

Abstract
The lipid-rich cell wall is a defining feature of Mycobacterium species. Individual cell wall components affect diverse mycobacterial phenotypes including colony morphology, biofilm formation, antibiotic resistance, and virulence. In this study, we describe a transposon insertion mutant of Mycobacterium smegmatis mc2155 that exhibits altered colony morphology and defects in biofilm formation. The mutation was localized to the lsr2 gene. First identified as an immunodominant T-cell antigen of Mycobacterium leprae, lsr2 orthologs have been identified in all sequenced mycobacterial genomes, and homologs are found in many actinomycetes. Although its precise function remains unknown, localization experiments indicate that Lsr2 is a cytosolic protein, and cross-linking experiments demonstrate that it exists as a dimer. Characterization of cell wall lipid components reveals that the M. smegmatis lsr2 mutant lacks two previously unidentified apolar lipids. Characterization by mass spectrometry and thin-layer chromatography indicate that these two apolar lipids are novel mycolate-containing compounds, called mycolyl-diacylglycerols (MDAGs), in which a mycolic acid (α- or α′-mycolate) molecule is esterified to a glycerol. Upon complementation with an intact lsr2 gene, the mutant reverts to the parental phenotypes and MDAG production is restored. This study demonstrates that due to its impact on the biosynthesis of the hydrophobic MDAGs, Lsr2 plays an important role in the colony morphology and biofilm formation of M. smegmatis.
The cell wall is a defining feature of mycobacteria. This complex, lipid-rich, hydrophobic structure is responsible for the acid-fast staining properties, distinctive colony morphology, and innate antibiotic resistance of Mycobacterium species (12, 25, 28). Among pathogens, including Mycobacterium tuberculosis, the causative agent of tuberculosis, cell wall components contribute to virulence, persistence within macrophages, and modulation of the host immune response (16, 45).
The cell wall forms an asymmetric lipid bilayer (25, 28). The inner leaflet is composed of mycolic acids that are covalently bound to arabinogalactan, which is further linked to peptidoglycan via a phosphodiester bridge (12). The outer leaflet contains a variety of lipid components (26, 28). In total, lipids comprise 60% (wt/wt) of the cell wall (12, 25). In addition to mycolic acids, various types of complex lipids are present in the cell wall. These include lipoglycans (e.g., lipoarabinomannan [LAM]), trehalose-containing glycolipids, phthiocerol dimycocerosates, phenolic glycolipids, and glycopeptidolipids (GPLs) (12, 25). The distribution of these lipids varies among mycobacterial species (12). Triacylglycerols (TAGs) are also present in the mycobacterial cell wall (35) and are thought to fill the gap between the meromycolate arm and the shorter α-chain of mycolic acids (28).
Different lipids appear to have different roles. For example, LAM from M. tuberculosis, but not the structurally distinct LAM of nonpathogenic mycobacteria, exhibits various immunomodulatory effects and may contribute to the survival and persistence of M. tuberculosis in host macrophages (32, 45). Surface glycolipids that contain multiple methyl-branched fatty acids, such as phthiocerol dimycocerosate and phenolic glycolipids, are known to play a role in the virulence and pathogenesis of M. tuberculosis (16, 46). GPLs contribute to biofilm formation (37, 38). Although the in vivo formation of biofilms by M. tuberculosis remains a matter of debate (21), it is well established that Mycobacterium smegmatis and other environmental mycobacteria such as M. avium subsp. avium, M. fortuitum, M. chelonae, and M. marinum are capable of biofilm formation (7, 14, 40). These mycobacteria are normal inhabitants of a wide variety of environmental reservoirs, including natural and municipal waters. Their ability to form biofilms suggests that mycobacterial populations can persist in a flowing system despite their slow growth. Biofilm-producing bacteria are less susceptible to antibiotics, which may exacerbate the impact of these pathogens on human health.
Although functions for many cell wall lipids have yet to be elucidated, a combination of biochemical and genetic approaches has provided much insight into the mechanism of cell wall biosynthesis. The availability of complete genome sequences for several Mycobacterium and related Corynebacterium species has confirmed that these organisms dedicate enormous resources to the cell wall synthesis, with ∼250 lipid metabolism genes identified in M. tuberculosis (15, 18). It is difficult to assign specific functions to all of these genes. However, the bacterial cell wall is the primary interface between the organism and the environment, such that alterations in the cell wall composition can have dramatic effects on many phenotypes, including colony morphology (2, 27, 47).
We have used a forward genetics approach to obtain colony morphology mutants of M. smegmatis mc2155. In this study, we describe a transposon insertion in the lsr2 gene. This mutant exhibits smooth colony morphology and defects in biofilm formation. The lsr2 mutation also impairs the biosynthesis of a novel mycolic acid-containing triacylglyerol compound.
MATERIALS AND METHODS
Bacterial strains, media, and growth conditions.
Wild-type (wt) Mycobacterium smegmatis strain mc2155 and its mariner insertion mutants were routinely grown in Middlebrook 7H9 broth or Middlebrook 7H11 agar (Difco) supplemented with 10% OADC (oleic acid, bovine serum albumin [fraction V], dextrose, and catalase; Difco). To analyze biofilm formation, M63 salts minimal medium supplemented with 0.2% glycerol, 0.5% Bacto-Casitone (BD), 1 mM MgSO4, and 0.7 mM CaCl2 was used. For Lsr2 localization assays, mycobacteria were grown in liquid Sauton medium. Antibiotics (Sigma) were added at the following concentrations: kanamycin, 25 μg/ml for mycobacteria and 50 μg/ml for Escherichia coli; hygromycin, 75 μg/ml for mycobacteria and 150 μg/ml for E. coli.
Generation and screening of M. smegmatis mc2155 ΦMycoMar insertion library.
The mariner-based transposon system ΦMycoMar T7 was utilized to generate a transposon insertion mutant library of M. smegmatis mc2155 as described previously (1, 2). Kanamycin-resistant (i.e., transposon-containing) colonies were patched onto Middlebrook 7H11 agar to obtain a library of 7,680 clones (i.e., 80 plates × 96 colonies per plate). Colonies with unusual morphology were identified by visual inspection.
Localization of the ΦMycoMar insertion.
The method used to localize and identify the transposon-disrupted gene has been described previously (1, 2). Briefly, total chromosomal DNA of the transposon insertion mutant was cleaved with BamHI and then self-ligated with T4 DNA ligase and transformed into competent E. coli DH5α λpir116 cells. The MycoMar element contains an R6K origin and an aph gene such that recircularized fragments containing the transposon are able to replicate as kanamycin-resistant plasmids. Plasmid DNA was isolated from Kmr E. coli transformants, and MycoMar-specific primers were used to determine the DNA sequence at the transposon/chromosomal junction. These DNA sequences were compared to the GenBank database and the M. smegmatis mc2155 genome database at the Institute for Genomic Research (http://www.tigr.com/) using the BLASTN algorithm. Nucleotide sequences were also analyzed with NTI Suite software (Informax).
