

Abstract
Staphylococcus aureus is the most common cause of hospital-acquired infection. In healthy hosts outside of the health care setting, S. aureus is a frequent colonizer of the human nose but rarely causes severe invasive infection such as bacteremia, endocarditis, or osteomyelitis. To identify genes associated with community-acquired invasive isolates, regions of genomic variability, and the S. aureus population structure, we compared 61 community-acquired invasive isolates of S. aureus and 100 nasal carriage isolates from healthy donors using a microarray spotted with PCR products representing every gene from the seven S. aureus sequencing projects. The core genes common to all strains were identified, and 10 dominant lineages of S. aureus were clearly discriminated. Each lineage carried a unique combination of hundreds of “core variable” (CV) genes scattered throughout the chromosome, suggesting a common ancestor but early evolutionary divergence. Many CV genes are regulators of virulence genes or known or predicted to be expressed on the bacterial surface and to interact with the host during nasal colonization and infection. Within each lineage, isolates showed substantial variation in the carriage of mobile genetic elements and their associated virulence and resistance genes, indicating frequent horizontal transfer. However, we were unable to identify any association between lineage or gene and invasive isolates. We suggest that the S. aureus gene combinations necessary for invasive disease may also be necessary for nasal colonization and that community-acquired invasive disease is strongly dependent on host factors.
Staphylococcus aureus is a persistent resident of the human nose in 20% of the population and intermittently carried by another 60% (16). Most carriers harbor a single strain (2). S. aureus is a common cause of minor skin and wound infections, but only rarely causes severe community-acquired invasive infections such as bacteremia, endocarditis, and osteomyelitis. In contrast, S. aureus is the most common cause of hospital-acquired infection, which often occurs in association with breaches of the skin and mucous membranes in the immunocompromised host (14). Hundreds of S. aureus virulence factors and putative virulence genes have been described, including those involved in adherence to human tissue, evasion of the immune response, toxin secretion, and regulation of virulence gene expression (29). Specific toxins have also been described that play a pivotal role in toxin-mediated disease such as toxic shock syndrome (toxic shock syndrome toxin-1, encoded by tst; enterotoxins B and C, encoded by seb, sec) (5), scalded skin syndrome (exfoliative toxins A and B, encoded by eta, etb) (19), food poisoning (enterotoxin A, encoded by sea) (5), and more recently hemolytic pneumonia and skin and soft tissue infection (Panton Valentine leukocidin [PVL], encoded by the lukS-PV and lukF-PV genes) (20). Many of these genes are variably present as a result of being carried on mobile genetic elements (MGE) (22). However, critical to the development of targeted or preventive strategies is the elucidation of which if any of these genes are important in invasive infection.
An important isolate collection associated with community-acquired invasive disease or carriage by healthy donors in the Oxford, United Kingdom, region (7) has been examined using multilocus sequence typing (MLST) (6), in which fragments of seven housekeeping genes were amplified and sequenced. Unique alleles at the seven loci were given an allelic number, and the allelic profile (string of seven integers) was used to define sequence type (ST) for each isolate. Isolates with an identical profile were considered to be clonal, and those with at least five of seven matching genes were considered to belong to the same clonal cluster (CC). Isolates clustered into 10 major CCs, none of which were associated with invasive disease (7). This argued against the presence of virulent genotypes but did not exclude the possibility that one or more variable genes were overrepresented in the invasive-isolate group. This possibility was examined during a study that defined the presence or absence of 33 putative virulence genes in this isolate collection using PCR (27). Seven genes were found to be present more commonly in invasive isolates, including eta and those encoding fibronectin binding protein A (fnbA), collagen binding protein (cna), serine-aspartate repeat containing protein E (sdrE), staphylococcal enterotoxin J (sej), gamma-hemolysin (hlg), and intracellular adhesin (ica). This suggested there were differences between isolates that did not correlate with lineage. It also suggested that some isolates are potentially more virulent than others.
Each S. aureus isolate is thought to carry hundreds of variable genes including many putative virulence determinants. The first S. aureus comparative-genomics studies using a microarray (covering 92% of the genes found in the S. aureus COL genome) estimated that 22% of the S. aureus genome was variable (8). Many of the variable genes are known or putative virulence and resistance genes carried on MGE, and these elements are likely to transfer horizontally among staphylococci (see reference 22 for a review). The accumulation of such MGE may result in the emergence of “superbugs” that are increasingly resistant and virulent (22). The whole-genome sequencing of seven isolates of S. aureus (1, 10, 12, 18; www.genome.ou.edu/staph.html) has allowed us to design, print, and validate a multistrain PCR product S. aureus microarray carrying PCR products for every gene identified from these projects, probably the most comprehensive microarray of its kind (33). Here we describe the use of the seven-strain S. aureus microarray to investigate the Oxford collection of community-acquired S. aureus isolates. The aims were to identify which regions of the S. aureus genome vary, investigate gene distribution in a typical S. aureus population, and perform a comprehensive search for differences between invasive and carriage isolates.
MATERIALS AND METHODS
Strains.
The S. aureus isolates have been previously described (4). Sixty-one isolates associated with community-acquired infection in the Oxford region were compared to 100 nasal-carriage isolates from healthy blood donors in the same Oxford region. The 100 carriage isolates were chosen at random from 180 isolates in the original study.
Microarrays.
