


Abstract
Listeria species are ubiquitous in the environment and often contaminate foods because they grow under conditions used for food preservation. Listeria monocytogenes, the human and animal pathogen, causes Listeriosis, an infection with a high mortality rate in risk groups such as immune-compromised individuals. Furthermore, L.monocytogenes is a model organism for the study of intracellular bacterial pathogens. The publication of its genome sequence and that of the non-pathogenic species Listeria innocua initiated numerous comparative studies and efforts to sequence all species comprising the genus. The Proteome database LEGER (http://leger2.gbf.de/cgi-bin/expLeger.pl) was developed to support functional genome analyses by combining information obtained by applying bioinformatics methods and from public databases to improve the original annotations. LEGER offers three unique key features: (i) it is the first comprehensive information system focusing on the functional assignment of genes and proteins; (ii) integrated visualization tools, KEGG pathway and Genome Viewer, alleviate the functional exploration of complex data; and (iii) LEGER presents results of systematic post-genome studies, thus facilitating analyses combining computational and experimental results. Moreover, LEGER provides an unpublished membrane proteome analysis of L.innocua and in total visualizes experimentally validated information about the subcellular localizations of 789 different listerial proteins.
INTRODUCTION
Listeria are Gram-positive, non-sporulating, rod-shaped bacteria occurring ubiquitously in soil and water. They are able to survive and grow under a wide variety of hostile conditions. The pathogenic species, in particular the human pathogen L.monocytogenes, are therefore a persistent problem as contaminants in the food industry (1). The infection called Listeriosis is a serious illness with a high mortality rate especially for immune-compromised patients, with clinical manifestations like septicaemia, meningoencephalitis and abortions (2) and has been estimated to be the second most common cause of death from food borne infections in the United States (3). L.monocytogenes is responsible for about 2500 cases of Listeriosis in the United States each year, with a hospitalization rate of 91% and a case fatality rate of 20% (4). Additional unique capabilities of L.monocytogenes are a driving force of further academical research: as a facultative intracellular human pathogen it allows the detailed analyses of fundamental cellular processes, such as the re-organization of the actin cytoskeleton or the innate immune responses (5,6). It, therefore, evolved into one of the most important model organisms for intracellular pathogens in the last decade (7). Moreover, it was recently applied as a vaccine shuttle system in cancer research (8,9).
Pathogenicity of Listeria is mediated by proteins that specifically interact with host proteins or structures. Almost all of these virulence factors described for L.monocytogenes exhibit two common features: (i) they are under the control of the positive transcriptional regulator of virulence factor A (PrfA) and (ii) are released into the environment or locate at the bacterial surface. Different types of surface proteins contribute to Listeria pathogenicity and its outstanding physiological capacity to adapt to variable extracellular and intracellular environments. Detailed investigations of defined subproteomes can provide information about protein localizations and processes that correlate with individual pathogenic or non-pathogenic lifestyles.
The genus Listeria comprises six species and is closely related to, for example, Bacillus, Streptococcus and Staphylococcus. The genome sequences of L.monocytogenes and the non-pathogenic species L.innocua were published by the European Listeria Genome Consortium in 2001 (10). A genome database ListiList (http://genolist.pasteur.fr/ListiList/) was established at the Institut Pasteur summarizing basically the annotation generated for the original publication that is now partially outdated. The German PathoGenoMik network and the Institut Pasteur cooperate in sequencing genomes of all other listerial species (http://www.genomik.uni-wuerzburg.de/). Thus, Listeria will probably be the first completely sequenced bacterial genus. While the original publication of the genomes from L.monocytogenes and L.innocua has catalyzed many new research activities comprising several approaches for systematic investigations by proteomics and transcriptomics (11–14), the expected sequencing of the complete genus will entail additional functional genome analyses.
