

Abstract
Bacterial surface proteins are important molecules in the infectivity and survival of pathogens. Surface proteins on gram-positive bacteria have been shown to attach via a transpeptidase, termed sortase, that cleaves an LPXTG sequence found close to the C termini of nearly all surface proteins on these bacteria. We previously identified a unique enzyme (LPXTGase) from Streptococcus pyogenes that also cleaves the LPXTG motif with a catalytic activity higher than that of sortase, suggesting that it plays an important role in the attachment process. We have now purified and characterized an LPXTGase from Staphylococcus aureus and found that it has both similar and unique features compared to the S. pyogenes enzyme. The S. aureus enzyme is glycosylated and contains unusual amino acids, like its streptococcal counterpart. Like the streptococcal enzyme, staphylococcal LPXTGase has an overrepresentation of amino acids found in the peptidoglycan, i.e., glutamine/glutamic acid, glycine, alanine, and lysine, and furthermore, we find that these amino acids are present in the enzyme at precisely the same ratio at which they are found in the peptidoglycan for the respective organism. This suggests that enzymes responsible for wall assembly may also play a role in the construction of LPXTGase.
A variety of cell surface proteins of gram-positive bacteria are covalently linked through their C termini to cell wall peptidoglycan. These proteins are indispensable for pathogenic bacteria to establish successful colonization and infection of host tissue and for their continued survival in the host environment. More specifically, they play a role in (i) adhesion and invasion, (ii) antiphagocytic activities, (iii) degradation of host cell surface components, thereby facilitating spread, and (iv) hydrolysis of molecules for nutrient utilization (3, 4, 7, 12, 24).
One common feature of peptidoglycan-linked surface proteins is the presence of an LPXTG sequence at the C terminus. In all cases, the genes for these surface proteins contain nucleotide sequences following the LPXTG-encoding sequence that code for a hydrophobic domain and a short charged tail (6). During surface translocation, the proprotein is cleaved at a site within or adjacent to the LPXTG sequence, and the newly created C terminus becomes covalently linked to the cell wall peptidoglycan (18, 19).
Schneewind et al. (21) purified and characterized from Staphylococcus aureus a specific transpeptidase, which they named sortase, that cleaves the peptide bond between threonine and glycine of the LPXTG sequence present in the precursor of protein A. In addition, they showed that sortase also mediates covalent linkage of the C-terminal threonine of protein A to the terminal glycine of the pentaglycine cross-bridge in the S. aureus cell wall peptidoglycan.
Recently we reported the discovery and characterization of a unique endopeptidase from Streptococcus pyogenes that cleaves the LPXTG sequence motif. The S. pyogenes endopeptidase, which we named LPXTGase, is an unusual enzyme in that it contains uncommon amino acids and carbohydrate, with the latter being essential for enzyme activity (15). In contrast to the sortase from S. aureus, S. pyogenes LPXTGase neither contains a sulfhydryl group active site nor requires hydroxylamine for enzyme activity. Significantly, LPXTGase from S. pyogenes cleaves the LPXTG sequence motif at a rate that is greater than two orders of magnitude higher than that of S. aureus sortase (15).
Since S. pyogenes and S. aureus both have at least one sortase molecule to process surface proteins for surface anchoring (2, 21), we were curious whether S. aureus also has an LPXTGase that cleaves the LPXTG sequence as in S. pyogenes. To resolve this question, we searched for an endopeptidase in S. aureus that is analogous to the LPXTGase of S. pyogenes, and we found that S. aureus indeed produces an LPXTG-specific endopeptidase that is very similar to S. pyogenes LPXTGase. However, we find that as with LPXTGase from S. pyogenes, theprimary amino acids used in the staphylococcal enzyme mirrorthose used in peptidoglycan assembly. The present report describes purification and characterization of S. aureus LPXTGase and comparisons with the streptococcal enzyme.
MATERIALS AND METHODS
Enzymes.
All enzymes used were purchased from Sigma. These were N-glycosidase F (catalog no. G-5166), β-glucosidase (catalog no. G-6906), β-galactosidase (catalog no. G-5635), β-N-acetylhexosaminidase (catalog no. A-7708), β-mannosidase (catalog no. M-9400), α-mannosidase (catalog no. M-7257), lysostaphin (catalog no. L-4402), and trypsin (catalog no. T-1426).
Other materials.
Silica gel thin-layer chromatography (TLC) plates (catalog no. M5729-6) and solvents were purchased from Fisher. Carboxymethyl glass beads (catalog no. G-3910), 5-bromo-4-chloro-3-indolyl phosphate/Nitro Blue Tetrazolium (catalog no. B-1911), and molecular weight markers blue dextran (catalog no. D-4772), carbonic anhydrase (catalog no. C-7025), cytochrome c (catalog no. C-7150), aprotinin (catalog no. A-3886), and vancomycin (catalog no. V-2001) were purchased from Sigma. Iodobeads (product no. 28665), fluorescein isothiocyanate (FITC) (catalog no. 46110), and bicinchoninic acid (BCA) protein assay reagent (product no. 23225) were purchased from Pierce.
Anti-LPXTGase antibody.
The antibody against LPXTGase from S. pyogenes was produced in rabbits (Covance) by a primary dose of 200 μg of protein emulsified in complete Freund's adjuvant followed by two monthly doses of 100 μg/ml in incomplete adjuvant. Animals were bled 7 days after the last boost.
Bacterial strain and culture.
S. aureus strain RN4220 was grown in 25 liters of Todd-Hewitt medium supplemented with 1% yeast extract in a carboy with stirring by a motor-driven impeller. Cells were harvested when the optical density of the culture at 600 nm reached 1.2. To harvest the cells, the culture was concentrated to about 2 liters with a Millipore Procon filtration apparatus, and cells were then pelleted by centrifugation for 10 min at 8,000 rpm using a Sorvall GSA rotor. The cell pellet (about 80 g [wet weight]) was suspended in 1 liter of 20 mM Tris-HCl buffer, pH 7.7, and the cells were pelleted again by centrifugation.
Cell lysis and preparation of crude extract.
The washed cell pellet was suspended in 800 ml of 30 mM Tris-HCl buffer, pH 7.7, and then 3 mg of lysostaphin was added, and cell lysis was achieved by mixing the solution for 1 h at 37°C. Digestion of the cell wall peptidoglycan by lysostaphin resulted in cytosolic and membrane vesicle release. The cell lysate was then centrifuged for 20 min. at 10,000 rpm with a GSA rotor, and the supernatant containing cytosol and membrane vesicles was collected. The pellet, consisting of cell ghosts and unlysed cells, was suspended in 300 ml of Tris-HCl buffer, pH 7.7, containing 0.2% Brij 35, and the suspension was stirred overnight at 4°C. The suspension was then centrifuged for 20 min at 10,000 rpm, and the supernatant, termed membrane extract, was collected.
Preparation of LPXTG peptide substrates.
