


Abstract
MODBASE (http://salilab.org/modbase) is a database of annotated comparative protein structure models for all available protein sequences that can be matched to at least one known protein structure. The models are calculated by MODPIPE, an automated modeling pipeline that relies on MODELLER for fold assignment, sequence–structure alignment, model building and model assessment (http:/salilab.org/modeller). MODBASE is updated regularly to reflect the growth in protein sequence and structure databases, and improvements in the software for calculating the models. MODBASE currently contains 3 094 524 reliable models for domains in 1 094 750 out of 1 817 889 unique protein sequences in the UniProt database (July 5, 2005); only models based on statistically significant alignments and models assessed to have the correct fold despite insignificant alignments are included. MODBASE also allows users to generate comparative models for proteins of interest with the automated modeling server MODWEB (http://salilab.org/modweb). Our other resources integrated with MODBASE include comprehensive databases of multiple protein structure alignments (DBAli, http://salilab.org/dbali), structurally defined ligand binding sites and structurally defined binary domain interfaces (PIBASE, http://salilab.org/pibase) as well as predictions of ligand binding sites, interactions between yeast proteins, and functional consequences of human nsSNPs (LS-SNP, http://salilab.org/LS-SNP).
INTRODUCTION
The genome sequencing efforts are providing us with complete genetic blueprints for hundreds of organisms, including humans. We are now faced with the challenge of assigning, investigating and modifying the functions of proteins encoded by these genomes. This task is generally facilitated by 3D structures of the proteins (1–3), which are best determined by experimental methods, such as X-ray crystallography and NMR-spectroscopy. The number of experimentally determined structures deposited in the PDB increased from 23 096 to 31 823 over the last 2 years (August 2005) (4). However, the number of sequences in comprehensive sequence databases, such as UniProt (5) and GenPept (6), continues to grow even more rapidly, increasing from 1.2 to 2 million over the last 2 years (August 2005). Therefore, protein structure prediction is essential to obtain structural information for sequences where no experimental structure is available.
The most accurate models are generally obtained by homology or comparative modeling (7–10), a method that is applicable if an experimental structure related to a given target sequence is available. The fraction of sequences for which comparative models can be obtained automatically has increased moderately from ∼57 to ∼60% over the last 2 years, reflecting the counteracting effects of structural genomics (11,12) and many new genomic sequences.
The process of comparative modeling usually requires the use of a number of programs to identify template structures, to generate sequence–structure alignments, to build the models and to evaluate them. In addition, various sequence and structure databases that are accessed by these programs are needed. Once an initial model is calculated, it is generally refined and ultimately analyzed in the context of many other related proteins and their functional annotations.
In this paper, we present MODBASE, a database of comparative protein structure models, and several associated databases and servers that facilitate these tasks for both expert and novice users. We highlight the improvements of MODBASE that were implemented since the last report (13), including updated modeling software, a completely redesigned user interface, incorporation of annotated human single point mutations, homology-based prediction of interacting proteins and improved access to information about structurally defined ligand binding sites and binary domain interfaces.
CONTENTS
Comparative modeling
Models in MODBASE are calculated using MODPIPE, our completely automated software pipeline for comparative modeling (14). MODPIPE can calculate comparative models for a large number of protein sequences, using many different template structures and sequence–structure alignments. MODPIPE relies on the various modules of MODELLER (15) for its functionality and is adapted for large-scale operation on a cluster of PCs using scripts written in PERL.
The templates used for model building consist of representative multiple structure alignments extracted from DBAli (16). These alignments were prepared by the SALIGN module of MODELLER (M. S. Madhusudan, M. A. Marti-Renom and A. Sali, manuscript in preparation), which implements a multiple structure alignment method similar to that in the program COMPARER (17). Sequence profiles are constructed for both the target sequences and the templates by scanning against the UniProt database of sequences, relying on the BUILD_PROFILE module of MODELLER (N. Eswar, M. S. Madhusudhan and A. Sali, manuscript in preparation). BUILD_PROFILE is an iterative database searching protocol that relies on local dynamic programming and a robust method of estimation of alignment significance. Sequence–structure matches are established by aligning the target sequence profile against the template profiles, using local dynamic programming implemented in the PROFILE_PROFILE_SCAN module of MODELLER (18). Significant alignments covering distinct regions of the target sequence are chosen for modeling. Models are calculated for each of the sequence–structure matches using MODELLER. The resulting models are then evaluated by a composite model assessment criterion that depends on the compactness of a model, the sequence identity of the sequence–structure match and statistical energy Z-scores (D. Eramian, M. -Y. Shen, A. Sali, M. Marti-Renom, manuscript in preparation).