Content and design of LEGER were developed to support current and upcoming comparative functional analyses and to relate experimental and bioinformatics results to the organization of genes in metabolic pathways or in the genome. Several tools are available to compare the structural organization of genomes (e.g. ERGO from IntegratedGenomics), but unfortunately neither summarize existing knowledge that can be derived from recent systematic ‘omic’ approaches nor provide an actual view of the most recent functional interpretations that can be found in literature. Besides, just one experimental database was published for L.monocytogenes before but is restricted (i) to a few 2D-PAGE reference maps of cytosolic fractions and (ii) surprisingly does not provide any portal for user queries (15).
The concept of LEGER overcomes these limitations and is based on three complementary topics: (i) LEGER provides a comprehensive information system that specifically focuses on the requirements of Listeria research incorporating functional descriptions from the original genome annotation to the most recent findings that can be found in literature. (ii) Customized result tables are extended by the integration of a KEGG and Genome Viewer for the functional and comparative exploration of complex datasets. (iii) LEGER currently comprises the experimental knowledge of nine systematic post-genome analyses that either define the physiological adaptation to hostile environmental conditions or reveal the subcellular localization of proteins. Particular attention was and will be paid to the integration of results derived from non-gel-based approaches, such as LC-MS/MS, that—contrary to traditional 2D-PAGE analyses—are unbiased by the biochemical properties of the investigated proteins and can provide representative proteome profiles for any type of subcellular localization. Comprehensive studies already exist for L.monocytogenes and L.innocua that took advantage of this type of proteome workflow and have characterized systematically proteins located at the peptidoglycan layer of the cell wall or in supernatant fractions (16,17). The LC-MS/MS-based analysis of membrane proteins from L.monocytogenes (18) was complemented by data newly generated for L.innocua (this study). Consequently, LEGER can refer to three experimentally validated subcellular proteomes of gene products, namely the secretome, the cell wall and the membrane.
IMPLEMENTATION
The program that interfaces the database via Internet consists of common gateway interface scripts written in PERL, including the BioPerl (v1.5) library (19). The database was designed and built following the relational model, and installed on a MySQL server (v4.0.21). The scripts are executed on an Apache2 WWW server with Linux as operating system. Several links in the dynamically generated web pages that form the interface make the navigation easy and user-friendly. The dynamic pages are linked to other databases, such as KEGG, UniProt and the NCBI database, and were optimized for Mozilla/Firefox browsers. The BLASTP procedure uses the NCBI BLAST v2.2.9 Toolkit (20). The peptide sequences from the L.monocytogenes strains F2365, F6854 and H7858 and predictions of Rho-independent transcription terminators and operons in bacterial genomes were downloaded from TIGR.
DATABASE CONTENT
The information system
Currently LEGER is focusing on annotations corresponding to the genomes of L.monocytogenes EGD-e (serotype 1/2a) and L.innocua (CLIP11262) as described in Ref. (10). LEGER was designed to support the generation of customized result tables as requested by many researchers (Figure 1). Four ways exist for the selection of genes/proteins from both species that can be used in disjunction: selection from menus, keyword queries, pre-selection of proteins found in post-genome studies and a text field option. Noteworthy, the text field option can be exploited as a simple text-mining tool as LEGER will detect all proteins mentioned in the text according to the nomenclature of bacterial gene names.
Figure 1.