LPXTG-containing peptides, KRQLPSTGETANPF(Y) derived from the streptococcal M protein and AQALPETGEENPF(Y) from staphylococcal protein A, were synthesized with a C-terminal tyrosine by the Rockefeller University Protein/DNA Technology Center. The terminal tyrosines were labeled with 125I using Iodobeads. The labeled peptides were purified on silica gel TLC plates using propanol-pyridine-water (2:1:1) as a running solvent mixture. The N termini of the labeled peptides were then linked to carboxymethyl glass beads by carbodiimide catalysis according to the manufacturer's instructions and as described previously (15).
Enzyme assay method.
Cleavage activity of the LPXTGase enzyme was determined using glass bead-bound KRQLPSTGETANPFY as a substrate, where the terminal tyrosine was labeled with 125I, as described previously (15). Briefly, aliquots of 2 to 40 μl of enzyme were introduced into Microfuge tubes, after which 30 mM Tris-HCl buffer, pH 7.7, containing 0.1% Brij was added to achieve a 40-μl volume. A 10-μl water suspension of bead-bound, 125I-labeled LPXTG peptide was added to the tubes, and the reaction mixtures were shaken at 37°C for 1 h. At the end of the reaction, 150 μl of distilled water was added to each tube and the tubes were vortexed. The tubes were then centrifuged, and a 150-μl aliquot of the supernatant was withdrawn and radioactivity counted. The release of cleaved, 125I-labeled peptide fragment from the beads was the measure of enzyme activity. The tubes with all components except for the enzyme served as background control.
Purification of LPXTGase.
To 800 ml of the cytosol fraction, Brij 35 was added to a final concentration of 0.1%. The cytosol fraction was then combined with the membrane extract (300 ml) and the mixture applied to a DEAE-cellulose column (20 by 5.1 cm), equilibrated with 20 mM Tris-HCl buffer, pH 6.8. After the mixture had entered the column, the column was eluted with 20 mM Tris-HCl buffer, pH 7.7, containing 0.1% Brij 35, until the UV absorption reached baseline, requiring about 600 ml of the buffer. The fall-through fraction and the column wash were combined and concentrated to 20 ml using an Amicon ultrafiltration apparatus fitted with a YM3 membrane with a 3-kDa size limit. One-half of the concentrated enzyme solution was applied to a Sephadex G50 column (60 by 4.3 cm) equilibrated with 20 mM Tris-HCl buffer, pH 7.7, containing 0.1% Brij 35; the column was then eluted with the same buffer, and 25-ml fractions were collected. For the enzyme preparations used in amino acid and sugar composition analysis, the DEAE-cellulose chromatography and gel filtration steps were repeated once again.
Biochemical characterization.
Enzyme concentration was determined using the BCA protein assay reagent, which measures peptide bonds. The pH optimum of the enzyme was determined as described previously (15).
Determination of molecular weight. (i) Gel filtration.
Three hundred microliters of the concentrated LPXTGase (about 170 μg protein) was mixed with 200 μl of molecular weight standard proteins, and the mixture was applied to a Sephadex G50 column (48 by 2.4 cm) equilibrated with 20 mM Tris-HCl buffer, pH 7.7, containing 0.1% Brij 35. The column was eluted with the same buffer solution, and 8-ml fractions were collected. The molecular markers were identified by UV absorption at 280 nm, and LPXTGase was identified by LPXTG peptide cleavage activity.
(ii) Mass spectroscopy.
The mass of S. aureus LPXTGase was also determined by the Rockefeller University Protein/DNA Technology Center using a Voyager DE-STR mass spectroscope.
Identification of enzyme reaction products.
About 12 μg of 125I-labeled (1.4 × 106 cpm) AQALPETGEENPFY peptide in 50 μl of distilled water was added to two separate microcentrofuge tubes. To one tube was added ∼6 μg of S. aureus LPXTGase in 50 μl of 60 mM Tris-HCl buffer, pH 8.0, containing 0.2% Brij, and to the second tube was added ∼6 μg of S. pyogenes LPXTGase in 50 μl of the same buffer solution. The reaction mixtures were shaken at 37°C, and at designated times 10-μl aliquots were removed and spotted onto silica gel TLC plates. The plates were developed with a solvent mixture consisting of ethylacetate-pyridine-acetic acid-water (60:30:9:24), and the reaction products were identified by autoradiography. Reaction products were eluted from the plates, and amino acid sequences were determined with an Applied Biosystems AB1 Procise 498 instrument by the Protein/DNA Technology Center of the Rockefeller University. The 125I-labeled KRQLPSTGETANPFY peptide, derived from streptococcal M protein, was cleaved by LPXTGase from both S. pyogenes and S. aureus to determine the cleavage location in the LPXTG motif of this peptide as described above.
Inactivation of S. aureus LPXTGase by glycosidases.
About 1 μg S. aureus LPXTGase in a 2-μl volume was added to six microcentrofuge tubes. To each of the first four tubes was added 0.1 U of β-glucosidase, β-mannosidase, α-mannoside, or N-acetylhexosaminidase in 8 μl of 20 mM Na-acetate buffer, pH 5.5, containing 0.1% Brij 35. To each of the next two microcentrofuge tubes containing LPXTGase was added 0.1 U of β-galactosidase or N-glycosidase F in 8 μl of 20 mM Tris-HCl buffer, pH 7.6, containing 0.1% Brij 35. The reaction mixtures were incubated at 37°C for 1 h. At the end of glycosidase treatment, 1 μl of 300 mM Tris-HCl buffer, pH 8.9, was added to the first four microcentrofuge tubes which contained acetate buffer, after which 30 μl of 30 mM Tris-HCl buffer, pH 8.0, containing 0.1% Brij was added to all microcentrofuge tubes. After adjusting the pH of reaction mixtures to about 8.0, about 1 μg of bead-bound 125I-labeled KRQLPSTGETANPFY (1.7 × 105 cpm) in 10 μl of distilled water was added to each tube, and the peptide cleavage reaction was carried out as described above for 1 h at 37°C.
DEAE-cellulose chromatography of deglycosylated S. aureus LPXTGase.
About450 μg of purified S. aureus LPXTGase was incubated for 32 h at 37°C with 20 units of N-glycosidase F (PNGase F) in 1 ml of 30 mM Tris-HCl buffer, pH 7.6, containing 0.1% Brij 35. The reaction products were then applied to a DEAE-cellulose column (12 by 1.4 cm) equilibrated with the same buffer solution. The column was eluted with 30 ml of the same buffer solution, and 2-ml fractions were collected. The column was then eluted successively with 0.07 M and 0.2 M KCl in the same buffer solution and finally with 0.4 N NaOH, and 2-ml fractions were collected. Two 400-μl aliquots from each fraction were transferred to microcentrofuge tubes and the volumes of the aliquots reduced to about 50 μl by means of SpeedVac. Then, 50 μl methanol was added to microcentrofuge tubes in order to precipitate excess KCl and buffer salt. The precipitates were removed by centrifugation, and clear supernatants containing products derived from LPXTGase were collected. The precipitates were dissolved in 50 μl distilled water, after which 50 μl methanol was added to each tube, and clear supernatants were collected by centrifugation. The volumes of combined supernatants were reduced to about 20 μl, and these were used for protein and carbohydrate analyses. For protein concentration determination, the BCA method was used. For carbohydrate detection, the concentrated fractions were spotted on a silica gel TLC plate, and the plate was sprayed with 0.5% thymol in ethanol-sulfuric acid (95/5 [vol/vol]), and color developed by heating the plate for 15 min at 110°C.