DNA was extracted using QIAGEN genomic-tip 100/G columns, and concentration was measured using the optical density at 260 nm. Bulk reference DNA was prepared from MRSA252 using the cesium chloride method (21). Four milligrams of test DNA was labeled using Cy3 dye and DNA polymerase I large fragment (Klenow; Invitrogen), and 4 mg of reference DNA was labeled using Cy5 dye. The two samples were pooled and hybridized to an S. aureus microarray overnight, before washing and scanning (33). Reference DNA is used to provide a known internal control for each of the spots, and DNA from a single isolate has technical advantages over multiple isolates or PCR product controls (33).
The S. aureus microarray has been described previously (33) and contains 3,623 PCR products representing every predicted open reading frame in the seven genome sequencing projects. The seven sequenced strains and their corresponding ST and CC types are MRSA252/ST36/CC30, an epidemic MRSA-16 from a hospitalized patient, Oxford, United Kingdom; N315/ST5/CC5, a methicillin-resistant S. aureus (MRSA) isolate from a hospitalized Japanese patient; Mu50/ST5/CC5, an MRSA isolate related to N315 with intermediate-level resistance to vancomycin; COL/ST250/CC8, an early MRSA strain from the United Kingdom; 8325/ST8/CC8, parent of the standard laboratory S. aureus strain; MW2/ST1/CC1, a community-acquired invasive MRSA from the United States; and MSSA476/ST1/CC1, a community-acquired invasive S. aureus isolate from Oxford, United Kingdom. A number of genes predicted to play a role in virulence show significant genomic variation between the sequenced strains. Typically, the majority of the gene is highly conserved (>97% homology) but a section of the gene, often with discrete boundaries, is highly divergent. These genes had multiple PCR products designed to target the different variant types and included those coding for accessory gene regulator (agr), coagulase (coa), a putative bacillus-like toxin (bceT), fibronectin binding proteins A and B (fnbA and fnbB), and a hemagglutinin-like protein (sasA) (33). PCR products were printed in duplicate on GAPS slides (Corning). DNA was fixed using UV light and blocked with bovine serum albumin prior to hybridization. Microarrays were scanned using an Affymetrix 428 scanner (33).
Data analysis.
BlueFuse for Microarrays 2.0 (BlueGnome, Cambridge, United Kingdom) was used to convert all scanned images to raw data for analysis. Data analysis was performed in GeneSpring 6.2 (Silicon Genetics) (33), while results from Predict Parameter were confirmed using the updated function in GeneSpring 7.0. Raw data were normalized as a single experiment in GeneSpring using LOWESS (locally weighted scatterplot smoothing) with 50% of the data used for smoothing and a control channel cutoff of 0.01. Condition tree clustering using the Spearman correlation was performed as a function in GeneSpring and used to cluster isolates using defined gene lists. Predict Parameter in GeneSpring 7.0 using Fisher's exact test and the Golub method was used to identify genes associated with invasive isolates.
The first condition tree using the Spearman correlation was constructed using every gene (PCR product) on the microarray and normalized microarray data (ratio of signal intensities of test divided by control) from all 161 isolates. The gene list was then adjusted to remove all MGE genes and then all core genes. MGE genes were identified manually from the annotated sequencing projects and included all those annotated and/or clearly carried on a bacteriophage, S. aureus pathogenicity island (SaPI), plasmid, transposon, staphylococcal cassette chromosome (SCC), or genomic island (GI). For COL and 8325 (unannotated at the time of microarray design), genomes were compared by the Artemis comparison tool (32) to the annotated isolates, and BLAST searches of specific genes were used to confirm MGE. Composite genomes of each MGE were constructed by identifying the microarray PCR product that best matched each gene in the MGE and listing them in order (33). Core genes were identified as those with a fluorescence intensity ratio (test isolate/reference isolate) between 0.5 and 2 in >95% of the isolates. Condition trees are presented using a color code for each gene based on the fluorescence intensity ratio, with yellow genes representing those found in both the test and reference, blue those found in the reference only, and red those found in the test only. Spots flagged as poor-quality data or with signals less than twofold above background are colored gray. Those with weak fluorescence in both channels appear close to white.
Microarray fluorescence intensity values are presented as a ratio of test over reference. In order to convert this information to “present” or “absent” for each gene, we tested a number of options (33). Firstly, ratios were converted to log2 values and cutoffs were set at either above 1, 1.5, 2, or 2.5 (“present”) or below −1, −1.5, −2, or −2.5 (“absent”). All four combinations generated gene lists that were tested. A second approach was to use GACK software to convert the data from each individual microarray to present or absent (15, 33). For all methods and for every gene, the proportion of genes present, absent, or indeterminate for the invasive isolates was compared to the carriage isolates using the chi-squared test. We applied Bonferroni and Benjamini and Hochberg false-discovery rate multiple testing corrections to reduce the number of false positives due to the large number of genes tested.
One hundred fifty genes of interest (known or putative virulence genes) (see Table S2 in the supplemental material) were manually called “present” or “absent” based on both signal intensity ratio and total intensity on a scatter plot, and in comparison to the sequenced strains as controls; this was performed by one person, who was unaware of the source of the isolate. In this case, absence or presence was called by eye using the following criteria: a gene present in MRSA252 was called present in the test isolate if it fell on or near the “median line” at a signal intensity comparable to that of the same gene in a known “positive” sequenced isolate and absent if it fell below the lower twofold line with a signal intensity comparable to that of the same gene in a known “negative” sequenced isolate. Similarly, if a gene is absent in MRSA252, it was called present in the test isolate if it fell above the upper twofold line at a signal intensity comparable to that of the same gene in a known “positive” sequenced isolate and absent if the signal in both channels was flagged by BlueFuse as poor quality (signal intensity less that twice the background). Results were then compared by chi-squared test and multiple testing corrections as above.