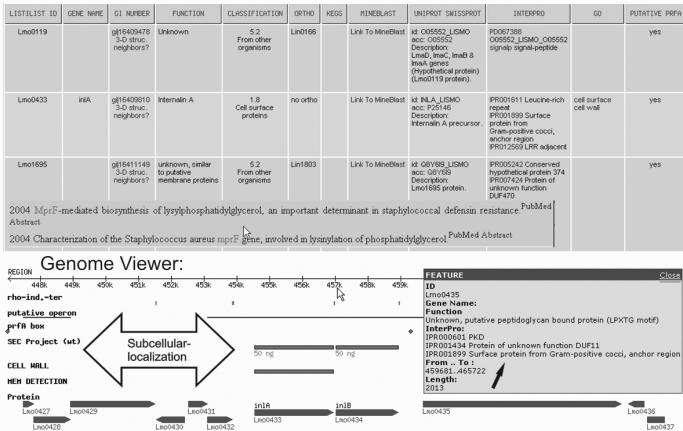
Customized result tables are complemented by MineBlast and visualization tools, such as the Genome Viewer. The MineBlast report (partial) reveals functional descriptions for Lmo1695 found in literature that are currently not available in genome annotations. The Genome Viewer displays the genomic organization around a selected gene (Lmo0433). Each gene symbol provides functional assignments within Pop-up windows (‘Feature’). Graphical tracks point to further computational and experimental information. Green bars above the gene symbols summarize validated knowledge about the subcellular localization of proteins. The Genome Viewer by default comparatively presents both Listeria genomes to highlight differential structures and protein abundances (data not shown).
The content of the result table can be defined by pre-selecting individual types of information that are grouped and presented by LEGER as part of four main topics, termed Annotation, Subcellular Localization, Protein Information and Comparative Genomics.
The topic ‘Annotation’ refers to information for the functional assignment of genes or proteins. Whereas ‘Description’ and ‘Classification’ represent the original annotation, the UniProt–Swiss-Prot entries give access to the data of curated databases to reveal inconsistencies accumulated in the past years. However, further powerful databases and services have to be considered for the generation of functional hypotheses and thus are included as part of this topic (e.g. Gene Ontology Annotation from UniProt and InterPro). With ‘SPS-Annotation’, the user receives information whether the selected genes/proteins were mentioned within a systematic post-genome study. This part requires manual updating and currently refers to the most important and comprehensive studies such as the investigation of PrfA-dependent gene expression (11), stress regulation and adaptation (12,13) and proteome studies aiming at subcellular reference maps derived from 2D-PAGE analyses (15,21). ‘MineBlast’ was recently published and can retrieve the most recent functional information for listerial proteins and their orthologues from other organisms in literature (22). MineBlast often can complement missing functional assignments with data that were published following the first annotation of a genome and thus alleviates a still highly manual task. A genome-wide MineBlast query was already completed and adapted result pages shortcut the alternative processing using the public NCBI services (BLAST and PubMed queries). ‘Genome View’ and ‘KEGG’ generate links to graphical presentations for every gene/protein in the result table. Adapted KEGG pathway pages display the distribution of proteins along biochemical pathways (Figure 2). The Genome Viewer displays the experimental results at the level of genome organization. Genes are visualized according to their length, order and orientation in the genome (Figure 1). Rho-independent terminator sites and operon predictions help to recognize putative transcriptional units. The Genome View is exceptionally qualified for the analysis of proteome profiles generated from defined subcellular localizations. Additional tracks, abbreviated as ‘Sec’, ‘Mem’ and ‘Cell wall’, indicate, by green colored bars above the gene symbols, whether a gene product was detected in non-gel-based proteome studies of the supernatant fraction, the membrane or the bacterial cell wall, respectively.
Figure 2.

The integrated KEGG Pathway Viewer facilitates the generation of functional hypotheses. A separate table summarizes affected pathways. Submitted genes or proteins are marked along metabolic or signaling pathway maps. For example, Lmo1986 and Lmo2006, two enzymes out of seventeen that are annotated within the ‘Pantothenate and CoA biosynthesis’ pathway were found to be co-regulated and mediate the first steps of this synthesis (see partial view of the corresponding map).
Numerous bioinformatics studies predict the subcellular localization of proteins and stimulate an intensive exchange of predictive and experimental data. The topic ‘Subcellular Localization’ refers to state-of-the-art procedures to forecast the presence of transmembrane domains (TMDs) and signal peptides. Two datasets from different approaches for the prediction of TMDs concerning their number, expected position within the peptide sequence and predicted topology at the membrane are integrated (23,24). Also available are data of a prediction of signal peptides for L.monocytogenes published recently by Trost et al. (16) ranking the results of seven approaches.