Reactivity of S. aureus LPXTGase with anti-S. pyogenes LPXTGase antibody.
About 4 μg (each) of S. pyogenes LPXTGase and S. aureus LPXTGase in 5 μl of 20 mM Tris buffer was spotted on a nitrocellulose sheet. The enzymes were then fourfold serially diluted in phosphate-buffered saline and spotted linearly on the sheet. The nitrocellulose sheet was then reacted with the S. pyogenes LPXTGase antibody, and after washing, the sheet was reacted with antirabbit antibody conjugated with alkaline phosphatase. After washing again, alkaline phosphatase activity was detected with 5-bromo-4-chloro-3-indolyl phosphate-Nitro Blue Tetrazolium as the substrate.
Analysis of enzyme-linked sugar species.
About 220 μg of purified S. aureus LPXTGase in 0.5 ml Tris-HCl buffer was digested with 10 U of N-glycosidase F as described above. The digestion products were mixed with 50 ml of distilled water, and the mixture was subjected to ultrafiltration using an YM3 membrane with a 3,000-Da size limit. The filtrate was lyophilized, and the dried material was dissolved in 0.2 ml of distilled water. The concentrated filtrate was then applied to a P2 gel filtration column (48 by 1.4 cm) equilibrated with distilled water, the column was eluted with distilled water, and 3-ml fractions were collected. Aliquots of 0.3 ml from each fraction were transferred to microcentrofuge tubes and volumes reduced to 20 μl by means of a SpeedVac. The concentrated column fractions were spotted on a silica gel TLC plate, and sugars were detected as described above. The remainder of the column fractions was analyzed for sugar composition by the Complex Carbohydrate Research Center of the University of Georgia, Athens, Ga.
Amino acid and sugar composition of S. aureus LPXTGase.
Amino acid composition of LPXTGase was determined by the Protein/DNA Technology Center of the Rockefeller University, and sugar composition was determined by M-Scan, Inc. (West Chester, PA), employing gas chromatography of trimethylsilylate derivatives of component sugars.
Effect of lysostaphin pretreatment of S. aureus LPXTGase on its enzyme activity.
Two-microgram samples of S. aureus LPXTGase in microcentrofuge tubes were incubated for 2 h at 37°C with various amounts of lysostaphin, ranging from 0 to 1 μg, in a 40-μl reaction volume of 30 mM Tris-HCl buffer, pH 7.6, containing 0.1% Brij 35. At the end of the incubation with lysostaphin, 1 μg of 125I-labeled, bead-bound LPXTG peptide suspended in 10 μl of distilled water was added to each tube, and LPXTG peptide cleavage activity was determined as described above.
Effect of trypsin pretreatment of S. aureus LPXTGase on its enzyme activity.
One hundred two micrograms of S. aureus LPXTGase was incubated with 6 μg of trypsin for 4 h at 37°C in a 200-μl volume of 30 mM Tris-HCl buffer, pH 8.0, containing 0.1% Brij 35. The reaction mixture was then subjected to gel filtration using a Sephadex G50 column (42 by 2.4 cm). To 40-μl aliquots from column fractions, a 10-μl water suspension of 1 μg bead-bound 125I-labeled LPXTG peptide was added, and the cleavage of LPXTG peptide was determined as described above. For comparison, an identical amount of untreated LPXTG peptide was subjected to gel filtration, and the cleavage reaction of column fractions was determined in the same manner.
Trypsin and lysostaphin digestion of intact and deglycosylated LPXTGase.
Aliquots of 300 μl containing 22 μg of LPXTGase from the second Sephadex G50 chromatography were added to three microcentrofuge tubes. To the second tube, 3 μg trypsin was added, and to the third tube, 3 μg lysostaphin was added, with no additions to the first tube. The tubes were shaken for 2 h at 37°C, and after reducing the volume of the mixtures to 60 μl using a SpeedVac, reaction products were frozen until used.
To examine the effect of proteases on deglycosyslated LPXTGase, a 900-μl aliquot of purified LPXTGase in a microcentrofuge tube was mixed with 15 U N-glycosidase F, and the mixture was incubated for 28 h at 37°C. N-glycosidase F-treated LPXTGase was then equally divided into three microcentrofuge tubes, after which 3 μg trypsin was added to the second tube, 3 μg lysostaphin was added to the third tube with no further additions to the first tube, and then the mixtures were incubated for 2 h at 37°C. The volume of untreated and protease-treated deglycosylated LPXTGase samples was reduced to 60 μl by SpeedVac.
To the six samples above, 8 μl of 1 M Na2CO3-NaHCO3, pH 9.5, and 70 μg FITC were added, and the mixtures were incubated in the dark for 2 h. At the end of the incubation, 14 volumes of ethanol was added to each tube, the enzyme precipitates were pelleted by centrifugation, and free FITC in the supernatant was discarded. The enzyme pellets were dissolved in 80 μl of water, and enzyme samples were precipitated again by adding 14 volumes of ethanol and pelleted by centrifugation. The enzyme pellets were dissolved in 30 μl of water and spotted on a silica gel TLC plate, and the reaction products were resolved by developing the plate with 70% methanol as a running solvent.
Detection of d-alanine in S. aureus LPXTGase.
S. aureus, strain 4220, was grown in 1 liter of Todd-Hewitt medium supplemented with 1% yeast extract, and cells were harvest by centrifugation at late exponential phase. Cell pellets were suspended in 1 liter of alanine minus synthetic medium, described previously (16). 100 μCi l-[14C]alanine was added to the cell suspension, and cells were grown until the optical density at 600 nm reached 1.3. [14C]alanine-labeled LPXTGase was purified as described above. The labeled enzyme was hydrolyzed with 6 N HCl and [14C]alanine purified, and configurations of [14C]alanine were determined by the d-amino acid oxidation method as described previously (16).
RESULTS
Purification of LPXTGase.
Previously we found that S. pyogenes LPXTGase did not bind to DEAE-cellulose, whereas most cytosolic proteins could (15). Assuming that S. aureus LPXTGase would be similar, we used DEAE-cellulose chromatography as the first step of enzyme purification. The crude extract (1.1 liter) consisting of the cytosol fraction and membrane extract was applied to a DEAE-cellulose column and the fall-through fraction collected. With the S. aureus enzyme, we applied a smaller amount of crude extract to a larger column (20 by 5.1 cm) than we did with the S. pyogenes enzyme, resulting in a clear eluate leaving the turbid membrane vesicles bound to the column. As seen for streptococcal LPXTGase, enzyme cleavage activity could not be detected either in the fall-through fraction or in the crude extract, which we attributed to the presence of a low-molecular-weight inhibitor of LPXTGase in the S. aureus crude extract (as seen with the streptococcal enzyme). The fall-through fraction and column wash were combined, and the volume was reduced to about 20 ml using an Amicon ultrafiltration apparatus fitted with a YM3 membrane having a 3-kDa size limit. When the concentrated enzyme preparation was chromatographed on a Sephadex G50 column, high LPXTG peptide cleavage activity eluted shortly after the void volume in a pattern consistent with the elution profile of S. pyogenes LPXTGase (not shown). As in the case of streptococcal LPXTGase, it appears that the low-molecular-weight enzyme inhibitor present in the fall-through fraction was separated from the enzyme during ultrafiltration and gel filtration, and the LPXTGase peak showed no UV absorption at 280 nm.