Cloning of lsr2.
The intact M. smegmatis lsr2 gene was amplified from strain mc2155 genomic DNA by PCR using the forward primer 5′-GATCTGAGCGTTGTTGATAG-3′ and the reverse primer 5′-GTACCTGCCGTCCACTCTAA-3′. To generate pLSR2, used for complementation experiments, the PCR product (652 bp) was first cloned into the vector pDrive (QIAGEN). Next, a BamHI-XbaI restriction fragment was subcloned into the E. coli-Mycobacterium shuttle vector pNBV1 (22). Plasmid pLSR2-HIS was constructed for expression of the histidine-tagged Lsr2 protein. The forward primer 5′-ACGGATCCGATCTGAGCGTTGTTGATAGAACC-3′ and the reverse primer 5′-GCTCTAGAAGCTTACTAGTGATGGTGATGGTGATGAGTTGCCGCGTGGAATGC-3′ were used for PCR amplification of lsr2. They contain, respectively, BamHI and HindIII sites (italics), which were used for cloning of the PCR product into pNBV1. Of note, the reverse primer was designed to replace the native lsr2 stop codon with six histidine codons followed by two new stop codons (underlined). All constructs were confirmed by restriction digestions and DNA sequencing. Standard electroporation protocols were used for transformation of pLSR2 and pLSR2-HIS into M. smegmatis. Transformants were selected on Middlebrook 7H11 agar containing hygromycin.
Pellicle formation and biofilm assays.
Pellicle formation was monitored by growing standing cultures of mycobacteria without shaking in either Middlebrook 7H9 medium or M63 medium without Tween 80 at 37°C for 48 h. Biofilm formation was assayed using polyvinyl chloride (PVC) or polystyrene microwell plates and the crystal violet staining method developed by Recht et al. (37, 38). Briefly, 1 ml of M63 medium was added to 24-well polystyrene (Costar) or 96-well PVC (BD) plates and inoculated with mycobacterial cells to an optical density at 600 nm of 0.05. The plates were incubated at room temperature on an orbital shaker set at 60 rpm for 48 h, washed with deionized water, stained with 1% crystal violet, and assayed for biofilm formation by spectrophotometric reading of the ethanol extract at 570 nm.
TLC analysis of cell wall lipids.
All thin-layer chromatography (TLC) analyses were performed on Silica Gel 60 plates (Whatman). Mycolic acids were extracted, methylated, and analyzed by TLC as previously described (47). The apolar and polar lipids were prepared from M. smegmatis cells (50 mg dry biomass) according to previously published procedures (2). These lipids were analyzed by two-dimensional TLC (2D-TLC) using the following solvent systems. Apolar lipids were developed with petroleum ether-ethyl acetate (98:2; three times) in the first dimension and petroleum ether-acetone (98:2) in the second dimension. Polar lipids were separated with chloroform-methanol-water (60:30:6) in the first dimension and chloroform-acetic acid-methanol-water (40:25:3:6) in the second dimension. Lipids were detected by charring with α-naphthol or 5% phosphomolybdic acid.
To specifically detect GPLs, a procedure previously described for GPL extraction and TLC analysis was used (13). Both acetylated and deacetylated GPLs were obtained and analyzed. In the case of acetylated GPLs, the deacetylation step performed by alkaline methanolysis was omitted. Samples were applied to silica gel plates and developed with the following three solvents: solvent I (chloroform-methanol-water [60:27:4]), solvent II (chloroform-methanol-water [65:25:4]), and solvent III (chloroform-methanol-water [60:16:2]). The GPL bands were visualized by spraying with 10% H2SO4 in ethanol and heating at 110°C.
MS analysis.
Preparative TLC was performed to isolate lipids of interest for analysis by mass spectrometry. Briefly, apolar lipids separated on TLC plates were located by staining with 0.01% ethanolic rhodamine 6G and viewed under long-wave (366 nm) UV light. The compounds were then scraped from the TLC plates and extracted with diethyl ether and evaporated to dryness. Samples were dissolved in CHCl3-MeOH (2:1) and subjected to mass spectrometry (MS) analysis at the Molecular Medicine Research Center, University of Toronto. Matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) and MALDI-PSD (post-source decay) spectra were acquired as described previously (2).
Electron microscopy.
Specimens for transmission electron microscopy were prepared as previously described (47). Briefly, an aliquot of cells grown in 7H9 broth (without Tween 80) containing approximately 5 × 107 cells was pelleted by centrifugation, fixed with Karnovsky's 4% paraformaldehyde-2.5% glutaraldehyde, and postfixed with 1% OsO4. Cells were dehydrated in a graded series of ethanol and embedded in Spurr's resin. Thin sections were cut with a Reichert Ultracut E microtome and poststained first in saturated uranyl acetate and then in Reynold's lead citrate. Electron microscopy was performed with a Hitachi H7000 transmission electron microscope operating at 75 kV (Microscopy Imaging Laboratory, University of Toronto).
Localization of Lsr2.
M. smegmatis mc2155 or MS 8444 expressing the His-tagged Lsr2 protein was grown in Sauton medium (500 ml) to an optical density at 600 nm of 0.5 to 0.8. Cells were pelleted by centrifugation, and the supernatants were collected and filtered (0.2 μm; Nalgene) to obtain cell-free culture filtrates (CF). The CF was desalted and concentrated to a final volume of 5 ml by centrifugation and filtration (Amicon, Millipore). Cell pellets were resuspended in phosphate-buffered saline containing a protease inhibitor cocktail (Roche) and divided into 1-ml aliquots, to which 0.5 g glass beads (Sigma) was added. Cells were disrupted by four 1-min pulses with 1-min intervals on ice using a Mini-Beadbeater (Biospec, Bartlesville, OK). The cell lysates were centrifuged, and the supernatant (∼5 ml) was collected and designated as cell lysate. The pellet, which contains cell membrane and cell wall components, was washed and resuspended in 100 μl phosphate-buffered saline. Equal amounts of protein (10 μg) from each fraction, i.e., CF, cell lysate, and cell membrane, were separated on 14% Tricine sodium dodecyl sulfate (SDS) gels and analyzed by Western blot using an anti-His antibody (Invitrogen).
Cross-linking of Lsr2.