Model datasets
Models in MODBASE are organized into a number of datasets. The largest dataset contains models of all sequences in the UniProt database that are detectably related to at least one known structure in the PDB. This dataset is freely accessible to academic scientists. Currently, it contains ∼3 million models for domains in 1.1 million out of the 1.8 million unique sequences in the UniProt database (July 5, 2005), with an average length of 235 residues per model. For example, there are models for domains in 32 985 human sequences, 22 880 sequences from Arabidopsis thaliana, 15 195 sequences from Drosophila melanogaster and 9691 sequences from Escherichia coli. Other datasets include models calculated for the New York Structural GenomiX Research Consortium, datasets calculated by MODWEB and various datasets associated with our other modeling projects.
Some of the older datasets were calculated with an earlier version of MODPIPE (19) based on single template structures and sequence–structure matches generated by PSI-BLAST (20) and IMPALA (21).
MODWEB
Closely connected to MODBASE is MODWEB, our comparative modeling web-server (http://salilab.org/modweb) (14). MODWEB accepts one or many sequences in the FASTA format and calculates their models using MODPIPE based on the best available templates from the PDB. Alternatively, MODWEB also accepts a protein structure as input and calculates models for all identifiable sequence homologs in the UniProt database. The latter mode is a useful tool for structural genomics efforts (22) to assess the impact of a newly determined protein structure on the modeling of sequences of unknown structure. It is also used to identify new members of sequence superfamilies with at least one member of known structure. The results of MODWEB calculations are available through the MODBASE interface as private datasets protected with passwords.
DBAli
DBAli (http://salilab.org/DBAli/) stores pairwise comparisons of all structures in the PDB calculated using the program MAMMOTH (23), as well as multiple structure alignments generated by the SALIGN module of MODELLER. DBAli is updated weekly. As of June 2005, DBAli contains more than 800 million pairwise comparisons and ∼8500 family-based multiple structure alignments for ∼22 000 non-redundant protein chains in the PDB. Several programs are used to provide additional information: (i) ModDom assigns domain boundaries from structure; (ii) ModClus allows the user to generate clusters of similar protein structures; and (iii) AnnoLyze and AnnoLite annotate the functions of proteins in DBAli. The DBAli tools help users to analyze the protein structure space by establishing relationships between protein structures and their fragments in a flexible and dynamic manner.
Predicted ligand binding sites
MODBASE stores a list of the binding sites of known structure for ∼100 000 ligands found in the PDB (24). The ligands include small molecules, such as metal ions, nucleotides, saccharides and peptides. Binding sites in all known structures are defined to consist of residues with at least one atom within 5 Å of any ligand atom. MODBASE also contains predicted binding sites on template structures that are inherited from any related known structure if at least 75% of the binding site residues are within 4 Å of the template residues in a global superposition of the two structures in DBAli and if at least 75% of the binding site residue types are invariant. The putative ligand binding sites in the models are then mapped via the target-template alignments. The putative ligand binding sites are stored as SITE records and the binding site membership frequency per residue is indicated in the B-factor column of the model coordinate files. A total of 65% of MODBASE models have at least one predicted binding site.