RESULTS
Fully annotated microarray data have been deposited in BμG@Sbase (accession number E-BUGS-33; http://bugs.sgul.ac.uk/E-BUGS-33) and also ArrayExpress (accession number E-BUGS-33). Visual inspection of the normalized data for 3,623 genes showed substantial variation between the 161 isolates. A condition tree constructed by Spearman correlation of all genes from all of the isolates was complex with few dominant lineages but some clustering of isolates corresponding to the MLST CC types (see Fig. S1 in the supplemental data). This tree showed that a substantial amount of variation between isolates was due to MGE genes.
Lineages.
Analysis followed a stepwise progression. Next, we constructed a condition tree using all genes apart from the MGE genes (total of 2,734 genes; see Fig. S2 in the supplemental material). Isolates clustered into major lineages, and a large number of genes were defined as core (present in >95% of isolates). This list included 52 core genes that were identified in the rapid COL annotation but were not identified as open reading frames in the published annotated whole-genome sequences, mostly due to their small size. All of the core genes were identified and removed from the gene list, leaving 728 genes that were termed “core-variable” (CV) genes; these genes are listed in Table S3 in the supplemental material. The condition tree constructed using these genes is shown in Fig. 1, along with the signals for a selection of 30 CV PCR products of interest which illustrate the typical variation seen. (A tree showing results for all 728 genes is shown in Fig. S3 in the supplemental material).
FIG. 1.

Conditional tree constructed using the Spearman correlation of 161 isolates and 728 CV genes. A selection of 30 CV PCR products is displayed representing insertions/deletions and gene variants of interest (see Fig. 2). Each vertical line represents an isolate, and its branch on the tree is colored according to MLST CC type, with the same colors repeated in the row of boxes at the bottom with corresponding CC numbers. Note that the six pale green clusters represent 13 “orphan” isolates that did not match an MLST CC and do not cluster with the most common lineages by microarray. *, two CC5 (ST6) isolates that do not cluster with the other CC5 (ST5) isolates, marked in dark green; †, two CC1 (ST188) isolates that do not cluster with the other CC1 isolates, marked in red. In the central region, each row of colored squares represents the hybridization signal to a microarray PCR product. The corresponding PCR products are listed in Fig. 2. The intensity of all the colors is an indicator of the total signal intensity, while the color is an indicator of test signal over reference signal ratio. Thus, PCR products colored yellow hybridized to both the test and reference (MRSA252) isolates, PCR products in blue hybridized to the reference strain only, and PCR products in red hybridized to the test strain only. Spots with fluorescent signals lower than two times the background are flagged and colored gray, indicating a gene absent in both the test and reference. Genes with white signals are very low intensity and regarded as negative for both isolates. The figure clearly shows CV genes that are present or absent in accordance with lineage, and the lineages correlate with MLST CC groups.
The tree clearly discriminates distinct lineages, and these closely match the MLST clonal clusters CC1, CC5, CC8, CC9, CC12, CC15, CC22, CC25, CC30, CC45, and CC51, so we have kept the same lineage nomenclature. Thirteen “orphan” isolates that were not assigned to a major CC by MLST were distributed throughout the tree (pale green) and did not cluster with any of the major CC groups. Two isolates called ST6, CC5 by MLST clustered separately from the ST5, CC5 isolates in the tree, suggesting they belong to a distinct lineage (Fig. 1). Similarly, two isolates called ST188, CC1 by MLST clustered separately from the other CC1 isolates, suggesting they are also of a distinct lineage (Fig. 1). It is interesting to note that common STs within each CC did not always cluster together. For example, ST39 isolates within CC30 do not cluster together by CV genes (see Fig. S3 in the supplemental material).
Figure 1 also shows the presence/variability of 30 select CV PCR products of interest. These genes are listed in Fig. 2, with a cartoon representation of which is found in each lineage. Some variation represents insertion or deletions of regions carrying between one and nine genes, such as sarT (staphylococcal accessory regulator T gene) and sasG (staphylococcal anchored to surface G gene). Other differences are due to divergent regions within a gene. For example, fnbA has a central region of approximately 145 bp showing variation between the sequenced isolates, and specific PCR products for those regions from strains MRSA252, N315, and 8325 were included on the microarray. Isolates of CC12 and CC51 do not hybridize with any of these primers, suggesting they carry a novel variant not found on the microarray; this is likely, as no isolate from these lineages has been sequenced.
FIG. 2.