Glutaraldehyde (1%) was added to 200 μl of the cell lysate prepared from strain mc2155 or MS 8444 expressing His-tagged Lsr2. Aliquots (∼40 μg protein) were removed at various time points (0, 2, 5, 15, 30, and 45 min), mixed with 2× Tricine SDS-polyacrylamide gel electrophoresis sample buffer, and incubated at 65°C for 10 min. These samples were separated on 14% Tricine SDS gels and analyzed by Western blot using an anti-His antibody.
Antibiotic sensitivity assay.
The MICs of isoniazid, ethambutol, novobiocin, rifampin, erythromycin, chloramphenicol, ampicillin, cefoxitin, and cephaloridine (Sigma) were determined by the broth dilution method (27).
RESULTS
Isolation of an MS 8444 mutant that exhibits altered colony morphology.
Colony morphology is a complex phenotype. Even subtle defects in cell wall components can modify the surface properties of individual cells, alter cell-to-cell interactions, and ultimately give rise to colonies with visibly different morphologies (2). We have generated a library of ∼8,000 M. smegmatis transposon insertion mutants and visually inspected them to identify clones with altered colony morphologies. More than 20 mutants have been isolated. Among these, one clone, designated MS 8444, exhibits the most dramatic change in morphology. When grown on 7H11 agar plates, MS 8444 colonies are very smooth, wet, and round, a phenotype in stark contrast to the dry, rough, and rugose morphology of the wt mc2155 parent strain (Fig. 1A and B).
FIG. 1.

Colony morphology of MS 8444 (A), mc2155 (B), MS 8444/pNBV1 (C), and MS 8444/pLSR2 (D). The MS 8444 mutant exhibits very smooth colony morphology (A and C), which is complemented by the lsr2 gene (D).
In 7H9 liquid medium, MS 8444 and mc2155 exhibit equivalent growth rates. However, the mutant aggregates less than the wt strain. The sensitivity of MS 8444 to various antibiotics including isoniazid, ethambutol, rifampin, erythromycin, chloramphenicol, novobiocin, ampicillin, cefoxitin, and cephaloridine remained unchanged (data not shown).
Identification of the lsr2 gene.
The MS 8444 loci disrupted by the transposon insertion were identified as described in Materials and Methods. DNA sequencing and analysis of the MycoMar/M. smegmatis chromosomal junction revealed that the transposon had inserted at a TA dinucleotide within the lsr2 gene (Fig. 2A). Lsr2 was first identified as an immunodominant T-cell antigen of M. leprae (24, 33, 41, 42). Sequence analysis revealed that lsr2 orthologs are present in all sequenced mycobacterial genomes, including M. tuberculosis, M. bovis, M. leprae, M. marinum, M. avium subsp. avium, and M. avium subsp. paratuberculosis (Fig. 2B). The sequence of Lsr2 is highly conserved among mycobacteria (>85% identity in amino acid sequences). Lsr2 homologs are also found in related actinomycetes, including Streptomyces coelicolor, Nocardia farcinica, and Rhodococcus equi. Interestingly, mycobacteriophages Omega and CJW1 each contain an Lsr2 homolog. The biological function of Lsr2 has not been described. Proteomic analysis showed that the expression of M. tuberculosis lsr2 (Rv3597c) increased under high iron concentrations (48). Microarray studies showed that M. tuberculosis lsr2 was up-regulated at high temperatures (44) and up-regulated after 24 h of nutrient starvation (10).
FIG. 2.

Genetic organization of the lsr2 region. (A) M. smegmatis lsr2 region and the corresponding genes in M. tuberculosis H37Rv. The transposon insertion within lsr2 is indicated. (B) Sequence alignment of Lsr2 from representative mycobacterial species.
Complementation of the MS 8444 mutant with M. smegmatis lsr2.
To confirm that the morphological phenotype of MS 8444 was due to the disruption of the lsr2 gene and not a polar effect caused by the transposon insertion, we cloned the intact lsr2 gene from the parental M. smegmatis strain into the shuttle vector pNBV1. The resulting plasmid, pLSR2, was transformed into MS 8444. Complementation with plasmid pLSR2, but not pNBV1, restored the wt colony morphology (Fig. 1C and D). This result confirms that the inactivation of lsr2 is responsible for the altered colony morphology of MS 8444.
The lsr2 mutant exhibits normal cell morphology.
Since the MS 8444 mutant exhibits dramatically different colony morphology, we examined whether individual mutant cells also differed from wt cells. Under the light microscope, MS 8444 cells are of normal size and shape (data not shown). The cell morphology and the cell wall architecture were further examined by transmission electron microscopy, and there is no apparent difference between the mutant and the wt cells (data not shown).
The lsr2 mutant is defective in pellicle and biofilm formation.
Since the lsr2 mutant forms drastically different colonies on agar plates and aggregates less than the wt strain in liquid media, we also examined whether it is defective in pellicle formation. Because of their high lipid content, mycobacteria grown in the absence of detergent (e.g., Tween 80) form a surface pellicle in standing liquid media. When the parental M. smegmatis mc2155 strain was grown in standing 7H9 medium without Tween 80 and shaking, significant pellicle growth appeared on the surface of the broth (Fig. 3A). In contrast, the lsr2 mutant was unable to form pellicles under the same experimental conditions (Fig. 3A). Pellicles have recently been recognized as a biofilm that assembles at the air-liquid interface (11). The pel mutants of Pseudomonas aeruginosa isolated by screening for colonies unable to form pellicles were defective in biofilm formation on PVC or polystyrene plates (19). A strong correlation between pellicle formation and the ability to form a biofilm on PVC was also found in Salmonella enterica serovar Enteritidis isolates from different origins (43).
FIG. 3.

Pellicle and biofilm formation. (A) Wild-type M. smegmatis mc2155 forms a thick pellicle at the air-liquid interface of the standing 7H9 culture. The MS 8444 mutant is defective in pellicle formation, but pellicle formation is restored upon complementation with an intact lsr2 gene. A similar result is obtained in biofilm medium (not shown). (B) Biofilm formation assayed using the crystal violet (CV) staining assay. Cells of mc2155 (filled bars), MS 8444 (crossed bars), and MS 8444/pLSR2 (empty bars) in biofilm media were grown on polyvinyl chloride and polystyrene plates for 48 h. Results are representative of at least three independent experiments.