PIBASE
PIBASE (http://salilab.org/pibase) is a comprehensive database of structurally defined interfaces between pairs of protein domains (25). It is composed of binary interfaces extracted from structures in the PDB and the Probable Quaternary Structure server PQS (26) using domain assignments from the Structural Classification of Proteins (27) and CATH (28) fold classification systems. PIBASE currently contains 158 915 interacting domain pairs between 105 061 domains from 2125 SCOP families. A diverse set of geometric, physiochemical and topologic properties are calculated for each complex, its domains, interfaces and binding sites. A subset of the interface properties is used to remove interface redundancy within PDB entries, resulting in 20 912 distinct domain–domain interfaces. The complexes are grouped into 989 topological classes based on their patterns of domain–domain contacts. The binary interfaces and their corresponding binding sites are categorized into 18 755 and 30 975 topological classes, respectively, based on the topology of secondary structure elements. PIBASE is a convenient resource for structural information on protein–protein interactions and is easily integrated with other databases. It is currently used by the DBAli function annotation module and the LS-SNP annotation system, and serves as a source of templates for the complex prediction module of MODBASE.
Predicted protein complexes
The composition and structure of protein complexes are predicted based on similarity to template complexes of known structure using comparative modeling. The structural models of the complexes are assessed with a statistical potential derived from binary domain interfaces obtained from PIBASE. The approach is applied to the 2434 yeast proteins with at least one structurally modeled domain, resulting in 3115 binary interaction predictions and 159 complex predictions of more than two proteins involving 506 and 71 proteins, respectively. The predictions are cross-referenced with the YeastGFP database of yeast protein subcellular localizations (29). A comparison of the predicted interactions to experimental results in the BIND database (30) reveals an overlap of ∼3%. The predictions are assigned confidence levels (Z-scores) that may be used to produce a list of testable hypotheses about interacting proteins. The estimated false positive rate at the Z-score threshold value of −1 is 16%.
LS-SNP
LS-SNP (http://www.salilab.org/LS-SNP) (31) is a database of annotated single nucleotide polymorphisms in human protein-coding exons that result in a changed amino acid residue type (non-synonymous SNPs or nsSNPs). The genomic locations of the SNPs were taken from the dbSNP database (32) and comprehensively mapped on the human proteins in the UniProt database via a collection of protein-to-mRNA and mRNA-to-genome alignments produced with the Known Genes algorithm (Fan Hsu, private communication). Using MODPIPE, we built comparative protein structure models for each significant alignment covering a distinct region of protein sequence (E-value cut-off 0.0001). We used the modeling results, in conjunction with PIBASE and the ligand tables of MODBASE, to infer which SNPs may destabilize protein quaternary structure or interfere with small molecule ligand binding. In addition, a support vector machine (33) that combines features of sequence, structure and evolutionary conservation was used to predict the SNPs that are associated with human disease. The resulting structural and functional annotations can be queried via the web interface from multiple viewpoints: single SNP, all SNPs in a gene or protein of interest, all nsSNPs in a genomic region of interest and all SNPs in a KEGG biochemical pathway (34). LS-SNP annotations are cross-linked with MODBASE and UCSC Genome Browser. The comparative models used in LS-SNP as well as details of the SNP annotations are available through MODBASE.
Case study: modeling of translated protein sequences from EST data
MODBASE contains a new dataset of protein structure models for the expressed sequence tags (ESTs) from the Vitis vinifera genome (dataset Grape-1). While there are no protein structures from V.vinifera in the PDB, our dataset contains 5594 reliable models for domains of the most probable translated reading frames of 3144 EST sequences obtained from the UNIGENE database (32). The structural modeling of protein sequences implied by the EST data presented new challenges, including selection of the most probable reading frame and consideration of the coverage of the ESTs and the template structure in order to estimate the coverage of the target sequence. There are at least two reasons for building protein structure models directly from EST data: (i) a larger fraction of transcript variants is probably considered for modeling than when starting with entries in a protein sequence database; and (ii) assessment of models built for inferred protein sequences can contribute to gene annotation efforts by helping to decide which ones are actually translated and folded into a stable structure (35).
ACCESS AND INTERFACE
The main access to MODBASE is through its web interface at http://salilab.org/modbase, by querying with Uniprot and GI identifiers, annotation keywords, PDB codes, datasets, sequence similarity to the modeled sequences (BLAST) and model specific criteria, such as model reliability, model size and target-template sequence identity. Additionally, it is possible to retrieve coordinate files, alignment files and ligand binding information in text files.