The topic ‘Protein information’ provides basic knowledge about expected physicochemical properties of proteins, such as their calculated hydrophobicity (GRAVY), molecular mass (MolMass) and isoelectric point (pI). This information can help to choose an appropriate biochemical method for the separation of the desired proteins a priori. These parameters can support the detection of proteins within gel systems or indicate by comparative analyses between calculated and observed values the occurrence of post-translational modifications. The number of cysteines ‘count Cysteines’ has to be considered in 2D-PAGE analyses and mass spectrometry since in some methods cysteins are a prerequisite for the quantitative characterization of proteins (DIGE and ICAT).
The topic ‘Comparative Genomics’ refers to information that is expected to correlate, in particular, with the saprophytic or pathogenic lifestyle and evolutionary adaptation of L.innocua and L.monocytogenes, respectively. LEGER can also reveal meaningful differences between orthologues. This was achieved by a genome-wide application of InterPro and a comparison of the results (Differences in InterPro). The binding of PrfA at a palindromic sequence within the promoter region is a typical feature of almost all described virulence factors so far. A systematic prediction of genes that are putatively regulated by PrfA using a straightforward mismatch analysis was published in 2001 (10). Approximately 10% of all genes from L.monocytogenes were supposed to be PrfA-dependently regulated (putative PrfA). To further improve the relevance of these predictions we performed a genome-wide weight matrix analysis based on RSAT (25) that takes into account more or less conserved nucleotides from PrfA boxes of known virulence factors (RSAT PrfA).
In conclusion, the information system LEGER comprises a well-organized collection of information beneficial for a variety of listerial post-genome studies. The external utilization of extracted data is guaranteed by an optional export to an Excel file (link below result table).
Integration of post-genome projects
While genome sequencing reveals the ‘blueprint of life’, the derived knowledge promotes hypotheses that have to be validated experimentally. Results generated by systematic approaches, such as proteomics, provide evidence that cannot be derived directly from genome sequences. Here, the investigation of physical interactions of proteins with other molecules and cellular components is of particular interest. Different mechanisms control and ascertain the translocation of proteins from ribosomes to their ‘working place’ indicating a direct correlation of protein localization and function. State-of-the-art proteome technologies—basically the combination of liquid chromatography with mass spectrometry—can give representative access to proteome profiles from different subcellular localizations that are largely unbiased by the biochemical properties of the proteins.
Three LC-MS/MS studies regarding L.monocytogenes and L.innocua were published focusing on detailed analyses of proteins that (i) are released into the bacterial environment (16), (ii) are closely attached to the peptidoglycan layer (17) and (iii) were detected in sucrose density gradient purified bacterial membranes (18). Whereas two of these studies comprise results from both species, the investigation of the membrane so far was restricted to L.monocytogenes. To complement this approach (and the presentation in LEGER) we analyzed L.innocua precisely according to the protocol of Wehmhoner et al. (18) and identified 244 different proteins. The results of the mentioned studies can be pre-selected individually in the information system. LEGER currently refers to 970 validated protein localizations corresponding to 789 different proteins. The results are visualized within the integrated Genome Viewer allowing an intuitive recognition of the subcellular localization of proteins (Figure 1).