To better quantify the defect of MS 8444, biofilm formation was assayed on both PVC and polystyrene plates using an M63-based liquid medium, which was previously used to assay biofilm formation of M. smegmatis (38). Growth experiments were repeated in this biofilm medium, and the parental and mutant strains exhibited equivalent growth rates and were indistinguishable when grown with shaking. However, when grown in standing biofilm medium without shaking, the mutant was unable to form pellicles, whereas the wt strain did (data not shown). Quantification of biofilm formation on PVC and polystyrene plates was performed by extracting the biofilm-associated crystal violet dye with ethanol and measuring the optical density at 570 nm (38). As shown in Fig. 3B, when grown in the M63-based medium, biofilm formation by the lsr2 mutant was significantly reduced (∼5-fold) compared to biofilm formation by the wt strain. A similar, though less-pronounced, result was obtained when biofilm formation was assayed using cultures grown in 7H9 broth (data not shown). Importantly, complementation of MS 8444 with plasmid pLSR2 restored pellicle and biofilm formation to wt levels (Fig. 3A and B). Taken together, these results indicate that lsr2 is involved in pellicle and biofilm formation.
Cell wall lipid analysis.
The changes in colony morphology and biofilm formation suggested that the lsr2 mutation affected some component of the MS 8444 cell wall. As such, we examined the cell wall composition of the parental and mutant strains. We first examined whether the composition of GPLs was changed in the mutant, as GPLs have been previously shown to be required for biofilm formation. An mps mutant of M. smegmatis lacking GPLs was defective in biofilm formation (38). It was further shown that acetylation of GPLs is important for biofilm formation (37). GPLs of the lsr2 mutant in both acetylated and deacetylated forms were extracted and analyzed by TLC according to a previously published procedure (13). The result showed that, unlike the mps or atf1 mutant of M. smegmatis, the lsr2 mutant exhibited normal GPLs. There is no detectable difference between the GPLs (either in acetylated or deacetylated form) of the mutant and the wt strains (Fig. 4A), indicating that the inability of the lsr2 mutant to form a biofilm was not due to the lack of or improper processing of GPLs.
FIG. 4.

TLC analysis of cell wall lipids. (A) TLC analysis of acetylated (−) and deacetylated (+) GPLs of wt strain mc2155 and lsr2 mutant MS 8444. (B) TLC analysis of cell wall-bound mycolic acids of the wt and mutant cells. TLC was developed with hexane-ethyl acetate (9:1). (C to E) 2D-TLC analysis of apolar lipids of mc2155, MS 8444, and MS 8444/pLSR2. TLC plates were developed using petroleum ether-ethyl acetate (98:2; three times) in the first dimension (1st) and petroleum ether-acetone (98:2) in the second dimension (2nd). The two previously unassigned lipids that are absent from the MS 8444 mutant are indicated by arrows and labeled as spots 1 and 2 (C and D). They are restored upon transformation with pLSR2 (E). (F) Spots 1 and 2 were purified from TLC plates, subjected to saponification and methylation, and then analyzed by TLC as described previously (27). TLC was developed with hexane-ethyl acetate (9:1). Mycolic acids of whole cells were analyzed in parallel. Lanes: 1, mycolic acid methyl esters of whole cells; 2, spot 1; 3, spot 2. Spot 1 contains α-mycolates. Spot 2 contains α′-mycolates and contaminants of α-mycolates from spot 1. α, α-mycolate methyl esters; α′, α′-mycolate methyl esters; E, epoxymycolate methyl esters; FAME, short-chain fatty acid methyl esters.
We next examined the composition of other cell wall lipids. Analysis of cell wall-bound mycolic acids by TLC showed that they are unchanged in the mutant compared to the wt strain (Fig. 4B). 2D-TLC analysis of polar lipids, which consists of phosphatidylinositol mannosides, diphosphatidylglycerol, phosphatidylethanolamine, and phosphatidylinositol, also did not reveal any differences (data not shown). However, examinations of apolar lipids revealed a distinct 2D-TLC profile for the lsr2 mutant (Fig. 4C and D). Two lipids present in the wt cell wall (Fig. 4C, spot 1 and 2, arrows) were not detected in the mutant cell wall (Fig. 4D). The identities of these lipids have not been reported previously in the literature. Moreover, the amount of TAGs in the mutant cell wall was increased, suggesting that the unassigned lipids may have metabolic relationships with TAGs. Complementation of MS 8444 with plasmid pLSR2 restored the ability of the mutant to produce these lipids (Fig. 4E), indicating that the disruption of lsr2 is responsible for the altered cell wall lipid composition.
To identify the two unknown apolar lipids, spots 1 and 2 were purified by preparative TLC and subjected to mass spectrometry analysis. Figure 5A shows the MALDI-TOF spectrum of spot 1, which consists of a group of molecular ions that differ in size by increments of 14 atomic mass units (amu), the molecular mass of -CH2-, suggesting that these ions belong to a homologous series differing only in the acyl chain length. Similarly, the MALDI-TOF spectrum of spot 2 revealed a series of molecular ions, m/z 1,396.30 to 1,508.35, differing by 28 amu (Fig. 5C). The main molecular ions of spots 1 and 2, m/z 1,660 and 1,424, respectively, were further analyzed by MALDI-PSD, and the resulting fragment ions are shown (Fig. 5B and D). We assume that these compounds contain a glycerol moiety since TAGs were accumulated in the mutant cells lacking spots 1 and 2, suggesting that they are related. The TAG spots were also analyzed by MALDI-TOF, which revealed a series of molecular ions, m/z 829.7 to 1,025.9, differing by 28 amu, and were identical in the mutant and wt cells (data not shown). The MS spectra of spots 1 and 2 can be explained by substituting a fatty acyl chain of TAGs with a mycolic acid molecule. M. smegmatis synthesizes three types of mycolic acids, α-, α′-, and epoxy-mycolates (9). The α-mycolates are full-length mycolic acids most frequently containing 78 and 79 carbons, whereas the α′-mycolates are shorter, most often 64 carbons in length (9). The molecular mass of mycolic acids deduced from the MS spectra of spots 1 and 2 agrees with the size of α- and α′-mycolates in M. smegmatis; for example, the molecular ion m/z 1,660 of spot 1 corresponds to α-mycolates containing 79 carbons, and the molecular ion m/z 1,424 of spot 2 corresponds to α′-mycolates containing 62 carbons (Fig. 5). Therefore, spot 1 represents compounds containing one α-mycolic acid molecule esterified to a diacylglycerol, while spot 2 represents compounds containing one α′-mycolic acid molecule esterified to a diacylglycerol. The presence of mycolic acids in these compounds was further confirmed by alkaline deacylation of purified spots 1 and 2, followed by methylation and TLC analysis (27), which showed that α- and α′-mycolic acids were present in spots 1 and 2, respectively (Fig. 4F). Based on these results, we propose that spot 1 and spot 2 compounds are mycolyl-diacylglycerols (MDAGs), in which a mycolic acid molecule (α- or α′-mycolate) is esterified to a glycerol moiety (Fig. 5A and C).