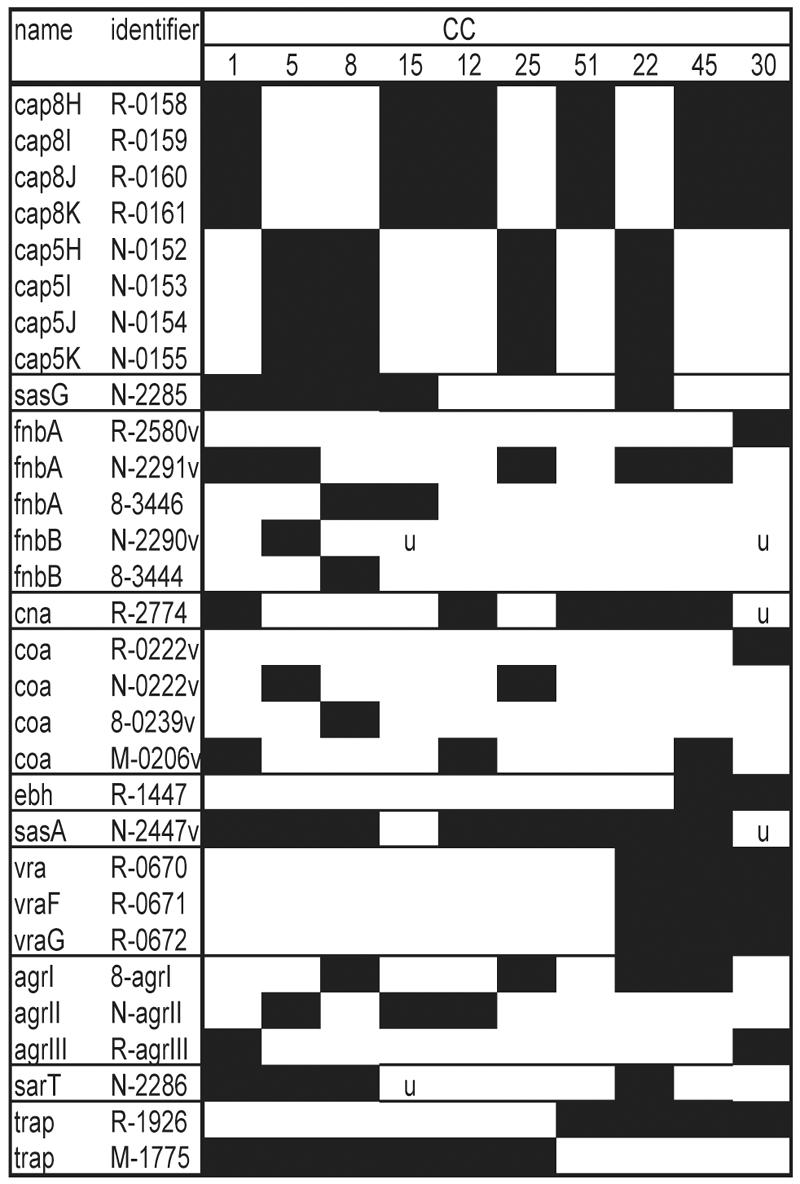
PCR products in Fig. 1. PCR products representing CV genes or gene variant regions are listed in the same order as Fig. 1 by putative gene product name and identifier or annotated gene number (R products are from MRSA252, N products are from N315, 8- products are from 8325-4, and the M product is from MW2). “v” denotes a PCR product designed to a specific variant region, and so multiple variant regions of some genes are included. A black box indicates that the gene is present in that CC. “u” indicates variation in gene distribution for that CC. For some genes that are found in all isolates, variant regions may not have been sequenced and therefore are not on the microarray, e.g., the coagulase gene variant region in isolates of CC15.
Many of the CV genes are known or predicted to be expressed on the S. aureus cell surface. capHIJK genes are necessary for S. aureus capsule production, and these four genes define major capsule types 5 and 8. sasG encodes an LPXTG cell wall-anchored protein that binds to nasal epithelial cells (31) and is closely related to the accumulation-associated protein of Staphylococcus epidermidis, implicated in biofilm production. fnbA, fnbB, and cna encode LPXTG proteins anchored to the cell wall that bind to host tissue (9). Coagulase (encoded by coa) is secreted by S. aureus and converts fibrinogen to fibrin, although some coagulase is retained on the cell surface (23). ebh encodes an enormous immunodominant surface-exposed protein that also binds host proteins (3), while hemagglutinin-like protein (encoded by sasA) has an LPXTG motif and is predicted to be surface anchored. Aside from the examples in Fig. 1 and 2, many other CV genes show variation associated with lineage and encode proteins predicted to be surface expressed, including those encoding proteins that bind host tissue (sdrD and sdrE) and immunodominant antigen B (isaB), peptidoglycan synthesis genes (mrp and fmhC), a cell wall hydrolase/autolysin gene (lytN), several oligopeptide transport genes, and genes encoding many putative lipoproteins and membrane proteins of unknown function. For example, the vra genes are an ABC transporter operon upregulated in intermediate-level-vancomycin- resistant isolates (17).
The CV genes also include the global virulence gene regulators agr, trap (target of RNAIII activator protein), and sarT, all known to regulate expression of surface proteins (see reference 26 for a review). Four known variants of agr exist (types I to IV), and each agr type regulates virulence genes in a different way (13). Each of the lineages was associated with agr types I to III, except all CC51 isolates. agrIV is predicted to have 87% homology with the agrI PCR product (over 796 bp), and no agrIV-specific spot was designed for the microarray. The agr region of a CC51 isolate (strain 3) was sequenced in this region, and it matched exactly to the published agrIV. It has been suggested that trap is involved in activation of the agr activator molecule, RNAIII (26). Two variant types exist and are 86% homologous. sarT (also known as sarH3) is one of several sarA-like regulators that form a complex network controlling expression of virulence genes in S. aureus (26). sarT is carried on an “islet” found in some strains and not others; this islet includes two accumulation-associated genes (aac), another sar homolog (sarH2), and a possible transposase gene.
Figure 3 shows that CV genes found in MRSA252 are randomly distributed throughout the chromosome. By microarray, this isolate has 1,954 genes hybridizing to core genes on the microarray (71.2%), 327 CV genes (11.9%), and 460 MGE genes (16.8%).
FIG. 3.

Representation of the MRSA252 genome with each gene colored according to whether it is a core gene or CV gene or whether it is found on an MGE. The figure was generated in GeneSpring using the same lists used for identifying CV genes. The outer circle represents genes on the forward coding strand, and the inner circle represents genes on the complementary strand.
