


Abstract
The completion of the sequencing of the human genome and the concurrent, rapid development of high-throughput proteomic methods have resulted in an increasing need for automated approaches to archive proteomic data in a repository that enables the exchange of data among researchers and also accurate integration with genomic data. PeptideAtlas (http://www.peptideatlas.org/) addresses these needs by identifying peptides by tandem mass spectrometry (MS/MS), statistically validating those identifications and then mapping identified sequences to the genomes of eukaryotic organisms. A meaningful comparison of data across different experiments generated by different groups using different types of instruments is enabled by the implementation of a uniform analytic process. This uniform statistical validation ensures a consistent and high-quality set of peptide and protein identifications. The raw data from many diverse proteomic experiments are made available in the associated PeptideAtlas repository in several formats. Here we present a summary of our process and details about the Human, Drosophila and Yeast PeptideAtlas builds.
INTRODUCTION
PeptideAtlas was initially designed to annotate eukaryotic genomes with peptide sequences obtained from mass spectrometry (MS) experiments. These peptide sequences can be collected using the procedure summarized in Figure 1. Sample proteins are proteolytically cleaved into peptides (usually by the enzyme trypsin although other proteases can also be used). The resulting peptide mixture is then subjected to chromatographic separation by strong cation exchange and reverse phase capillary chromatography. The resulting peptide pools are then analyzed by ESI-MS/MS. The database search program SEQUEST™ (1) is used to assign a peptide sequence to the MS/MS spectra. The confidence of these peptide assignments is then evaluated using PeptideProphet (2), which assigns a probability P of being correct to each top-scoring peptide sequence identification. All the experimental data products, including PeptideProphet probability scores, are loaded into SBEAMS—Proteomics, a proteomics analysis database built as a module under the Systems Biology Experiment Analysis Management System (SBEAMS) framework (http://www.sbeams.org/).
Figure 1.

PeptideAtlas analysis pipeline for the annotation of genomes with high-quality peptide sequences derived from high-throughput 2D-LC-MS/MS analysis of biological samples.
The identified peptide sequences are then mapped onto their respective genome sequence. First a modified BLAST (3) algorithm is used (with the parameters adapted for searching small peptides: -E 1 -W 2 -M PAM30 -G 9 -e 10 -K 50 -b 50 -F F) to exactly match each peptide to an organism's reference protein database; next, exact and complete matches are used to infer a peptide's chromosomal coordinates; finally, the results are loaded into the PeptideAtlas relational database. We denote each execution of the mapping process as a ‘build’. The database schema (available at project website http://www.peptideatlas.org) can accommodate multiple PeptideAtlas builds, that is, it can handle a variety of organisms and a variety of reference protein sequence sets. Visualization of the results is then achieved using the Distributed Annotation System (DAS) (4) in conjunction with the Ensembl genome browser (5).
Owing to growing interest from the scientific community, other PeptideAtlas builds were added to the initial human PeptideAtlas (6), including a build for human plasma/serum (7) and for other species such as Drosophila melanogaster and yeast Saccharomyces cerevisiae. A summary of the currently available builds is shown in Figure 2.
Figure 2.

Plots: cumulative number of MS/MS spectra versus cumulative number of distinct peptides (all spectra and peptides correspond to peptide identifications with probabilities ≥0.9). The figure will show saturation when new spectra do not yield new peptide identifications. Tables: statistics for atlas builds (number of experiments, number of spectra that yielded a peptide identification with probability P ≥ 0.9, number of distinct peptides identified, number of proteins identified and percentage of all genes to which the proteins map).
FEATURES OF PeptideAtlas
The current Human build of PeptideAtlas (April 2005) contains peptide sequences identified in 90 proteomic experiments, in which proteins were extracted from various cell and tissue types. This number of experiments represents a considerable increase compared with the original build (6) and comprises published as well as a large number of (yet) unpublished human datasets from various cell types, such as T cells, B cells, lymphocytes, lymphoblasts, hepatocytes, intestinal cells, hepatoma cells and others. A full listing of all the experiments and samples currently in PeptideAtlas can be found at the project website. The raw data for all published or released datasets are also provided in a repository there. In the April 2005 build, 3.3 million MS/MS spectra were searched and yielded 35 391 distinct peptides with PeptideProphet probability P ≥ 0.9 that were mapped onto 11 115 of the human Ensembl proteins (version 30.35c; March 22, 2005). These proteins represent unique proteins or splice forms from 30% of human genes in Ensembl.