The output of a search is displayed on pages with varying amounts of information about the modeled sequences, template structures, alignments and functional annotations. An example for the output of a search resulting in one model is shown in Figure 1. A ribbon diagram of the model with the highest target-template sequence identity is displayed by default, together with details of the modeling calculation. Ribbon thumbprints of additional models for this sequence link to corresponding pages with more information. The ribbon diagrams are generated on the fly using Molscript (36) and Raster-3D (37). Additionally, cross-references to various databases, including PDB, UniProt, SwissProt/TrEMBL, PubMed and the UCSC Genome Browser, are provided. A pull-down menu provides links to additional functionality: the ligand binding module, the SNP module, retrieval of coordinate and alignment files, and the Molecular Modeling System Chimera (38) that allows the user to display template and model coordinates together with their alignment. Other MODBASE pages provide overviews of more than one sequence or structure. All MODBASE pages are interconnected to facilitate easy navigation between the different modules. Models are also directly accessible from other databases, including the SwissProt/TrEMBL sequence pages, PIR's iProClass, EBI's InterPro, the UCSC Genome Browser and PubMed (LinkOut).
Figure 1.
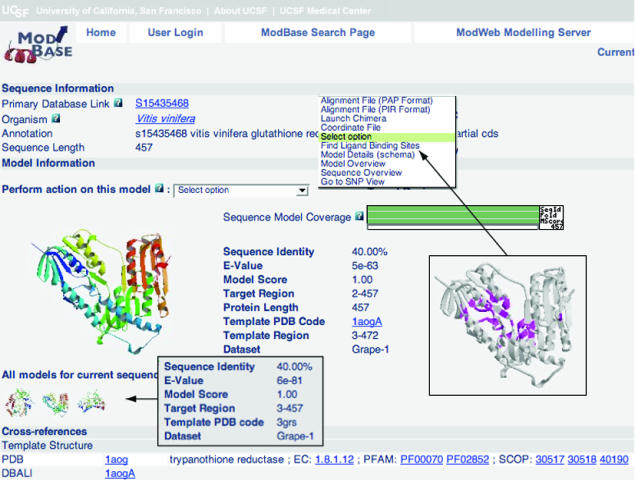
MODBASE Model Details page (example S15435468 from the Grape-1 dataset): This page provides links to all models for a specific sequence. A ribbon diagram of the primary model, database annotations and modeling details are displayed. Links to additional models for different target regions or models from other datasets are displayed as thumbprints. The pull-down menu provides access to alternative MODBASE views and other types of information (if available), such as data about SNPs and putative ligand binding sites.
FUTURE DIRECTIONS
MODBASE will be updated monthly to reflect the growth of the sequence and structure databases, as well as improvements in the methods and software used for calculating the models.
CITATION
Users of MODBASE are requested to cite this article in their publications.
Acknowledgments
We are grateful to Tom Ferrin, Daniel Greenblatt, Conrad Huang and Tom Goddard for CHIMERA and contributing to the MODBASE/CHIMERA interface. For linking to MODBASE from their databases, we thank David Haussler, Jim Kent (UCSC Genome Browser), Amos Bairoch (SwissProt/TrEMBL), Rolf Apweiler (InterPro), Patsy Babbitt (SFLD), and Kathy Wu (PIR/iProClass). The project has been supported by NIH/NIGMS R01 GM 54762, NIH/NIGMS P50 GM62529, NIH/NIGMS U54 GM074945, NIH/NCI R33 CA84699, NIH GM 08284 (DE), FONDEF G02S1001 (FM), the Sandler Family Supporting Foundation, Sun Academic Equipment Grant EDUD-7824-020257-US, an IBM SUR grant and an Intel computer hardware gift. Funding to pay the Open Access publication charges for this article was provided by NIH/NIGMS U54 GM074945.
Conflict of interest statement. None declared.