MGE.
The amount of variation in MGE was truly remarkable, even within lineages. For each MGE distributed among the 161 isolates, there were two types of variation. Firstly, distribution patterns of each MGE are assumed to reflect mobility of the MGE. For example, MGE strongly associated with lineage are thought to be stable (infrequently lost or acquired) and distributed mostly by vertical transmission to daughter cells. An MGE that is randomly distributed is likely to be transferred horizontally. This is because a hypothesis of infrequent transfer of each MGE and subsequent multiplication of isolates is not supported because of the extensive variation of other MGE between isolates, even within the same lineage. MGE that are horizontally transferred but conspicuously absent from certain lineages are thought to have some restrictions on horizontal transmission (18). The second type of variation occurring within each MGE was typically seen as conservation of short mosaic fragments of an MGE but not the rest of the element. From the sequencing projects, there is evidence of substantial homologous recombination within MGE, such that each MGE is composed of multiple short mosaic fragments that are randomly spread through other MGE of the same type (e.g., phage or SaPI). Therefore, the presence of only a fragment of an MGE in an isolate by microarray is likely to indicate the carriage of a whole MGE, with the remaining fragments either not represented on the microarray or represented by other short mosaic fragments on the microarray.
Bacteriophages were the most widespread and variable MGE. Some phages are clearly transferred horizontally, while others such as the φ3 group are more stable. Significant recombination (as seen by conserved mosaic fragments) was common. Virulence genes carried on phages include lukS-PV and lukF-PV, sea, the staphylokinase gene (sak), the gene encoding chemotaxis inhibitory protein (chips), and eta. lukS-PV and lukF-PV are carried on φ2 (MW2) in strain MW2, but of the seven lukS-PV- and lukF-PV-positive isolates identified by microarray in this collection, only one carried φ2 genes. Thus, PVL genes are likely to be carried on an unrelated phage in these isolates. Similarly, sea and chips are found on φ3 phage in the sequenced isolates, but by microarray these genes are sometimes found in isolates that do not carry many φ3 genes. The nine eta-positive isolates were all from unsequenced lineages (mostly CC51), and carried few phage genes that hybridized to the microarray.
At least two SaPI genes were found in 152 (94%) of the isolates, showing that SaPIs are widespread. SaPIs are typically 15 kb, carry an integrase gene related to bacteriophage integrase genes, integrate at specific sites, and can transfer horizontally at very high frequency with the help of specific bacteriophages (21). Some SaPIs are clearly quite stable such as SaPI4(MRSA252), with the majority of the SaPI found in 34 of the 51 CC30 isolates (67%). SaPIs related to SaPI(N315), carrying the tst gene, were common in CC30 isolates, yet are clearly missing in some CC30 isolates including MRSA252. Fragments of these SaPIs were also found in other lineages, suggesting horizontal transfer.
A number of plasmid genes were widespread in the collection, indicating frequent horizontal transfer but with some restriction. The integrated plasmid in MRSA252 was found in 48 of 51 CC30 isolates, suggesting it is stable and rarely lost. Integrated conjugative elements were also relatively stable. The transposon Tn552, encoding β-lactamase resistance, was widespread, but Tn554, encoding erythromycin resistance, was rare. Surprisingly, only three isolates carried an SCC-like element. One of these isolates carried an SCCmecIV element, and was CC22, indicating it was an epidemic methicillin-resistant MRSA-15, typical of hospital isolates in the United Kingdom. The other two isolates included the sequenced MSSA476 and carried a putative fusidic acid resistance gene. Insertion sequences were common but were associated strongly with lineages, suggesting horizontal transfer is rare.
The S. aureus genomic islands GIα and GIβ are variable, but variation corresponded strongly to CC. Thus they seem stable and more like CV genes. However, some variation in GIβ in the CC30 lineage occurs only in the eight ST39 isolates. This suggests that a stable variant of this region may be associated with a subgroup of CC30, although members of the ST39 group do not cluster together in a condition tree constructed using the CV genes.
Genes associated with invasive isolates.
GeneSpring Predict Parameter failed to identify any gene that was convincingly or statistically different between the invasive and carriage groups. The cutoff and GACK methods for assigning genes as present or absent did not identify any gene that was significantly associated with invasive isolates. Using a visual confirmation of the 150 known or putative virulence genes that were manually called present or absent, no gene was found to be statistically associated with invasive isolates. The PVL locus is thought to be associated with invasive community-acquired isolates (20), particularly those that are methicillin resistant, but was found in only seven isolates of this study, six of which were invasive. This association was not statistically significant, due to the low incidence of the gene, and shows that PVL is not responsible for invasive disease in this collection.
If each CC has a unique complement of virulence genes, it could be that any gene truly associated with invasive isolates is masked by this bias. Only one CC group contained sufficient isolates to investigate this hypothesis: CC30, which contains 51 isolates, including 19 associated with invasive disease. Using Predict Parameter, log2 cutoff method and manual calling, no gene was found to be associated with the invasive isolates.
DISCUSSION
The seven S. aureus sequencing projects suggested the genome consisted of core genes and accessory genes found on MGE (22). Here, we identify a third group of genes that show substantial variation between isolates but are typically stable and transferred vertically. These CV genes are scattered throughout the genome and make up approximately 10 to 12% of any genome. Our findings suggest that the common ancestor of human S. aureus isolates, represented by a backbone of 1,954 core genes, has over time acquired CV genes which may subsequently recombine or be lost. Of the billions of possible combinations, the progeny of only 10 major lineages have become established in the human nose.