FIG. 5.

MALDI-TOF and MALDI-PSD analysis. (A) Positive MALDI mass spectrum of spot 1 from Fig. 4C. (B) MALDI-PSD spectrum of the parent [M + H]+ at m/z 1,660 from A. (C) Positive MALDI mass spectrum of spot 2 from Fig. 4C. (D) MALDI-PSD spectrum of the parent [M + H]+ at m/z 1,424 from C. The deduced structures of spots 1 and 2 are also shown. M.W., molecular mass.
Lsr2 is localized in the cytosol and forms a dimer.
Lsr2 is a small and basic protein (molecular mass of 12.5 kDa and pI of 9.6 for M. smegmatis Lsr2). Sequence analysis did not reveal any known motifs or recognized domains. Lsr2 could play a structural role in the cell wall architecture such that its absence results in changed colony morphology and defects in pellicle and biofilm formation. Alternatively, Lsr2 could play an indirect (e.g., regulatory) role by controlling enzymes involved in cell wall lipid metabolisms, which results in altered cell wall lipid composition and consequently altered colony morphology and defective biofilm formation. To test these possibilities, we first determined the localization of Lsr2. A plasmid, pLSR2-HIS, which expresses the His-tagged Lsr2, was constructed and transformed into both mc2155 and MS 8444. Transformation of pLSR2-HIS into MS 8444 restored the wt colony morphology (data not shown), indicating that the His-tagged Lsr2 is fully functional. Cell fractionation followed by Western blot analysis using an anti-His monoclonal antibody showed that Lsr2 was found in the cytosolic fraction (Fig. 6A), consistent with a recent proteomic analysis in which Lsr2 of M. leprae was found only in the cytosol (29). Lsr2 was not present in the culture filtrate or the cell membrane/cell wall fractions (Fig. 6A), suggesting that Lsr2 is not associated with the cell surface.
FIG. 6.
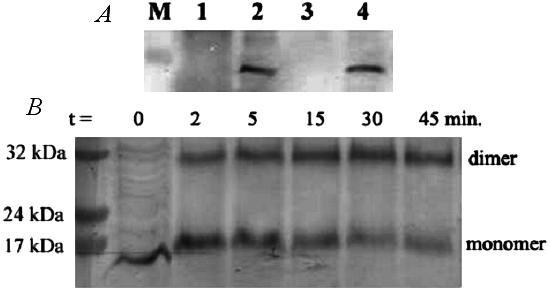
Localization and dimerization of Lsr2. (A) Localization of Lsr2. Cells of M. smegmatis mc2155 expressing His-tagged Lsr2 were fractionated and analyzed by Western blot using an anti-His antibody. Lane 1, cell-free culture supernatant; lane 2, crude cell lysate; lane 3, cell membrane/cell wall fraction; lane 4, cytosolic fraction. A similar result was obtained with MS 8444/pLSR2-HIS (not shown). (B) Crossing-linking of Lsr2. Glutaraldehyde (1%) was added to the cytosolic fraction of mc2155/pLSR2-HIS. Aliquots were removed at the indicated time points and analyzed by Western blotting with an anti-His antibody. Lsr2 forms a dimer under the experimental conditions. A similar result was obtained with MS 8444/pLSR2-HIS (data not shown).
Although no known motifs could be detected, the highly basic nature of Lsr2 is suggestive of nucleic acid binding capability. In addition, Lsr2 is homologous to mycobacteriophage proteins such as Gp39, Gp61, and Gp206, proteins with a putative DNA binding function (36). We suggest that Lsr2 could function as a regulatory protein. To test this possibility, we determined whether Lsr2 forms a dimer, which is characteristic of many nucleic acid binding proteins. The cytosolic fraction of mc2155/pLSR2-HIS was treated with 1% glutaraldehyde. Glutaraldehyde covalently cross-links proteins in close proximity, thereby allowing the detection of oligomers. Aliquots of this mixture at different time points after the addition of glutaraldehyde were analyzed by Western blotting. As shown in Fig. 6B, Lsr2 dimers appeared 2 min into the cross-linking reaction, and levels increased slightly with time, whereas amounts of the Lsr2 monomer gradually decreased over the same period. This result indicates that Lsr2 exists in the cytosol of mycobacterial cells as a dimer.
DISCUSSION
In this study, we described MS 8444, an lsr2 mutant of M. smegmatis. This mutant was isolated by screening for transposon insertion mutants of M. smegmatis mc2155 that exhibited an altered colony morphology. The lsr2 mutant exhibits smooth colony morphology and is defective in pellicle and biofilm formation. The mutant cell wall also lacks two previously unrecognized apolar lipids, which we have identified as MDAGs. All MS 8444 mutant phenotypes revert to the wild type upon the introduction of an intact lsr2 gene. Taken together, these results indicate that Lsr2 plays an important role in the colony morphology and biofilm formation of M. smegmatis, presumably by controlling the production of hydrophobic MDAGs.
Mycolic acids are long-chain α-alkyl-β-hydroxy fatty acids that are unique to mycobacteria and closely related genera (9, 25). M. smegmatis contains three types of mycolic acids, α-, α′-, and epoxy-mycolates (9). Mycolic acids are known to exist in mycobacterial cells in two basic forms: covalently linked to arabinogalactan of the cell wall or esterified to a trehalose in forms such as trehalose dimycolate (TDM) and trehalose monomycolate, which can be extracted in organic solvents. The cell wall-bound mycolic acids play a structural role, important for the integrity of the mycobacterial cell wall (26, 28). TDM, also called cord factor, has been implicated in the pathogenesis of mycobacterial diseases (9). We now describe a third form of mycolic acids, MDAGs, in which a mycolic acid (α- or α′-mycolate) is esterified to a glycerol moiety. Disruption of lsr2 by transposon mutagenesis blocked the synthesis of MDAGs and resulted in the accumulation of TAGs, suggesting a metabolic relationship between these two molecules: MDAGs could be synthesized by the transesterification of TAGs. Kremer et al. recently described a structurally related molecule, meromycolyl-diacylglycerol, in mycobacteria, in which a meromycolate, the precursor of full-length mycolic acid, is esterified to a glycerol (23). Given the diverse roles of mycolic acids and the large numbers of lipid metabolic enzymes revealed by mycobacterial genome sequencing, it is not surprising that some new lipid species remain to be uncovered.