Figure 2 also shows the cumulative number of distinctly identified peptides as a function of the number of spectra with PeptideProphet probability P ≥ 0.9 identifications. When most peptides that can be observed with the technology used are catalogued, this curve is expected to flatten—each additional dataset should then yield few new gene matches. However, most curves continue to increase steeply with the exception of yeast, where flattening is evident due to a large number of similar experiments. The last yeast experiment to be added showed a significant increase in contributed peptides as a new free-flow electrophoresis fractionation technique was used, allowing many previously unidentified peptides to be detected.
Repository function. It is our intent to make publicly available for download as much of the raw data that we use to build the PeptideAtlas as possible. This includes datasets that have been previously published or otherwise released by the data producers. Unpublished raw datasets that were used to build PeptideAtlas are kept private until publication or release by the authors. There are currently ∼75 human and yeast experiments available for download in the repository, totaling over 85 GB of downloads. We provide for download the raw MS/MS files, mzXML format MS/MS files (8), full SEQUEST search results, PeptideProphet results as well as the final ProteinProphet (9) output file. Simple sample descriptions and links to related publications are also provided.
Database function. The results from the PeptideAtlas builds can be downloaded or browsed by users via the PeptideAtlas web interface, as depicted in Figure 3. The database functionality is provided by SBEAMS. The interface allows users to select and browse experimentally observed peptides that match available constraints. There is a summary page at the peptide level, displaying attributes and mappings for a single peptide sequence, and also a summary page at the protein level, displaying attributes of a specified protein and which peptides map to it. Visualization of the results is achieved using the DAS in conjunction with the Ensembl genome browser. DAS allows sequence annotations to be added by multiple third-party annotators and viewed on an as-needed basis in the Ensembl genome browser.
Figure 3.
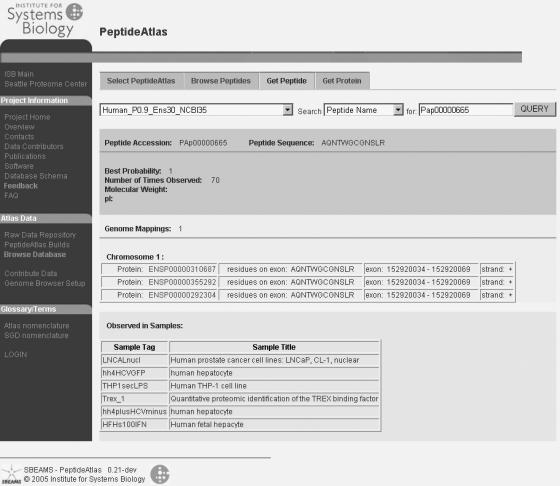
Peptide view in the PeptideAtlas web-based database interface. For the specified peptide (by accession number or sequence) a summary is displayed indicating individual attributes of the peptide, protein and genome mapping information, and the samples in which it has been observed. Different information is available in other views. Additionally there are two-way links to the Ensembl genome browser that allow users to see PeptideAtlas peptides as tracks in the genome browser and link out for more information. Additional information is available at our website.
Users can view the most current ISB Human PeptideAtlas tracks in the Ensembl genome browser by following the instructions on the website. Once the PeptideAtlas tracks have been defined, the peptide coordinate URLs in our web database interfaces link to a view of the peptide in the Ensembl genome browser. From that genome view, one can also link back to the PeptideAtlas database by clicking on the peptide link.
STATISTICS FOR PeptideAtlas
Reliably estimating the false positive error rates in an automated fashion is critical: large-scale datasets generated by high-throughput methods inherently contain results with a large number of false identifications (10). PeptideAtlas uses a program called PeptideProphet (9) to remove the majority of false positive identifications. PeptideProphet computes a probability that an assignment of an MS/MS spectrum to a peptide sequence is correct based on the database search scores, the difference between the measured and theoretical peptide masses, the expected and found number of termini for the type of enzymatic cleavage used and a variety of other factors. Probabilities computed by PeptideProphet have been shown to be accurate in the entire probability range and, therefore, can be used to filter out the probabilities that fall below a certain threshold (2). We provide options for users to browse or download versions of the PeptideAtlas built with several P thresholds.