The lineages identified here correlate very strongly with those generated by MLST. This confirms the usefulness of both methods in classifying isolates of S. aureus into the major lineages. MLST detects point mutations in core housekeeping genes, and it seems likely that these mutations accumulate slowly and are passed to daughter cells in the lineage, just as the CV insertions/deletions and gene variants detected by microarray are passed on. It is interesting that MLST detects further differences (ST) within each CC that are not corroborated by microarray. This could be due to the enormous amount of variation seen in the S. aureus genome, so that relying on only a few markers places increased weight on minor differences. Within lineages, most of the variation between isolates detected by microarray was due to the acquisition or loss of MGE, and this variation was substantial. Most other S. aureus typing methods are skewed by MGE genes which effectively mask the lineages, including pulse-field gel electrophoresis and amplified fragment length polymorphisms (11, 24).
Our results show that variation between lineages is due to a range of insertions, deletions, and variant regions in hundreds of S. aureus genes and, in particular, microbial surface components recognizing adhesive matrix molecules (MSCRAMMs) as well as key regulators that control their expression. S. aureus binding to tissue during infection is thought to be a key step in pathogenesis, primarily mediated by MSCRAMMs binding to fibrinogen, fibronectin, collagen, and other components of the host extracellular matrix (9). MSCRAMMs are also candidate molecules for vaccines and other immunotherapies to prevent colonization and infection (30). If each bacterial lineage presents a unique surface architecture to the human host, presumably each lineage interacts with the host in unique ways. Furthermore, isolates from only four of the dominant lineages have been sequenced, so there are likely to be variations unique to the other lineages that are currently unidentified. The investigation of these variants and how they interact with the host should enhance our understanding of host-pathogen interactions and the development of therapies.
The normal habitat of S. aureus is as a commensal of the human nose, where it binds to nasal epithelial cells and mucin. Many bacterial factors are implicated in nasal colonization including fnbA, fnbB, cna, sasG, and capsule (28, 31). These genes are core variable by microarray. Most colonized humans carry only one lineage of S. aureus (2, 28), and this may be due to specific host factors, as a host cleared of a colonizing strain will preferentially recolonize with the same strain, even when inoculated with a mixture of strains (25). It therefore seems likely that different lineages preferentially colonize particular hosts. Although the factors responsible for host variation remain speculative, this host variation in the nose may have driven the evolution of the 10 lineages.
There were no consistent differences in gene content that could be used to distinguish between invasive and carriage isolates in this study. The isolate collection used here represents a subset of those used in a study in which seven genes (fnbA, cna, sdrE, sej, eta, hlg, and ica) were more commonly detected by PCR in invasive isolates, and the combination of all seven genes was even more predictive (27). However, using the microarray, these genes were not confirmed as associated with invasive isolates either alone or in combination. fnbA, cna, and sdrE were CV genes by microarray, while all 161 isolates were positive for hlg and ica. The possibility that the previous finding based on PCR (27) was due to variation in sequence around the primer binding sites cannot be discounted.
If each lineage expresses a distinct combination of surface structures that have been implicated in virulence, it seems remarkable that no lineage seems any more virulent that the others. If each lineage is essentially equally virulent, perhaps the presence and variability in these individual genes are not as important for disease as initially thought, and the key is gene combinations. If we also consider that only 10 lineages are found in the human nose, it raises the possibility that these lineages carry the necessary virulence gene combinations for successful colonization of the nose. Therefore, it could be that the genes necessary for virulence are the ones that allow nasal carriage and that the ability to cause invasive disease is mostly dependent on host factors.
The ability of particular toxins to render an isolate virulent is established for toxin-mediated diseases such as toxic shock syndrome, food poisoning, and scalded skin syndrome. Recent studies have suggested that PVL is associated with necrotizing pneumonia in children and outbreaks of severe skin infection in healthy people (20). Melles et al. (24) also suggested an association with this toxin and arthritis and abscess isolates. PVL genes were rare in our collection, and although an association with invasive isolates was seen, it was not significant. No evidence for the association with any other toxin with invasive isolates in this study was seen.
In conclusion, we find no evidence that certain genes or lineages are associated with invasive isolates in the community setting. However, it is possible that some genes or lineages are associated with particular types of invasive disease, e.g., bacteremia, osteomyelitis, and pneumonia, and specific isolate collections will be needed to address this question. It is also possible that this strain collection is not typical of strains carried in other parts of the United Kingdom or the world, or that S. aureus populations change over time, and further studies will be needed to confirm this. While we generated an enormous amount of data and identified substantial differences between isolates, it could be that virulence is due to the expression of one or more important genes under appropriate in vivo conditions. Testing for this will be complicated by identifying appropriate conditions for S. aureus growth.
Despite the enormous variation seen between S. aureus isolates and the considerable amount of genetic exchange between isolates, we have no evidence that this variation influences pathogenesis. Future studies may show that variation is important for nasal carriage. The key to understanding S. aureus pathogenesis may lie in the identification of host factors that contribute to colonization, and subsequent susceptibility to community-acquired infection.
Supplementary Material
Acknowledgments
We thank the St George's Charitable Trust for funding consumables for this project. The Bacterial Microarray Group is supported by a grant from The Wellcome Trust under its Functional Genomics Resources Initiative (grant number 062511). S.J.P. is a Wellcome Trust Career Development Fellow in Clinical Tropical Medicine.