Molecular mass of S. aureus LPXTGase.
Figure 1 shows the elution profiles of proteins of known molecular mass and S. aureus LPXTGase. From this, the molecular mass of LPXTGase was estimated to be 14 kDa, which is the same as the molecular mass of S. pyogenes LPXTGase, also estimated by gel filtration. However, Fig. 2 shows that the molecular mass of the S. aureus enzyme is only 7,807.4 Da as determined by mass spectroscopy.
FIG. 1.

Determination of molecular mass of S. aureus LPXTGase by gel filtration. Purified S. aureus LPXTGase was mixed with proteins of known molecular mass, and the mixture was subjected to gel filtration on a Sephadex G 50 column. LPXTGase and molecular mass markers were located by LPXTG peptide cleavage activity and UV absorption at 280 nm, respectively, as described in Materials and Methods.
FIG. 2.

Determination of molecular mass of S. aureus LPXTGase by mass spectroscopy. Molecular mass of S. aureus LPXTGase was determined by the Rockefeller University Protein/DNA Technology Center using a Voyager DE-STR mass spectroscope.
pH optimum.
As can be seen in Fig. 3, S. aureus LPXTGase exhibits a broad pH optimum ranging from pH 7 to pH 10, consistent with that of S. pyogenes LPXTGase.
FIG. 3.

pH optimum of S. aureus LPXTGase. LPXTGase activity was determined at 50 mM concentrations of various buffers as described in Materials and Methods. The buffers employed were Mes-NaOH for pHs 5, 6, 6.5, and 7, Tris-HCl for pHs 7.5, 8, 8.5, and 9, NH4OH-HCl for pH 10, and trimethylamine-HCl for pHs 11 and 12.
Inhibition of LPXTGase activity by salts.
Because we previously found that streptococcal LPXTGase could be inhibited by certain salts (15), we tested whether the pattern of inhibition was the same for the staphylococcal enzyme. Table 1 shows that the effects of various salts on the activities of LPXTGase from S. aureus and S. pyogenes are remarkably similar. For both enzymes, divalent cations showed higher inhibition than monovalent cations. The enzymes from both sources were also inhibited by hydroxylamine. Interestingly, ammonium bicarbonate slightly stimulated the cleavage activity of LPXTGase from both organisms. At 20 mM, sodium sulfate inhibited, whereas ammonium sulfate slightly stimulated, LPXTGase activities, indicating that the ammonium ion is slightly stimulatory. Also, sodium sulfate inhibited, whereas sodium bicarbonate slightly stimulated, LPXTGase activity, indicating that the carbonate ion stimulates LPXTGase activity. Ammonium bicarbonate, which contains both stimulatory ions, showed the highest stimulatory effect.
TABLE 1.
Effects of salts on the activities of LPXTGases from S. aureus and S. pyogenes
| Salt (concn [mM]) | Peptide cleaved, cpm (% of control)
|
|
|---|---|---|
| S. aureus | S. pyogenes | |
| Sodium chloride (100) | 6,190 (14.1) | 6,720 (16.2) |
| Potassium chloride (100) | 5,820 (13.2) | 8,608 (20.7) |
| Ammonim acetate (100) | 27,900 (63.6) | 22,380 (53.9) |
| Hydroxylamine, HCI (100) | 12,160 (27.4) | 11,200 (27.0) |
| Calcium chloride (20) | 8,220 (18.6) | 9,706 (23.4) |
| Magmesium chloride (20) | 6,160 (14.0) | 8,615 (20.7) |
| Potassium phosphate (20) | 23,920 (54.2) | NDa |
| Sodium phosphate (20) | ND | 20,646 (49.7) |
| Sodium sulfate (20) | 22,920 (52.0) | 23,454 (56.5) |
| Ammonum sulfate (20) | 49,930 (113.4) | 42,040 (101.2) |
| Sodium bicarbonate (20) | 54,080 (122.8) | 52,610 (126.7) |
| Ammonum bicarbonate (20) | 70,360 (159.8) | 58,890 (141.8) |
| EDTA (5) | 12,360 (28.2) | 2,147 (5.2) |
| Control; no salt | 44,040 (100) | 41,530 (100) |
ND, not determined.
Enzyme reaction product.
To identify where in the LPXTG motif the S. aureus enzyme cleaves, we subjected the LPXTG-containing peptide of protein A, AQALPETGEENPFY, to S. aureus LPXTGase, and the reaction products were examined. Forcomparison, the same peptide was cleaved with S. pyogenes LPXTGase, and the results are shown in Fig. 4A. As can be seen, the S. aureus enzyme cleaves after threonine in the peptide, yielding a radioactive GEENPFY product. In contrast, the S. pyogenes enzyme cleaves the same peptide at two positions, the first after threonine and the second before the last glutamic acid, yielding two radioactive products, GEENPFY and ENPFY. Given the same quantity of starting material, overall, the S. pyogenes enzyme appears to yield more total product in the cleavage reaction.
FIG. 4.

A. Cleavage of LPXTG-containing protein A fragment by LPXTGase from S. aureus and by LPXTGase from S. pyogenes. Free AQALPETGEENPFY, in which the terminal tyrosine was labeled with 125I, was incubated with LPXTGase from S. aureus or from S. pyogenes for various lengths of time. Aliquots of reaction products were withdrawn at designated times and were examined by silica gel TLC as described in Materials and Methods. B. Cleavage of LPXTG-containing M protein fragment by LPXTGase from S. aureus and by LPXTGase from S. pyogenes. Free KRQLPSTGETANPFY, in which the terminal tyrosine was labeled with 125I, was incubated with LPXTGase from S. aureus or from S. pyogenes for various lengths of time. Aliquots of reaction products were withdrawn at designated times and were examined by silica gel TLC as described in Materials and Methods.
We also subjected the LPXTG-containing segment of the streptococcal M protein, KRQLPSTGETANPFY, to both S. aureus and S. pyogenes LPXTGases. As seen in Fig. 4B, both enzymes cleave the streptococcal substrate after serine and glutamic acid, yielding two radioactive products, TGETANPFY and TANPFY. Again, the S. pyogenes enzyme appears to be more active than the S. aureus enzyme in cleaving this peptide. A comparison of Fig. 4A and B reveals that both the S. aureus and S. pyogenes enzymes appear to be more active towards the M protein substrate than the protein A peptide.
Inactivation of S. aureus LPXTGase by glycosidases.