The MDAGs identified in this study also represent an unusual TAG-related molecule. Mycobacterium (6, 17, 20, 30) and related actinomycetes such as Nocardia (5), Rhodococcus (3, 4), and Streptomyces (34) accumulate large amounts of TAGs, which are thought to serve as a storage reservoir for energy and carbon (6). By analogy to these molecules, MDAGs may represent a storage or carrier for mycolic acids. The mycolate residues may then be available for transfer to the cell wall or TDM under adverse environmental conditions such as a nutrient-poor, nonreplicating state. Consistent with this notion, it was shown recently that the diacylglycerol acetyltransferases involved in TAG biosynthesis were up-regulated in M. tuberculosis stationary-phase cultures and that TAGs accumulated in dormant cultures (17).
Our current study indicates that the newly identified MDAGs play a role in colony morphology and biofilm formation of mycobacteria. Colony morphology is a complex phenotype influenced by the ability of cells to interact with one another. The loss of hydrophobic MDAGs from MS 8444 is consistent with the smooth colony phenotype observed on solid media and the defect in pellicle formation observed in liquid media. A similar smooth colony phenotype was recently described for an fbpA mutant of M. smegmatis. Disruption of the fbpA gene, encoding a mycolyltransferase, resulted in a 45% reduction of TDM in the mutant, but cell wall-bound mycolic acids were not affected (31). Although TDM remains unchanged in our MS 8444 mutant (data not shown), the lsr2 and fbpA studies both indicate that mycolic acids esterified either to trehaloses or glycerols impact cell surface hydrophobicity and colony morphology.
The loss of MDAGs in the mutant cell wall, which reduces the cell surface hydrophobicity, is expected to impact biofilm formation. A biofilm is an assemblage of microbial cells that is associated with a surface and is enclosed in an extracellular polymeric substance matrix (11). The first step of biofilm formation is the attachment of individual cells from the planktonic condition to a surface. Bacterial cell surface hydrophobicity, the presence of fimbriae and flagella, and the production of extracellular polymeric substance all influence the rate and extent of attachment in the process of biofilm formation. In M. smegmatis, the failure to synthesize or properly acetylate GPLs is associated with defects in both sliding motility and the formation of biofilm on polyvinyl chloride (37, 38). In M. avium, changes in GPL content are also associated with variations in colony morphology. Rough, in contrast to smooth, variants were found to be devoid of polar GPLs (8). The absence of these GPLs is thought to increase cell surface hydrophobicity. This is supported by the finding that in liquid cultures of M. avium, rough variants were overrepresented in the surface pellicle (8). Conversely, a decrease of cell surface hydrophobicity in the lsr2 mutant due to the lack of the hydrophobic MDAGs could explain the inability of the mutant to form surface pellicles. A recent study showed that the deletion of the M. smegmatis upk gene, which encodes the undecaprenyl phosphokinase involved in peptidoglycan synthesis, both impairs biofilm formation and alters colony morphology (39). The structural basis for these observations remained unknown since the peptidoglycan of the mutant cell wall appears to be unchanged. In the present study, electron microscopy revealed no differences between MS 8444 and the parental M. smegmatis strain. A thorough examination of cell wall components by TLC showed no differences in cell wall-bound mycolic acids, GPLs, or polar lipids. The lsr2 mutation specifically affected the production of MDAGs.
Lsr2 is highly conserved among mycobacteria. First described in M. leprae as an immunodominant T-cell antigen (24), Lsr2 orthologs are present in all sequenced mycobacterial genomes, including pathogens of the M. tuberculosis complex. The high level of sequence identity among the Lsr2 homologs suggests that they perform a similar function. Transcription of lsr2 of M. tuberculosis was found to be up-regulated under high-iron conditions (48) as well as at high temperatures (44). However, growth of the M. smegmatis lsr2 mutant and parental strains are equivalent under high (500 mg/liter)- or low (500 μg/liter)-iron conditions and at elevated temperatures (45°C) (data not shown). This suggests that Lsr2 is not involved in iron metabolism. Additional growth experiments indicate that the mutant and wt strains are equally resistant to a broad range of antibiotics. Only when grown on solid media, or in standing liquid culture without shaking, are the differences between the lsr2 mutant and the parent strain readily apparent.
Despite the effect of the lsr2 mutation on cell wall lipid biosynthesis, localization experiments indicate that the Lsr2 protein is cytosolic and not associated with the cell wall. These data are in agreement with a proteome scan of M. leprae, which also revealed Lsr2 to be cytosolic (29). Database searches failed to identify any proteins of known function that are homologous to Lsr2, and no structural motifs associated with lipid metabolism enzymes are found in the Lsr2 sequence. As such, we propose that Lsr2 is not a structural component of the cell wall but that it has a regulatory role that affects lipid production, possibly by controlling gene expression of enzymes involved in the synthesis of MDAGs. Although Lsr2 does not have a classical DNA binding domain, it is arginine rich (e.g., 12.3% for M. smegmatis Lsr2), highly basic, positively charged, and potentially able to interact with negatively charged phosphate groups of nucleic acids. Moreover, cross-linking experiments indicate that, like many nucleic acid binding proteins, Lsr2 exists as a dimer in vivo. Additional work is required to define how Lsr2 mediates MDAG biosynthesis and to elucidate the role of these novel mycobacterial lipids.
Acknowledgments
This work is supported by Canadian Institutes of Health research grant MOP-15107, and a grant from the National Sanitarium Association of Canada (to J.L.).