We thank Denise Waldron for sequencing the agrIV region.
Footnotes
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Baba, T., F. Takeuchi, M. Kuroda, H. Yuzawa, K. Aoki, A. Oguchi, Y. Nagai, N. Iwama, K. Asano, T. Naimi, H. Kuroda, L. Cui, K. Yamamoto, and K. Hiramatsu. 2002. Genome and virulence determinants of high virulence community-acquired MRSA. Lancet 359:1819-1827. [DOI] [PubMed] [Google Scholar]
- 2.Cespedes, C., B. Said-Salim, M. Miller, S. H. Lo, B. N. Kreiswirth, R. J. Gordon, P. Vavagiakis, R. S. Klein, and F. D. Lowy. 2005. The clonality of Staphylococcus aureus nasal carriage. J. Infect. Dis. 191:444-452. [DOI] [PubMed] [Google Scholar]
- 3.Clarke, S. R., L. G. Harris, R. G. Richards, and S. J. Foster. 2002. Analysis of Ebh, a 1.1-megadalton cell wall-associated fibronectin-binding protein of Staphylococcus aureus. Infect. Immun. 70:6680-6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Day, N. P., C. E. Moore, M. C. Enright, A. R. Berendt, J. M. Smith, M. F. Murphy, S. J. Peacock, B. G. Spratt, and E. J. Feil. 2001. A link between virulence and ecological abundance in natural populations of Staphylococcus aureus. Science 292:114-116. [DOI] [PubMed] [Google Scholar]
- 5.Dinges, M. M., P. M. Orwin, and P. M. Schlievert. 2000. Exotoxins of Staphylococcus aureus. Clin. Microbiol. Rev. 13:16-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Enright, M. C., N. P. Day, C. E. Davies, S. J. Peacock, and B. G. Spratt. 2000. Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J. Clin. Microbiol. 38:1008-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feil, E. J., J. E. Cooper, H. Grundmann, D. A. Robinson, M. C. Enright, T. Berendt, S. J. Peacock, J. M. Smith, M. Murphy, B. G. Spratt, C. E. Moore, and N. P. Day. 2003. How clonal is Staphylococcus aureus? J. Bacteriol. 185:3307-3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fitzgerald, J. R., D. E. Sturdevant, S. M. Mackie, S. R. Gill, and J. M. Musser. 2001. Evolutionary genomics of Staphylococcus aureus: insights into the origin of methicillin-resistant strains and the toxic shock syndrome epidemic. Proc. Natl. Acad. Sci. USA 98:8821-8826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foster, T. J., and M. Hook. 1998. Surface protein adhesins of Staphylococcus aureus. Trends Microbiol. 6:484-488. [DOI] [PubMed] [Google Scholar]
- 10.Gill, S. R., D. E. Fouts, G. L. Archer, E. F. Mongodin, R. T. Deboy, J. Ravel, I. T. Paulsen, J. F. Kolonay, L. Brinkac, M. Beanan, R. J. Dodson, S. C. Daugherty, R. Madupu, S. V. Angiuoli, A. S. Durkin, D. H. Haft, J. Vamathevan, H. Khouri, T. Utterback, C. Lee, G. Dimitrov, L. Jiang, H. Qin, J. Weidman, K Tran, K. Kang, I. R. Hance, K. E. Nelson, and C. M. Fraser. 2005. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J. Bacteriol. 187:2426-2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grundmann, H., S. Hori, M. C. Enright, C. Webster, A. Tami, E. J. Feil, and T. Pitt. 2002. Determining the genetic structure of the natural population of Staphylococcus aureus: a comparison of multilocus sequence typing with pulsed-field gel electrophoresis, randomly amplified polymorphic DNA analysis, and phage typing. J. Clin. Microbiol. 40:4544-4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holden, M. T. G., E. J. Feil, J. A. Lindsay, S. J. Peacock, N. P. Day, M. C. Enright, T. J. Foster, C. E. Moore, L. Hurst, R. Atkin, A. Barron, N. Bason, S. D. Bentley, C. Chillingworth, T. Chillingworth, C. Churcher, L. Clark, C. Corton, A. Cronin, J. Doggett, L. Dowd, T. Feltwell, Z. Hance, B. Harris, H. Hauser, S. Holyroyd, K. Jagels, K. D. James, N. Lennard, A. Line, R. Mayes, S. Moule, K. Mungall, D. Ormond, M. A. Quail, E. Raddinowitsch, K. Rutherford, M. Sanders, S. Sharp, M. Simmonds, K. Stevens, S. Whitehead, B. G. Barrell, and J. Parkhill. 2004. Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proc. Natl. Acad. Sci. USA 101:9786-9791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jarraud, S., G. J. Lyon, A. M. Figueiredo, L. Gerard, F. Vandenesch, J. Etienne, T. W. Muir, and R. P. Novick. 2000. Exfoliatin-producing strains define a fourth agr specificity group in Staphylococcus aureus. J. Bacteriol. 182:6517-6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones, R. N. 2003. Global epidemiology of antimicrobial resistance among community-acquired and nosocomial pathogens: a five-year summary from the SENTRY antimicrobial surveillance program (1997-2001). Semin. Respir. Crit. Care Med. 24:121-134. [DOI] [PubMed] [Google Scholar]