REFERENCES
- 1.Domingues F.S., Koppensteiner W.A., Sippl M.J. The role of protein structure in genomics. FEBS Lett. 2000;476:98–102. doi: 10.1016/s0014-5793(00)01678-1. [DOI] [PubMed] [Google Scholar]
- 2.Brenner S.E., Levitt M. Expectations from structural genomics. Protein Sci. 2000;9:197–200. doi: 10.1110/ps.9.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Skolnick J., Fetrow J.S., Kolinski A. Structural genomics and its importance for gene function analysis. Nat. Biotechnol. 2000;18:283–287. doi: 10.1038/73723. [DOI] [PubMed] [Google Scholar]
- 4.Westbrook J., Feng Z., Jain S., Bhat T.N., Thanki N., Ravichandran V., Gilliland G.L., Bluhm W., Weissig H., Greer D.S., et al. The Protein Data Bank: unifying the archive. Nucleic Acids Res. 2002;30:245–248. doi: 10.1093/nar/30.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bairoch A., Apweiler R., Wu C.H., Barker W.C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M., et al. The Universal Protein Resource (UniProt) Nucleic Acids Res. 2005;33:D154–D159. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benson D.A., Karsch-Mizrachi I., Lipman D.J., Ostell J., Wheeler D.L. GenBank. Nucleic Acids Res. 2005;33:D34–D38. doi: 10.1093/nar/gki063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker D., Sali A. Protein structure prediction and structural genomics. Science. 2001;294:93–96. doi: 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- 8.Wallner B., Elofsson A. All are not equal: a benchmark of different homology modeling programs. Protein Sci. 2005;14:1315–1327. doi: 10.1110/ps.041253405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hillisch A., Pineda L.F., Hilgenfeld R. Utility of homology models in the drug discovery process. Drug Discov. Today. 2004;9:659–669. doi: 10.1016/S1359-6446(04)03196-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marti-Renom M.A., Stuart A.C., Fiser A., Sanchez R., Melo F., Sali A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 11.Bonanno J.B., Almo S.C., Bresnick A., Chance M.R., Fiser A., Swaminathan S., Jiang J., Studier F.W., Shapiro L., Lima C.D., et al. New York-Structural GenomiX Research Consortium (NYSGXRC): a large scale center for the protein structure initiative. J. Struct. Funct. Genomics. 2005;6:225–232. doi: 10.1007/s10969-005-6827-0. [DOI] [PubMed] [Google Scholar]
- 12.Xie L., Bourne P.E. Functional coverage of the human genome by existing structures, structural genomics targets, and homology models. PLoS Comput. Biol. 2005;1:e31. doi: 10.1371/journal.pcbi.0010031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pieper U., Eswar N., Braberg H., Madhusudhan M.S., Davis F.P., Stuart A.C., Mirkovic N., Rossi A., Marti-Renom M.A., Fiser A., et al. MODBASE, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 2004;32:D217–D222. doi: 10.1093/nar/gkh095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eswar N., John B., Mirkovic N., Fiser A., Ilyin V.A., Pieper U., Stuart A.C., Marti-Renom M.A., Madhusudhan M.S., Yerkovich B., et al. Tools for comparative protein structure modeling and analysis. Nucleic Acids Res. 2003;31:3375–3380. doi: 10.1093/nar/gkg543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 16.Marti-Renom M.A., Ilyin V.A., Sali A. DBAli: a database of protein structure alignments. Bioinformatics. 2001;17:746–747. doi: 10.1093/bioinformatics/17.8.746. [DOI] [PubMed] [Google Scholar]
- 17.Zhu Z.Y., Sali A., Blundell T.L. A variable gap penalty function and feature weights for protein 3-D structure comparisons. Protein Eng. 1992;5:43–51. doi: 10.1093/protein/5.1.43. [DOI] [PubMed] [Google Scholar]
- 18.Marti-Renom M.A., Madhusudhan M.S., Sali A. Alignment of protein sequences by their profiles. Protein Sci. 2004;13:1071–1087. doi: 10.1110/ps.03379804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pieper U., Eswar N., Stuart A.C., Ilyin V.A., Sali A. MODBASE, a database of annotated comparative protein structure models. Nucleic Acids Res. 2002;30:255–259. doi: 10.1093/nar/30.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schaffer A.A., Wolf Y.I., Ponting C.P., Koonin E.V., Aravind L., Altschul S.F. IMPALA: matching a protein sequence against a collection of PSI-BLAST-constructed position-specific score matrices. Bioinformatics. 1999;15:1000–1011. doi: 10.1093/bioinformatics/15.12.1000. [DOI] [PubMed] [Google Scholar]