REFERENCES
- 1.Alexander, D. C., J. R. Jones, and J. Liu. 2003. A rifampin-hypersensitive mutant reveals differences between strains of Mycobacterium smegmatis and presence of a novel transposon, IS1623. Antimicrob. Agents Chemother. 47:3208-3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexander, D. C., J. R. Jones, T. Tan, J.M. Chen, and J. Liu. 2004. PimF, a mannosyltransferase of mycobacteria, is involved in the biosynthesis of phosphatidylinositol mannosides and lipoarabinomannan. J. Biol. Chem. 279:18824-18833. [DOI] [PubMed] [Google Scholar]
- 3.Alvarez, H. M., R. Kalscheuer, and A. Steinbuchel. 2000. Accumulation and mobilization of storage lipids by Rhodococcus opacus PD630 and Rhodococcus ruber NCIMB 40126. Appl. Microbiol. Biotechnol. 54:218-223. [DOI] [PubMed] [Google Scholar]
- 4.Alvarez, H. M., F. Mayer, D. Fabritius, and A. Steinbuchel. 1996. Formation of intracytoplasmic lipid inclusions by Rhodococcus opacus strain PD630. Arch. Microbiol. 165:377-386. [DOI] [PubMed] [Google Scholar]
- 5.Alvarez, H. M., M. F. Souto, A. Viale, and O. H. Pucci. 2001. Biosynthesis of fatty acids and triacylglycerols by 2,6,10,14-tetramethyl pentadecane-grown cells of Nocardia globerula 432. FEMS Microbiol. Lett. 200:195-200. [DOI] [PubMed] [Google Scholar]
- 6.Alvarez, H. M., and A. Steinbuchel. 2002. Triacylglycerols in prokaryotic microorganisms. Appl. Microbiol. Biotechnol. 60:367-376. [DOI] [PubMed] [Google Scholar]
- 7.Bardouniotis, E., H. Ceri, and M. E. Olson. 2003. Biofilm formation and biocide susceptibility testing of Mycobacterium fortuitum and Mycobacterium marinum. Curr. Microbiol. 46:28-32. [DOI] [PubMed] [Google Scholar]
- 8.Barrow, W. W., and P. J. Brennan. 1982. Isolation in high frequency of rough variants of Mycobacterium intracellulare lacking C-mycoside glycopeptidolipid antigens. J. Bacteriol. 150:381-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barry, C. E., R. E. Lee, K. Mdluli, A. E. Sampson, B. G. Schroeder, R. A. Slayden, and Y. Yuan. 1998. Mycolic acids: structure, biosynthesis and physiological functions. Prog. Lipid Res. 37:143-179. [DOI] [PubMed] [Google Scholar]
- 10.Betts, J. C., P. T. Lukey, L. C. Robb, R. A. McAdam, and K. Duncan. 2002. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 43:717-731. [DOI] [PubMed] [Google Scholar]
- 11.Branda, S. S., S. Vik, L. Friedman, and R. Kolter. 2005. Biofilms: the matrix revisited. Trends Microbiol. 13:20-26. [DOI] [PubMed] [Google Scholar]
- 12.Brennan, P. J., and H. Nikaido. 1995. The envelope of mycobacteria. Annu. Rev. Biochem. 64:29-63. [DOI] [PubMed] [Google Scholar]
- 13.Brennan, P. J., M. Souhrada, B. Ullom, J. K. McClatchy, and M. B. Goren. 1978. Identification of atypical mycobacteria by thin-layer chromatography of their surface antigens. J. Clin. Microbiol. 8:374-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carter, G., M. Wu, D. C. Drummond, and L. E. Bermudez. 2003. Characterization of biofilm formation by clinical isolates of Mycobacterium avium. J. Med. Microbiol. 52:747-752. [DOI] [PubMed] [Google Scholar]
- 15.Cole, S. T., R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, K. Eiglmeier, S. Gas, C. E. Barry, F. Tekaia, K. Badcock, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. Davies, K. Devlin, T. Feltwell, S. Gentles, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, A. Krogh, J. McLean, S. Moule, L. Murphy, K. Oliver, J. Osborne, M. A. Quail, M. A. Rajandream, J. Rogers, S. Rutter, K. Seeger, J. Skelton, R. Squares, S. Squares, J. E. Sulston, K. Taylor, S. Whitehead, and B. G. Barrell. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537-544. [DOI] [PubMed] [Google Scholar]
- 16.Cox, J. S., B. Chen, M. McNeil, and W. R. J. Jacobs. 1999. Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature 402:79-83. [DOI] [PubMed] [Google Scholar]
- 17.Daniel, J., C. Deb, V. S. Dubey, T. D. Sirakova, B. Abomoelak, H. R. Morbidoni, and P. E. Kolattukudy. 2004. Induction of a novel class of diacylglycerol acyltransferases and triacylglycerol accumulation in Mycobacterium tuberculosis as it goes into a dormancy-like state in culture. J. Bacteriol. 186:5017-5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dover, L. G., A. M. Cerdeno-Tarraga, M. J. Pallen, J. Parkhill, and G. S. Besra. 2004. Comparative cell wall core biosynthesis in the mycolated pathogens, Mycobacterium tuberculosis and Corynebacterium diphtheriae. FEMS Microbiol. Rev. 28:225-250. [DOI] [PubMed] [Google Scholar]
- 19.Friedman, L., and R. Kolter. 2004. Genes involved in matrix formation in Pseudomonas aeruginosa PA14 biofilms. Mol. Microbiol. 51:675-690. [DOI] [PubMed] [Google Scholar]
- 20.Garton, N. J., H. Christensen, D. E. Minnikin, R. A. Adegbola, and M. R. Barer. 2002. Intracellular lipophilic inclusions of mycobacteria in vitro and in sputum. Microbiology 148:2951-2958. [DOI] [PubMed] [Google Scholar]
- 21.Ha, K. Y., Y. G. Chung, and S. J. Ryoo. 2005. Adherence and biofilm formation of Staphylococcus epidermidis and Mycobacterium tuberculosis on various spinal implants. Spine 30:38-43. [DOI] [PubMed] [Google Scholar]
- 22.Howard, N. S., J. E. Gomez, C. Ko, and W. R. Bishai. 1995. Color selection with a hygromycin-resistance-based Escherichia coli-mycobacterial shuttle vector. Gene 166:181-182. [DOI] [PubMed] [Google Scholar]
- 23.Kremer, L., C. de Chastellier, G. Dobson, K. J. Gibson, P. Bifani, S. Balor, J. P. Gorvel, C. Locht, D. E. Minnikin, and G. S. Besra. 2005. Identification and structural characterization of an unusual mycobacterial monomeromycolyl-diacylglycerol. Mol. Microbiol. 57:1113-1126. [DOI] [PubMed] [Google Scholar]
- 24.Laal, S., Y. D. Sharma, H. K. Prasad, A. Murtaza, S. Singh, S. Tangri, R. S. Misra, and I. Nath. 1991. Recombinant fusion protein identified by lepromatous sera mimics native Mycobacterium leprae in T-cell responses across the leprosy spectrum. Proc. Natl. Acad. Sci. USA 88:1054-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu, J., C. E. Barry, and H. Nikaido. 1999. Cell wall: physical structure and permeability, p. 220-239. In C. Ratledge and J. W. Dale (ed.), Mycobacteria: molecular biology and virulence. Blackwell Science, Oxford, United Kingdom.