- 15.Kim, C. C., E. A. Joyce, K. Chan, and S. Falkow. 2002. Improved analytical methods for microarray-based genome-composition analysis. Genome Biol. 3:research0065.1-research0065.17. [Online.] http://genomebiology.com/2002/3/11/research/0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kluytmans, J., A. van Belkum, and H. Verbrugh. 1997. Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clin. Microbiol. Rev. 10:505-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuroda, M., K. Kuwahara-Arai, and K. Hiramatsu. 2000. Identification of the up- and down-regulated genes in vancomycin-resistant Staphylococcus aureus strains Mu3 and Mu50 by cDNA differential hybridization method. Biochem. Biophys. Res. Commun. 269:485-490. [DOI] [PubMed] [Google Scholar]
- 18.Kuroda, M., T. Ohta, I. Uchiyama, T. Baba, H. Yuzawa, I. Kobayashi, L. Cui, A. Oguchi, K. Aoki, Y. Nagai, J. Lian, T. Ito, M. Kanamori, H. Matsumaru, A. Maruyama, H. Murakami, A. Hosoyama, Y. Mizutani-Ui, N. K. Takahashi, T. Sawano, R. Inoue, C. Kaito, K. Sekimizu, H. Hirakawa, S. Kuhara, S. Goto, J. Yabuzaki, M. Kanehisa, A. Yamashita, K. Oshima, K. Furuya, C. Yoshino, T. Shiba, M. Hattori, N. Ogasawara, H. Hayashi, and K. Hiramatsu. 2001. Whole genome sequencing of methicillin-resistant Staphylococcus aureus. Lancet 357:1225-1240. [DOI] [PubMed] [Google Scholar]
- 19.Lee, C. Y., J. J. Schmidt, A. D. Johnson-Winegar, L. Spero, and J. J. Iandolo. 1987. Sequence determination and comparison of the exfoliative toxin A and toxin B genes from Staphylococcus aureus. J. Bacteriol. 169:3904-3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lina, G., Y. Piemont, F. Godail-Gamot, M. Bes, M. O. Peter, V. Gauduchon, F. Vandenesch, and J. Etienne. 1999. Involvement of Panton-Valentine leukocidin-producing Staphylococcus aureus in primary skin infections and pneumonia. Clin. Infect. Dis. 29:1128-1132. [DOI] [PubMed] [Google Scholar]
- 21.Lindsay, J. A., A. Ruzin, H. F. Ross, N. Kurepina, and R. P. Novick. 1998. The gene for toxic shock toxin is carried by a family of mobile pathogenicity islands in Staphylococcus aureus. Mol. Microbiol. 29:527-543. [DOI] [PubMed] [Google Scholar]
- 22.Lindsay, J. A., and M. T. G. Holden. 2004. Staphylococcus aureus: superbug, super genome? Trends Microbiol. 12:378-385. [DOI] [PubMed] [Google Scholar]
- 23.McDevitt, D., P. Vaudaux, and T. J. Foster. 1992. Genetic evidence that bound coagulase of Staphylococcus aureus is not clumping factor. Infect. Immun. 60:1514-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Melles, D. C., R. F. Gorkink, H. A. Boelens, S. V. Snijders, J. K. Peeters, M. J. Moorhouse, P. J. van der Spek, W. B. van Leeuwen, G. Simons, H. A. Verbrugh, and A. van Belkum. 2004. Natural population dynamics and expansion of pathogenic clones of Staphylococcus aureus. J. Clin. Investig. 114:1732-1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nouwen, J., H. Boelens, A. van Belkum, and H. Verbrugh. 2004. Human factor in Staphylococcus aureus nasal carriage. Infect. Immun. 72:6685-6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Novick, R. P. 2003. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol. Microbiol. 48:1429-1449. [DOI] [PubMed] [Google Scholar]
- 27.Peacock, S. J., C. E. Moore, A. Justice, M. Kantzanou, L. Story, K. Mackie, G. O'Neill, and N. P. Day. 2002. Virulent combinations of adhesin and toxin genes in natural populations of Staphylococcus aureus. Infect. Immun. 70:4987-4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peacock, S. J., I. de Silva, and F. D. Lowy. 2001. What determines nasal carriage of Staphylococcus aureus? Trends Microbiol. 9:605-610. [DOI] [PubMed] [Google Scholar]
- 29.Projan, S. J., and R. P. Novick. 1997. The molecular basis of pathogenicity, p. 55-81. In K. Crossley and G. Archer (ed.), The staphylococci in human disease. Churchill Livingston, New York, N.Y.
- 30.Rivas, J. M., P. Speziale, J. M. Patti, and M. Hook. 2004. MSCRAMM—targeted vaccines and immunotherapy for staphylococcal infection. Curr. Opin. Drug Discov. Dev. 7:223-227. [PubMed] [Google Scholar]
- 31.Roche, F. M., M. Meehan, and T. J. Foster. 2003. The Staphylococcus aureus surface protein SasG and its homologues promote bacterial adherence to human desquamated nasal epithelial cells. Microbiology 149:2759-2767. [DOI] [PubMed] [Google Scholar]
- 32.Rutherford, K., J. Parkhill, J. Crook, T. Horsnell, P. Rice, M. A. Rajandream, and B. Barrell. 2000. Artemis: sequence visualization and annotation. Bioinformatics 16:944-945. [DOI] [PubMed] [Google Scholar]
- 33.Witney, A. A., G. L. Marsden, M. T. G. Holden, R. A. Stabler, S. E. Husain, J. K. Vass, P. D. Butcher, J. Hinds, and J. A. Lindsay. 2005. Design, validation, and application of a seven-strain Staphylococcus aureus PCR product microarray for comparative genomics. Appl. Environ. Microbiol. 71:7504-7514. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.