We have shown previously that sugars are an integral part of S.pyogenes LPXTGase and certain sugars are essential for LPXTGase activity. The striking similarity of the LPXTGase enzymes from S. aureus and S. pyogenes in their chromatographic behavior and LPXTG peptide cleavage reactions prompted us to examine whether sugars are also essential for the S. aureus enzyme activity. S. aureus LPXTGase was therefore incubated with various glycosidases prior to testing for cleavage activity. As seen in Fig. 5, and similar to the case with S. pyogenes LPXTGase, pretreatment of the staphylococcal enzyme with N-glycosidase F β-glucosidase, α-mannosidase, β-mannosidase, or β-N-acetylhexosaminidase greatly reduced LPXTGase activity, but β-galactosidase did not.
FIG. 5.

Inactivation of LPXTGase activity by glycosidases. About 1 μg of LPXTGase was preincubated for 1 h with 0.1 U of each glycosidases, after which LPXTG peptide cleavage activities of glycosidase-treated LPXTGase were determined. Detailed experimental conditions are described in Materials and Methods.
The possibility that protease contaminants in the commercial glycosidases might be responsible for the inactivation of LPXTGase was considered. We therefore incubated 125I-labeled bovine serum albumin with the glycosidases that were used in the inactivation experiments. Only N-acetylhexosaminidase showed a trace amount of protease activity, while all other glycosidases showed no protease activity (results not shown).
DEAE-cellulose chromatography of deglycosylated LPXTGase.
Because LPXTGases from S. pyogenes and S. aureus do not bind to DEAE-cellulose even though these enzymes contain several acidic amino acids, we considered the possibility that the carbohydrate moieties of these enzymes shield the acidic amino acids, thereby preventing exposure of the acidic amino acid residues to DEAE-cellulose. To test this possibility, we examined the behavior of deglycosylated S. aureus LPXTGase on DEAE-cellulose chromatography, and the result is shown in Fig. 6. About 20% of LPXTGase (as judged by protein content) eluted in the fall-through fractions, and these fractions remained glycosylated and retained full enzyme activity. About 20% of N-glycosidase F-treated LPXTGase eluted with 0.07 M KCl, and this fraction of LPXTGase remained glycosylated, but enzyme activity was greatly reduced. The remaining 60% of the N-glycosidase F-treated LPXTGase was tightly bound to DEAE-cellulose, and some of it could be eluted with 0.2 M KCl, while the remainder could be eluted only with 0.4 N NaOH. The DEAE-cellulose-bound fractions did not retain any sugar as determined by the thymol/sulfuric acid method and were completely devoid of enzyme activity, confirming that carbohydrate is an integral part of LPXTGase and essential for enzyme activity and that acidic amino acids are shielded by carbohydrate.
FIG. 6.

DEAE-cellulose chromatography of deglycosylated S. aureus LPXTGase. Purified S. aureus LPXTGase was digested with N-glycosidase F and chromatographed on a DEAE-cellulose column, and protein and carbohydrate in the column fractions were determined as described in Materials and Methods.
Reactivity of anti-streptococcus LPXTGase antibody with staphylococcal enzyme.
Since LPXTGase from S. aureus and S. pyogenes showed similarities in molecular weight, pH optima, salt effects, and glycosidase sensitivity and both enzymes lead to similar reaction products, we tested whether they share structural similarities. To assess this, we examined whether S. aureus LPXTGase reacts with an anti-S pyogenes LPXTGase antibody. As shown in Fig. 7, the antiserum raised against S. pyogenes LPXTGase cross-reacted with S. aureus LPXTGase, although somewhat more weakly than with the streptococcal enzyme, indicating the two enzymes share at least one epitope.
FIG. 7.

Cross-reaction of S. aureus LPXTGase with anti-S. pyogenes antibody. Four micrograms (each) of S. pyogenes LPXTGase and S. aureus LPXTGase in 5 μl of Tris-HCl buffer was spotted on the first column of a nitrocellulose sheet. The enzymes were fourfold serially diluted with phosphate-buffered saline and spotted on the subsequent columns. The nitrocellulose sheet was reacted with anti-S. pyogenes antibody raised in rabbit, after which the sheet was reacted with anti-rabbit antibody conjugated with alkaline phosphatase. Alkaline phosphatase activities of spots were determined as described in Materials and Methods.
Characterization of the sugars of LPXTGase.
Although LPXTGase activity was reduced with a variety of glycosidases, it is uncertain whether the carbohydrate moiety is composed of a single and a unique species of oligosaccharide that is linked to a specific asparagine of LPXTGase or a population of heterogeneous sugars linked to various asparagines. For this purpose, we subjected the carbohydrate released from the enzyme to gel filtration using a Biogel P2 column. Oligosaccharride(s) with a molecular mass of 1,500 Da or larger eluted at the void volume, and most of the remainder eluted at the region of mono-, di-, and trisaccharides. Very small amounts of sugar eluted after the void volume but before the trisaccharide region. Examination of the sugar composition of the P2 column fractions revealed that the oligosaccharide(s) that eluted at the void volume contained xylose, mannose, galactose, and glucose in mole ratios of 18.2:7.4:15.0:59.4, while tri-, di-, and monosaccharide fractions contained only galactose and glucose with respective mole ratios of 19.2:80.9, 29.7:70.3, and 27.5:72.5. These results indicate that the carbohydrate of LPXTGase consists of a heterogeneous population of sugars.
Amino acid and sugar compositions of S. aureus LPXTGase.
Table 2 shows amino acid composition of S. aureus LPXTGase. For comparison the amino acid composition of S. pyogenes LPXTGase, which we determined previously (15), is also shown. S. aureus LPXTGase contains 11 species of common amino acids and a number of uncommon amino acids, which were resolved as unknown peaks by the amino acid analyzer. The same 11 known amino acid species were also found in S. pyogenes LPXTGase. In all, S. aureus LPXTGase contains 70 residues of common amino acids with an aggregate mass of 5,758 Da. As with S. pyogenes LPXTGase, the S. aureus molecule contains neither aromatic amino acids nor sulfur-containing amino acids. Of the total of 70 residues, four amino acid species, namely glutamine/glutamic acid, alanine, glycine, and lysine, amount to 57 residues or 81% of the total residues. Except for glycine, the remaining four amino acid species were also the most abundant in S. pyogenes LPXTGase, constituting 75% of its total residues. Interestingly, the major difference in composition between the streptococcal and staphylococcal enzymes is in the quantities of alanine and glycine, which parallel these amino acids present in the peptidoglycan cross-bridge of their respective cell walls, i.e., streptococci having a dialanine and staphylococci having a pentaglycine. Table 3 compares the ratios of these four amino acids in LPXTGase and in the cell wall peptidoglycan of S. aureus and S. pyogenes.
TABLE 2.
Comparison of amino acid compositions of LPXTGases from S. aureus and S. pyogenes
| Amino acid | No. of residues
|
|
|---|---|---|
| S. aureus | S. pyogenes | |
| Asn/Asp | 3 | 5 |
| Gln/Glu | 7 | 10 |
| Ser | 2 | 3 |
| Thr | 1 | 1 |
| Gly | 32 | 5 |
| Ala | 13 | 24 |
| Pro | 3 | 3 |
| Val | 1 | 1 |
| Leu | 1 | 1 |
| Ile | 1 | 1 |
| Lys | 6 | 7 |
| Unknown | ? | ? |
TABLE 3.
