. Sci. USA 99:7906-7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Niswender, C. M., S. C. Copeland, K. Herrick-Davis, R. B. Emeson, and E. Sanders-Bush. 1999. RNA editing of the human serotonin 5-hydroxytryptamine 2C receptor silences constitutive activity. J. Biol. Chem. 274:9472-9478. [DOI] [PubMed] [Google Scholar]
- 47.Niswender, C. M., K. Herrick-Davis, G. E. Dilley, H. Y. Meltzer, J. C. Overholser, C. A. Stockmeier, R. B. Emeson, and E. Sanders-Bush. 2001. RNA editing of the human serotonin 5-HT2C receptor: alterations in suicide and implications for serotonergic pharmacotherapy. Neuropsychopharmacology 24:478-491. [DOI] [PubMed] [Google Scholar]
- 48.Paulsson, J. 2004. Summing up the noise in gene networks. Nature 427:415-418. [DOI] [PubMed] [Google Scholar]
- 49.Rueter, S., and R. Emeson. 1998. Adenosine-to-inosine conversion in mRNA, p. 343-361. In H. Grosjean and R. Benne (ed.), Modification and editing of RNA. ASM Press, Washington, D.C.
- 50.Rueter, S. M., T. R. Dawson, and R. B. Emeson. 1999. Regulation of alternative splicing by RNA editing. Nature 399:75-80. [DOI] [PubMed] [Google Scholar]
- 51.Ryter, J. M., and S. C. Schultz. 1998. Molecular basis of double-stranded RNA-protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J. 17:7505-7513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sansam, C. L., K. S. Wells, and R. B. Emeson. 2003. Modulation of RNA editing by functional nucleolar sequestration of ADAR2. Proc. Natl. Acad. Sci. USA 100:14018-14023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schaub, M., and W. Keller. 2002. RNA editing by adenosine deaminases generates RNA and protein diversity. Biochimie 84:791-803. [DOI] [PubMed] [Google Scholar]
- 54.Seeburg, P. H., and J. Hartner. 2003. Regulation of ion channel/neurotransmitter receptor function by RNA editing. Curr. Opin. Neurobiol. 13:279-283. [DOI] [PubMed] [Google Scholar]
- 55.Simpson, M. L., C. D. Cox, and G. S. Sayler. 2003. Frequency domain analysis of noise in autoregulated gene circuits. Proc. Natl. Acad. Sci. USA 100:4551-4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Slavov, D., and K. Gardiner. 2002. Phylogenetic comparison of the pre-mRNA adenosine deaminase ADAR2 genes and transcripts: conservation and diversity in editing site sequence and alternative splicing patterns. Gene 299:83-94. [DOI] [PubMed] [Google Scholar]
- 57.Sodhi, M. S., P. W. J. Burnet, A. J. Makoff, R. W. Kerwin, and P. J. Harrison. 2001. RNA editing of the 5-HT2C receptor is reduced in schizophrenia. Mol. Psychiatry 6:373-379. [DOI] [PubMed] [Google Scholar]
- 58.Vissel, B., G. A. Royle, B. R. Christie, H. H. Schiffer, A. Ghetti, T. Tritto, I. Perez-Otano, R. A. Radcliffe, J. Seamans, T. Sejnowski, J. M. Wehner, A. C. Collins, S. O'Gorman, and S. F. Heinemann. 2001. The role of RNA editing of kainate receptors in synaptic plasticity and seizures. Neuron 29:217-227. [DOI] [PubMed] [Google Scholar]
- 59.Wagner, R. W., C. Yoo, L. Wrabetz, J. Kamholz, J. Buchhalter, N. F. Hassan, K. Khalili, S. U. Kim, B. Perussia, F. A. McMorris, and K. Nishikura. 1990. Double-stranded RNA unwinding and modifying activity is detected ubiquitously in primary tissues and cell lines. Mol. Cell. Biol. 10:5586-5590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang, Q., J. Khillan, P. Gadue, and K. Nishikura. 2000. Requirement of the RNA editing deaminase ADAR1 gene for embryonic erythropoiesis. Science 290:1765-1768. [DOI] [PubMed] [Google Scholar]
- 61.Wang, Q., M. Miyakoda, W. Yang, J. Khillan, D. L. Stachura, M. J. Weiss, and K. Nishikura. 2004. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J. Biol. Chem. 279:4952-4961. [DOI] [PubMed] [Google Scholar]
- 62.Xu, X., C. Li, L. Garrett-Beal, D. Larson, A. Wynshaw-Boris, and C.-X. Deng. 2001. Direct removal in the mouse of a floxed neo gene from a three-loxP conditional knockout allele by two novel approaches. Genesis 30:1-6. [DOI] [PubMed] [Google Scholar]
- 63.Yang, J. H., X. Luo, Y. Nie, Y. Su, Q. Zhao, K. Kabir, D. Zhang, and R. Rabinovici. 2003. Widespread inosine-containing mRNA in lymphocytes regulated by ADAR1 in response to inflammation. Immunology 109:15-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang, W., Q. Wang, K. L. Howell, J. T. Lee, D. S. Cho, J. M. Murray, and K. Nishikura. 2005. ADAR1 RNA deaminase limits short interfering RNA efficacy in mammalian cells. J. Biol. Chem. 280:3946-3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang, W., Q. Wang, S. J. Kanes, J. M. Murray, and K. Nishikura. 2004. Altered RNA editing of serotonin 5-HT2C receptor induced by interferon: implications for depression associated with cytokine therapy. Mol. Brain Res. 124:70-78. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.