- 26.Liu, J., C. E. Barry, G. S. Besra, and H. Nikaido. 1996. Mycolic acid structure determines the fluidity of the mycobacterial cell wall. J. Biol. Chem. 271:29545-29551. [DOI] [PubMed] [Google Scholar]
- 27.Liu, J., and H. Nikaido. 1999. A mutant of Mycobacterium smegmatis defective in the biosynthesis of mycolic acids accumulates meromycolates. Proc. Natl. Acad. Sci. USA 96:4011-4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu, J., E. Y. Rosenberg, and H. Nikaido. 1995. Fluidity of the lipid domain of cell wall from Mycobacterium chelonae. Proc. Natl. Acad. Sci. USA 92:11254-11258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marques, M. A., B. J. Espinosa, E. Xavier de Silveira, M. C. Pessolani, A. Chapeaurouge, J. Perales, K. M. Dobos, J. T. Belisle, J. S. Spencer, and P. J. Brennan. 2004. Continued proteomic analysis of Mycobacterium leprae subcellular fractions. Proteomics 4:2942-2953. [DOI] [PubMed] [Google Scholar]
- 30.McCarthy, C. M. 1984. Free fatty acid and triglyceride content of Mycobacterium avium cultured under different growth conditions. Am. Rev. Respir. Dis. 129:96-100. [DOI] [PubMed] [Google Scholar]
- 31.Nguyen, L., S. Chinnapapagari, and C. J. Thompson. 2005. FbpA-dependent biosynthesis of trehalose dimycolate is required for the intrinsic multidrug resistance, cell wall structure, and colonial morphology of Mycobacterium smegmatis. J. Bacteriol. 187:6603-6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nigou, J., M. Gilleron, M. Rojas, L. F. Garcia, M. Thurnher, and G. Puzo. 2002. Mycobacterial lipoarabinomannans: modulators of dendritic cell function and the apoptotic response. Microbes Infect. 4:945-953. [DOI] [PubMed] [Google Scholar]
- 33.Oftung, F., K. E. Lundin, R. Meloen, and A. S. Mustafa. 1999. Human T cell recognition of the Mycobacterium leprae LSR antigen: epitopes and HLA restriction. FEMS Immunol. Med. Microbiol. 24:151-159. [DOI] [PubMed] [Google Scholar]
- 34.Olukoshi, E. R., and N. M. Packter. 1994. Importance of stored triacylglycerols in Streptomyces: possible carbon source for antibiotics. Microbiology 14:931-943. [DOI] [PubMed] [Google Scholar]
- 35.Ortalo-Magne, A., A. Lemassu, M. A. Laneelle, F. Bardou, G. Silve, P. Gounon, G. Marchal, and M. Daffe. 1996. Identification of the surface-exposed lipids on the cell envelopes of Mycobacterium tuberculosis and other mycobacterial species. J. Bacteriol. 178:456-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pedulla, M. L., M. E. Ford, J. M. Houtz, T. Karthikeyan, C. Wadsworth, J. A. Lewis, D. Jacobs-Sera, J. Falbo, J. Gross, N. R. Pannunzio, W. Brucker, V. Kumar, J. Kandasamy, L. Keenan, S. Bardarov, J. Kriakov, J. G. Lawrence, W. R. J. Jacobs, R. W. Hendrix, and G. F. Hatfull. 2003. Origins of highly mosaic mycobacteriophage genomes. Cell 113:171-182. [DOI] [PubMed] [Google Scholar]
- 37.Recht, J., and R. Kolter. 2001. Glycopeptidolipid acetylation affects sliding motility and biofilm formation in Mycobacterium smegmatis. J. Bacteriol. 183:5718-5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Recht, J., A. Martinez, S. Torello, and R. Kolter. 2000. Genetic analysis of sliding motility in Mycobacterium smegmatis. J. Bacteriol. 182:4348-4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rose, L., S. H. Kaufmann, and S. Daugelat. 2004. Involvement of Mycobacterium smegmatis undecaprenyl phosphokinase in biofilm and smegma formation. Microbes Infect. 6:965-971. [DOI] [PubMed] [Google Scholar]
- 40.September, S. M., V. S. Brozel, and S. N. Venter. 2004. Diversity of nontuberculoid Mycobacterium species in biofilms of urban and semiurban drinking water distribution systems. Appl. Environ. Microbiol. 70:7571-7573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh, S., P. J. Jenner, N. P. Narayan, G. Ramu, M. J. Colston, H. K. Prasad, and I. Nath. 1994. Critical residues of the Mycobacterium leprae LSR recombinant protein discriminate clinical activity in erythema nodosum leprosum reactions. Infect. Immun. 62:5702-5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh, S., N. P. Narayanan, P. J. Jenner, G. Ramu, M. J. Colston, H. K. Prasad, and I. Nath. 1994. Sera of leprosy patients with type 2 reactions recognize selective sequences in Mycobacterium leprae recombinant LSR protein. Infect. Immun. 62:86-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Solano, C., B. Garcia, J. Valle, C. Berasain, J. M. Ghigo, C. Gamazo, and I. Lasa. 2002. Genetic analysis of Salmonella enteritidis biofilm formation: critical role of cellulose. Mol. Microbiol. 43:793-808. [DOI] [PubMed] [Google Scholar]
- 44.Stewart, G. R., L. Wernisch, R. Stabler, J. A. Mangan, J. Hinds, K. G. Laing, D. B. Young, and P. D. Butcher. 2002. Dissection of the heat-shock response in Mycobacterium tuberculosis using mutants and microarrays. Microbiology 148:3129-3138. [DOI] [PubMed] [Google Scholar]
- 45.Strohmeier, G. R., and M. J. Fenton. 1999. Roles of lipoarabinomannan in the pathogenesis of tuberculosis. Microbes Infect. 1:709-717. [DOI] [PubMed] [Google Scholar]
- 46.Tsenova, L., E. Ellison, R. Harbacheuski, A. L. Moreira, N. Kurepina, M. B. Reed, B. Mathema, C. E. Barry, and G. Kaplan. 2005. Virulence of selected Mycobacterium tuberculosis clinical isolates in the rabbit model of meningitis is dependent on phenolic glycolipid produced by the bacilli. J. Infect. Dis. 192:98-106. [DOI] [PubMed] [Google Scholar]
- 47.Wang, L., R. A. Slayden, C. E. Barry, and J. Liu. 2000. Cell wall structure of a mutant of Mycobacterium smegmatis defective in the biosynthesis of mycolic acids. J. Biol. Chem. 275:7224-7229. [DOI] [PubMed] [Google Scholar]
- 48.Wong, D. K., B. Y. Lee, M. A. Horwitz, and B. W. Gibson. 1999. Identification of fur, aconitase, and other proteins expressed by Mycobacterium tuberculosis under conditions of low and high concentrations of iron by combined two-dimensional gel electrophoresis and mass spectrometry. Infect. Immun. 67:327-336. [DOI] [PMC free article] [PubMed] [Google Scholar]