Ratios of glutamic acid/glutamine, alanine, glycine, and lysine in the LPXTGases and peptidoglycan of S. aureus and S. pyogenes
| Amino acid | Ratio
|
|||
|---|---|---|---|---|
|
S. aureus
|
S. pyogenes
|
|||
| LPXTGase | Peptidoglycan | LPXTGase | Peptidoglycan | |
| Glutamic acid/glutamine | 1.2 | 1.0 | 1.4 | 1.0 |
| Alanine | 2.3 | 2.0 | 3.4 | 4.0 |
| Glycine | 5.0 | 5.0 | ||
| Lysine | 1.0 | 1.0 | 1.0 | 1.0 |
According to the analytical data, 1 mol of S. aureus LPXTGase contains 2 mol each of glucose, galactose, and mannose, one mole of N-acetylglucosamine, one-half mole of rhamnose, and trace amounts of arabinose, fucose, and xylose. The aggregate mass of 7.5 residues of sugars results in 1,230 Da per mole. Thus, the combined mass of known amino acids and sugars amounts to 6,960 Da. Since the mass of enzyme as determined by mass spectroscopy is 7,804, the differential mass of 844 Da may be attributable to the uncommon amino acids present in the enzyme.
Presence of d-alanine in S. aureus LPXTGase.
Because S. pyogenes LPXTGase contains d-alanine (16), we tested for the presence of d-alanine in S. aureus LPXTGase by subjecting [14C]alanine from 14C-labeled S. aureus LPXTGase to a d-amino acid oxidase reaction, which deaminates alanine to form pyruvic acid products (16). As shown in Fig. 8, d-amino acid oxidase converted nearly half of 14C-alanine into pyruvic acid. As observed earlier for the d-amino acid oxidase reaction products of [14C]alanine prepared from 14C-labeled LPXTGase of S. pyogenes, a polymeric form of pyruvic acid (just above alanine) was also noted.
FIG. 8.
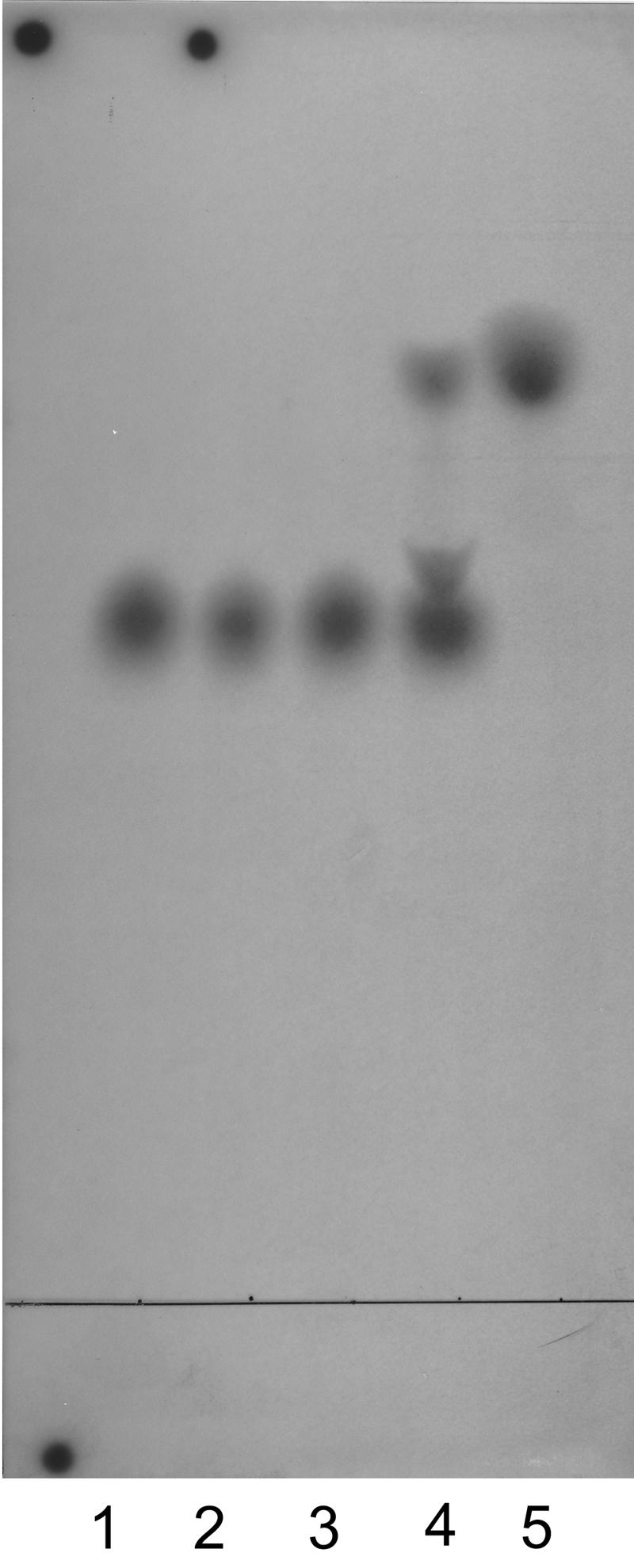
Presence of d-alanine in S. aureus LPXTGase. 14C-labeled S. aureus LPXTGase was prepared, and [14C]alanine was purified from 6 N HCl hydrolysate of the 14C-labeled enzyme. [14C]alanine was subjected to a d-amino acid oxidase reaction, and reaction products were separated on silica gel TLC plate using chloroform-methanol-water (1:2:1) as a running solvent. Radioactive products were detected by radioautography. Lane 1: l-alanine. Lane 2: l-alanine treated with d-amino acid oxidase. Lane 3: alanine from LPXTGase. Lane 4: alanine from LPXTGase treated with d-amino acid oxidase. Lane 5: pyruvic acid.
Resistance of S. aureus LPXTGase to proteolysis.
We observed previously that trypsin treatment of the S. pyogenes LPXTGase, which contains seven lysine residues, yielded only two tryptic fragments. We therefore examined whether the S. aureus LPXTGase is also resistant to proteolysis. Since the S. aureus LPXTGase contains 6 lysine residues and 30 glycine residues, we incubated the enzyme with an excess amount of trypsin to cleave at the lysines or lysostaphin to cleave the glycines. A 4-h incubation of 102 μg of S. aureus LPXTGase with 6 μg of trypsin resulted in practically no reduction in LPXTGase activity, and the elution profiles of trypsin-treated and control enzymes were identical (results not shown). Preincubation of 2 μg LPXTGase with up to 1 μg of lysostaphin for 2 h showed no reduction at all of LPXTGase activity (results not shown).