FUTURE DIRECTIONS
PeptideAtlas is a first step toward the goal of fully annotating and validating eukaryotic genomes by using experimentally observed protein products. PeptideAtlas provides a process and a framework to accommodate proteome information generated by high-throughput proteomics technologies and is able to efficiently disseminate experimental data in the public domain. Its significance continues to grow as more data are submitted from diverse experiments, using different cellular compartments and enrichments methods. PeptideAtlas also provides a resource for the development of new avenues of research. The datasets will provide a rich source of data for computational scientists to develop and test new algorithms for proteomic analysis, gene-discovery and splice variant prediction.
The need for public proteomics data repositories is recognized (11) and we intend PeptideAtlas to continue to grow as a public database and resource. We strongly encourage researchers to contribute their own MS/MS data to the PeptideAtlas project. In the near future, we will make builds for organisms such as mouse, Arabidopsis thaliana and Halobacterium sp. NRC-1, and continue to make subsets such as the Human Plasma PeptideAtlas (7). Also in the near future we hope to provide an interface to access representative spectra of peptides, and will provide a way to retrieve information on peptide modifications (such as phosphorylation, etc.).
Acknowledgments
This project has been funded in part with funds from the National Heart, Lung, and Blood Institute; National Institutes of Health under contract no. N01-HV-28179. Funding to pay the Open Access publication charges for this article was provided by Nestlé.
Conflict of interest statement. None declared.
REFERENCES
- 1.Eng J., McCormack A.L., Yates J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 2.Keller A., Nesvizhskii A.I., Kolker E., Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 3.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 4.Dowell R.D., Jokerst R.M., Day A., Eddy S.R., Stein L. The distributed annotation system. BMC Bioinformatics. 2001;2:7. doi: 10.1186/1471-2105-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hubbard T., Andrews D., Caccamo M., Cameron G., Chen Y., Clamp M., Clarke L., Coates G., Cox T., Cunningham F., et al. Ensembl 2005. Nucleic Acids Res. 2005;33:D447–D453. doi: 10.1093/nar/gki138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Desiere F., Deutsch E.W., Nesvizhskii A.I., Mallick P., King N.L., Eng J.K., Aderem A., Boyle R., Brunner E., Donohoe S., et al. Integration with the human genome of peptide sequences obtained by high-throughput mass spectrometry. Genome Biol. 2005;6:R9. doi: 10.1186/gb-2004-6-1-r9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deutsch E.W., Eng J.K., Zhang H., King N.L., Nesvizhskii A.I., Lin B., Lee H., Yi E.C., Osssola R., Aebersold R. Human Plasma PeptideAtlas. Proteomics. 2005;5:3497–3500. doi: 10.1002/pmic.200500160. [DOI] [PubMed] [Google Scholar]
- 8.Pedrioli P.G., Eng J.K., Hubley R., Vogelzang M., Deutsch E.W., Raught B., Pratt B., Nilsson E., Angeletti R.H., Apweiler R., et al. A common open representation of mass spectrometry data and its application to proteomics research. Nat. Biotechnol. 2004;22:1459–1466. doi: 10.1038/nbt1031. [DOI] [PubMed] [Google Scholar]
- 9.Nesvizhskii A.I., Keller A., Kolker E., Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 10.Nesvizhskii A.I., Aebersold R. Analysis, statistical validation and dissemination of large-scale proteomics datasets generated by tandem MS. Drug Discov. Today. 2004;9:173–181. doi: 10.1016/S1359-6446(03)02978-7. [DOI] [PubMed] [Google Scholar]
- 11.Prince J.T., Carlson M.W., Wang R., Lu P., Marcotte E.M. The need for a public proteomics repository. Nat. Biotechnol. 2004;22:471–472. doi: 10.1038/nbt0404-471. [DOI] [PubMed] [Google Scholar]