- 22.Chance M.R., Fiser A., Sali A., Pieper U., Eswar N., Xu G., Fajardo J.E., Radhakannan T., Marinkovic N. High-throughput computational and experimental techniques in structural genomics. Genome Res. 2004;14:2145–2154. doi: 10.1101/gr.2537904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ortiz A.R., Strauss C.E., Olmea O. MAMMOTH (matching molecular models obtained from theory): an automated method for model comparison. Protein Sci. 2002;11:2606–2621. doi: 10.1110/ps.0215902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stuart A.C., Ilyin V.A., Sali A. LigBase: a database of families of aligned ligand binding sites in known protein sequences and structures. Bioinformatics. 2002;18:200–201. doi: 10.1093/bioinformatics/18.1.200. [DOI] [PubMed] [Google Scholar]
- 25.Davis F.P., Sali A. PIBASE: a comprehensive database of structurally defined protein interfaces. Bioinformatics. 2005;21:1901–1907. doi: 10.1093/bioinformatics/bti277. [DOI] [PubMed] [Google Scholar]
- 26.Henrick K., Thornton J.M. PQS: a protein quaternary structure file server. Trends Biochem. Sci. 1998;23:358–361. doi: 10.1016/s0968-0004(98)01253-5. [DOI] [PubMed] [Google Scholar]
- 27.Andreeva A., Howorth D., Brenner S.E., Hubbard T.J., Chothia C., Murzin A.G. SCOP database in 2004: refinements integrate structure and sequence family data. Nucleic Acids Res. 2004;32:D226–D229. doi: 10.1093/nar/gkh039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pearl F., Todd A., Sillitoe I., Dibley M., Redfern O., Lewis T., Bennett C., Marsden R., Grant A., Lee D., et al. The CATH Domain Structure Database and related resources Gene3D and DHS provide comprehensive domain family information for genome analysis. Nucleic Acids Res. 2005;33:D247–D251. doi: 10.1093/nar/gki024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghaemmaghami S., Huh W.K., Bower K., Howson R.W., Belle A., Dephoure N., O'Shea E.K., Weissman J.S. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 30.Alfarano C., Andrade C.E., Anthony K., Bahroos N., Bajec M., Bantoft K., Betel D., Bobechko B., Boutilier K., Burgess E., et al. The Biomolecular Interaction Network Database and related tools 2005 update. Nucleic Acids Res. 2005;33:D418–D424. doi: 10.1093/nar/gki051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karchin R., Diekhans M., Kelly L., Thomas D.J., Pieper U., Eswar N., Haussler D., Sali A. LS-SNP: large-scale annotation of coding non-synonymous SNPs based on multiple information sources. Bioinformatics. 2005;21:2814–2820. doi: 10.1093/bioinformatics/bti442. [DOI] [PubMed] [Google Scholar]
- 32.Wheeler D.L., Barrett T., Benson D.A., Bryant S.H., Canese K., Church D.M., DiCuccio M., Edgar R., Federhen S., Helmberg W., et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2005;33:D39–D45. doi: 10.1093/nar/gki062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vapnik V., Chapelle O. Bounds on error expectation for support vector machines. Neural Comput. 2000;12:2013–2036. doi: 10.1162/089976600300015042. [DOI] [PubMed] [Google Scholar]
- 34.Ogata H., Goto S., Sato K., Fujibuchi W., Bono H., Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gopal S., Schroeder M., Pieper U., Sczyrba A., Aytekin-Kurban G., Bekiranov S., Fajardo J.E., Eswar N., Sanchez R., Sali A., et al. Homology-based annotation yields 1,042 new candidate genes in the Drosophila melanogaster genome. Nature Genet. 2001;27:337–340. doi: 10.1038/85922. [DOI] [PubMed] [Google Scholar]
- 36.Kraulis P.J. MOLSCRIPT: a program to produce both detailed and schematic plorts of protein structures. J. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]
- 37.Merritt E.A., Bacon D.J. Raster3D: photorealistic molecular graphics. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 38.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera - A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]