It is possible, however, that the proteases used may have nicked the LPXTGase enzyme and yet the nicked fragments remained together on gel filtration, retaining enzyme activity. It is also possible that the protease cleavage site(s) on LPXTGase might be shielded by carbohydrate, and the deglycosylated core protein may be susceptible to proteases. To examine these possibilities, we treated both LPXTGase holoenzyme and its deglycosylated core protein with trypsin or lysostaphin and chromatographed the reaction products on a silica gel thin-layer plate. We turned to silica gel TLC to resolve the reaction products, because both intact and deglycosylated enzymes migrate very little in sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and deglycosylated enzyme aggregates and elutes from Sephadex G50 column at void volume. As may be seen in lanes 1, 2, and 3 of Fig. 9, intact enzyme and both trypsin- and lysostaphin-treated intact enzymes remained at the origin and no faster-migrating smaller fragments were noted. Deglycosylated enzyme migrated a little more than halfway along the TLC plate (lane 4), as did both trypsin- and lysostaphin-treated deglycosylated enzymes (lanes 5 and 6), and no faster-moving fragments were detected, indicating that even deglycosylated LPXTGase could not be cleaved by trypsin or lysostaphin. An unusual conformation or structural feature may account for this resistance.
FIG. 9.

Resistance of LPXTGase to proteolysis. About 22 μg each of intact or deglycosylated S. aureus LPXTGase was incubated with either trypsin or lysostaphin as described in Materials and Methods. Untreated and protease-treated intact or deglycosylated LPXTGase samples were labeled with FITC, and the labeled products were chromatographed on silica gel thin-layer plates as described in Materials and Methods. Trypsin and lysostaphin remained at the origin under the same TLC conditions.
DISCUSSION
We succeeded in purifying an endopeptidase from S. aureus that is remarkably similar to S. pyogenes LPXTGase in physical properties and enzyme reaction. Like S. pyogenes, it is puzzling why S. aureus also has evolved at least two different enzymes, namely, sortase and LPXTGase, to cleave the LPXTG sequence motif of precursor proteins that are to be linked to the cell wall peptidoglycan. However, functional redundancy does not seem uncommon for some enzymes involved in cell wall construction. For example, in E. coli both penicillin binding proteins 1a and 1b catalyze identical transpeptidase reactions, which comprise cleavage of the peptide bond between the penultimate d-alanine and the terminal d-alanine of the N-acetylmuramyl pentapeptide, with concomitant linkage of the penultimate d-alanine with the meso-diaminopimelic acid of the adjacent pentapeptide of the nascent peptidoglycan (9, 13, 20, 23). However, E. coli penicillin binding proteins 2, 3, 4, and 5 possess only endopeptidase or carboxypeptidase activities, which cleave the peptide bond between the penultimate d-alanine and the terminal d-alanine of murein peptide, without transpeptidase activity (8).
In this regard, Kharat and Tomasz (14) examined the effect of a sortase (srtA) knockout mutation in Streptococcus pneumoniae on the localization of β-galactosidase and neuraminidase, both LPXTG motif-containing cell surface enzymes. They showed that in wild-type S. pneumoniae, two-thirds of the β-galactosidase was cell associated while one-third was released into the culture medium. In contrast, one-third of the enzyme was cell associated and two-thirds released in the srtA mutant. The observation that the anchoring of β-galactosidase is reduced but not completely abolished in the srtA mutants suggests the existence of a mechanism distinct from sortase A that may also act to anchor LPXTG-containing proteins to the cell wall. Furthermore, the enhanced release of β-galactosidase and neuraminidase into the culture medium in the srtA mutants must be explained. Because removal of the C-terminal charged amino acids and membrane-spanning hydrophobic sequence of the precursors of cell wall-linked proteins would facilitate release of these enzymes into the culture medium, the observed enhanced release of these enzymes in mutants suggests the existence of an alternative cleaving mechanism for the LPXTG motif in these S. pneumoniae enzymes, and LPXTGase could serve this function.
S. aureus LPXTGase, like its streptococcal counterpart, does not bind DEAE-cellulose in spite of the presence of acidic amino acids. This is likely due to the fact that the peptide backbone of the enzyme is shielded by carbohydrate, thus preventing its interaction with the charged groups on the DEAE-cellulose. Figure 6 indeed shows that partially deglycosylated LPXTGase binds weekly to DEAE-cellulose and elutes with 0.07 M KCl, while completely deglycosylated LPXTGase core protein binds tightly to DEAE-cellulose and elutes with 0.2 M KCl or only with 0.4 N NaOH in multiple peaks. Since the elution pattern from Sephadex G50 chromatography showed that deglycosylated LPXTGase formed variously sized aggregates (results not shown), we suspect that the multiple peaks are likely due to the aggregate formation of deglycosylated LPXTGase.
The molecular mass of S. aureus LPXTGase when measured by gel filtration was 14 kDa, but it was half that (7,804 Da) when determined by mass spectroscopy. The excess in molecular mass observed in gel filtration suggests that the enzyme may be rod shaped with an extended Stokes radius of gyration. The rod-shaped silk fibroin is basically alternating glycine and alanine, with serine residues occasionally substituting alanine (1). The fact that glycine and alanine residues account for 63% of the total amino acid residues of S. aureus LPXTGase is compatible with the notion that a large segment of its primary structure folds into a stretched, rod-like beta sheet.
With both AQALPETGEENPFY and KRQLPSTGETANPFY as peptide substrates, the S. pyogenes enzyme exhibited greater cleavage activity than the S. aureus enzyme. One possible reason for this is that S. pyogenes may elaborate a larger number of total cell surface proteins than S. aureus, requiring a more-active LPXTGase for this processing. Interestingly, LPXTGases from both organisms were more active toward the M protein LPXTG sequence than the protein A LPXTG sequence. For efficient bacterial proliferation, it may be necessary that LPXTGase be more active towards an LPXTG sequence of a high-copy-number cell surface protein, such as streptococcal M protein. However, we cannot rule out that staphylococcal protein A may also have a copy number comparable to that of the M protein. The difference in specific activities of the S. pyogenes and S. aureus enzymes is not likely due to a difference in purity, since we used comparably pure or nearly pure enzyme for these experiments.
The presence of unusual amino acids and d-alanine, along with an overrepresentation of four amino acid species, namely, glutamine/glutamic acid, glycine, alanine, and lysine, suggests that a large segment of the protein backbone of S. aureus LPXTGase may be constructed nonribosomally. It is noteworthy that these four predominant amino acid species of S. aureus LPXTGase are precisely the four amino acids that make up the peptide involved in cross-linking adjacent glycan chains of the peptidoglycan. The ratio of these four amino acid species in the enzyme is 1.2:5.0:2.3:1.0, which is remarkably close to the 1:5:2:1 ratio for these amino acids in the peptidoglycan of S. aureus. In S. pyogenes LPXTGase, the ratio of glutamine/glutamic acid:alanine:lysine was 1.4:3.4:1.0, which closely approximates the ratio of 1:4:1 for these three amino acid species in the streptococcal peptidoglycan. Therefore, it is tempting to speculate that the ligases MurC, MurD, MurE, and MurF (10, 11, 23) that are involved in the synthesis of the N-acetyl muramyl pentapeptide, l-Ala-d-Glu-l-Lys-d-Ala-d-Ala, as well as FmhB, FemA, and FemB (5, 22, 23), which sequentially add glycines to the ɛ-amino group of lysine of lipid II-associated muramyl pentapeptide, may in some way be involved in the construction of a large segment of S. aureus LPXTGase. This may be partly supported by our experiments showing the resistance of deglycosylated streptococcal (15) and staphylococcal LPXTGase to the action of trypsin, suggesting that the lysines in LPXTGase may be linked via their ɛ-amino groups. However, it does not explain the resistance to lysostaphin, which could be due to conformational features of LPXTGase. Furthermore, close examination of the S. aureus or S. pyogenes genomes failed to reveal a DNA sequence(s) with homologs to the nonribosomal peptide synthetases found in bacillus species involved in the synthesis of peptide antibiotics (17), supporting the notion that wall assembly enzymes may be involved in LPXTGase assembly.
REFERENCES
- 1.Asakura, T., R. Sugino, T. Okumura, and Y. Nakazawa. 2002. The role of irregular unit, GAAS, on the secondary structure of Bombyx mori silk fibroin studied with 13C CP/MAS NMR and wide angle X-ray scattering. Protein Sci. 11:1873-1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnett, T. C., and J. R. Scott. 2002. Differential recognition of surface proteins in Streptococcus pyogenes by two sortase gene homologs. J. Bacteriol. 184:2181-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bongaerts, R. J. M., H.-P. Heinz, U. Hadding, and G. Zysk. 2000. Antigenicity, expression and molecular characterization of surface-located pullulanase of Streptococcus pneumoniae. Infect. Immun. 68:7141-7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cue, D., P. E. Dombeck, H. Lam, and P. P. Cleary. 1998. Streptococcus pyogenes serotype M1 encodes multiple pathways for entry into human epithelial cells. Infect. Immun. 66:4593-4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ehlert, K., W. Schröder, and H. Labischinski. 1997. Specific FemA and FemB for different glycine residues: FemB cannot substitute for FemA in staphylococcal peptidoglycan pentaglycine side chain formation. J. Bacteriol. 179:7573-7576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fischetti, V. A., V. Pancholi, and O. Schneewind. 1990. Conservation of hexapeptide sequence in the anchor region of the surface proteins of gram positive cocci. Mol. Microbiol. 4:1603-1605. [DOI] [PubMed] [Google Scholar]
- 7.Fischetti, V. A., R. D. Horstmann, and V. Pancholi. 1995. Location of the complement factor H binding site on streptococcal M protein. Infect. Immun. 63:149-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goffin, C., and J. M. Ghuysen. 1998. Multimodular penicillin-binding proteins: an enigmatic family of orthologs and paralogs. Microbiol. Mol. Biol. Rev. 62:1079-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishino, F., K. Mitsui, and M. Matsuhashi. 1980. Dual enzyme activities of cell wall peptidoglycan transglycosylase and penicillin-sensitive transpeptidase in purified preparations of Escherichia coli penicillin-binding protein 1A. Biochem. Biophys. Res. Commun. 97:287-293. [DOI] [PubMed] [Google Scholar]
- 10.Ito, E., and J. L. Strominger. 1962. Enzymatic synthesis of the peptide in bacterial uridine nucleotides. I. Enzymatic addition of L-alanine, D-glutamic acid, and L-lysine. J. Biol. Chem. 237:2689-2695. [Google Scholar]
- 11.Ito, E., and J. L. Strominger. 1962. Enzymatic synthesis of the bacterial uridine nucleotides. II. Enzymatic synthesis and addition of D-alanyl-D-alanine. J. Biol. Chem. 237:2696-2703. [Google Scholar]
- 12.Janulczyk, R., F. Iannelli. A. G. Sjoholm, G. Pozzi, and L. Björck. 2000. Hic, a novel surface protein of Streptococcus pneumoniae that interferes with complement function. J. Biol. Chem. 275:37257-37263. [DOI] [PubMed] [Google Scholar]
- 13.Kamiryo, T., and M. Matsuhashi. 1972. Biosysnthesis of the cross-linking peptides in the cell wall peptidoglycan of Staphylococcus aureus. J. Biol. Chem. 247:6306-6311. [PubMed] [Google Scholar]
- 14.Kharat, A. S., and A. Tomasz. 2003. Inactivation of the srtA gene affects localization of surface proteins and decreases adhesion of Streptococcus pneumoniae to human pharyngeal cells in vitro. Infect. Immun. 71:2758-2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee, S. G., V. Pancholi, and V. A. Fischetti. 2002. Characterization of a unique glycosylated anchor endopeptidase that cleaves the LPXTG motif of cell surface proteins of gram positive bacteria. J. Biol. Chem. 277:46912-46922. [DOI] [PubMed] [Google Scholar]
- 16.Lee, S. G., and V. A. Fischetti. 2003. Presence of D-alanine in an endo-peptidase from Streptococcus pyogenes. J. Biol. Chem. 278:46649-46653. [DOI] [PubMed] [Google Scholar]
- 17.Mootz, H. D., and M. A. Marahiel. 1997. The tyrocidine biosynthesis operon of Bacillus brevis: complete nucleotide sequence and biochemical characterization of functional internal adenylation. J. Bacteriol. 179:6843-6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Navarre, W. W., and O. Schneewind. 1994. Proteolytic cleavage and cell wall anchoring at the LPXTG motif of surface proteins in gram positive bacteria. Mol. Micribiol. 14:115-121. [DOI] [PubMed] [Google Scholar]
- 19.Navarre, W. W., and O. Schneewind. 1999. Surface proteins of gram-positive bacteria and mechanisms of their targeting cell wall envelope. Microbiol. Mol. Biol. Rev. 63:174-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suzuki, H., Y. van Heijenoort, T. Tamura, J. Mizoguchi, Y. Hirota, and J. van Heijenoort. 1980. In vitro peptidoglycan polymerization catalyzed by penicillin binding protein 1b of Escherichia coli K-12. FEBS Lett. 110:287-293. [DOI] [PubMed] [Google Scholar]
- 21.Thon-That, H., G. Liu, S. K. Mazmanian, K. F. Faull, and O. Schneewind. 1999. Purification and characterization of sortase, the transpeptidase that cleaves surface proteins of Staphylococcus aureus at the LPXTG motif. Proc. Natl. Acad. Sci. USA 96:12424-12429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tshierske, M., C. Mori, S. Rohrer, K. Ehlert, K. I. Shaw, and B. Berger-Bächi. 1999. Identification of three additional femAB-like open reading frames in Staphylococcus aureus. FEMS Microbiol. Lett. 171:97-102. [DOI] [PubMed] [Google Scholar]
- 23.van Heijenoort, J. 2001. Recent advances in the formation of the bacterial peptidoglycan monomer unit. Nat. Prod. Rep. 18:503-519. [DOI] [PubMed] [Google Scholar]
- 24.Wang, J. R., and M. W. Stinton. 1994. M protein mediates streptococcal adhesion to Hep2 cells. Infect. Immun. 62:442-448. [DOI] [PMC free article] [PubMed] [Google Scholar]