LEGER is a portal for functional genome studies (concluding remarks)
Functional genome databases have to provide qualified information for at least two types of research: (i) systematic approaches, such as proteome studies that generate new functional hypotheses and (ii) individual projects striving to validate existing hypotheses, both aiming at the functional assignment of individual proteins as the pivotal task. LEGER integrates knowledge that is relevant for listerial research but was so far distributed among numerous public resources (e.g. InterPro, KEGG and GO). The comparison between the original annotation by Glaser et al. and entries retrieved from the manually curated UniProt–Swiss-Prot reveals both inconsistencies and new functional insights (e.g. Figure 1, Lmo0119). The ratio of functionally unassigned genes in the genomes of three recently sequenced pathogenic strains of L.monocytogenes strains is significantly lower compared with the reference EGD-e strain published in 2001 (26). In addition to the content of integrated databases, LEGER provides two independent services to improve the accuracy of functional citations: (i) The annotations of all five published listerial genomes (one L.innocua and four L.monocytogenes strains) can be inspected by an integrated BLASTP service that can always be accessed from the left frame. (ii) MineBlast can find and present most recent literature on homologous proteins and alleviate the functional assignment of listerial genes, such as lmo1695 [Figure 1 and (22)].
However, comparative ‘omic’ studies naturally group results based on the acquired experimental parameters. Co-regulated, co-stimulated or co-localized proteins are likely to belong to regulons, physiological pathways or molecular machineries. LEGER supports the generation of functional hypotheses by providing visualization tools, such as the KEGG pathway viewer. Users can submit any group of genes or proteins exhibiting a common feature (e.g. up- or down-regulated) and immediately will recognize which physiological and signaling pathways are affected (Figure 2). LEGER integrates proteome projects systematically and allows the exploration of data across projects, such as demonstrated for the subcellular localization of proteins. We suppose that data integration of post-genome studies will play a pivotal role in the understanding of the unique capacity of listerial species to adapt to adverse conditions. Consequently, we currently collect studies (see topic ‘Annotation’) that are related to the characterization of different stress regulons and prepare their graphical presentation in LEGER (contributions are welcome). The planned updates will also follow the recommendations for the publication of functional genome databases (27).
Acknowledgments
This work was funded by the German Bundesministerium für Bildung und Forschung (BMBF) ‘Verbundvorhaben: Intergenomics—Bioinformatische Modellierung der Wechselwirkung von Genomen’ (031U110A/031U210A). Funding to pay the Open Access publication charges for this article was provided by the GBF.
Conflict of interest statement. None declared.
REFERENCES
- 1.Nightingale K.K., Windham K., Wiedmann M. Evolution and molecular phylogeny of Listeria monocytogenes isolated from human and animal listeriosis cases and foods. J. Bacteriol. 2005;187:5537–5551. doi: 10.1128/JB.187.16.5537-5551.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vazquez-Boland J.A., Kuhn M., Berche P., Chakraborty T., Dominguez-Bernal G., Goebel W., Gonzalez-Zorn B., Wehland J., Kreft J. Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 2001;14:584–640. doi: 10.1128/CMR.14.3.584-640.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McLauchlin J., Mitchell R.T., Smerdon W.J., Jewell K. Listeria monocytogenes and listeriosis: a review of hazard characterisation for use in microbiological risk assessment of foods. Int. J. Food Microbiol. 2004;92:15–33. doi: 10.1016/S0168-1605(03)00326-X. [DOI] [PubMed] [Google Scholar]
- 4.Mead P.S., Slutsker L., Dietz V., McCaig L.F., Bresee J.S., Shapiro C., Griffin P.M., Tauxe R.V. Food-related illness and death in the United States. Emerg. Infect. Dis. 1999;5:607–625. doi: 10.3201/eid0505.990502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Welch M.D., Rosenblatt J., Skoble J., Portnoy D.A., Mitchison T.J. Interaction of human Arp2/3 complex and the Listeria monocytogenes ActA protein in actin filament nucleation. Science. 1998;281:105–108. doi: 10.1126/science.281.5373.105. [DOI] [PubMed] [Google Scholar]
- 6.Joseph S.B., Bradley M.N., Castrillo A., Bruhn K.W., Mak P.A., Pei L., Hogenesch J., O'connell R.M., Cheng G., Saez E., et al. LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell. 2004;119:299–309. doi: 10.1016/j.cell.2004.09.032. [DOI] [PubMed] [Google Scholar]
- 7.Alonso A., Garcia-del Portillo F. Hijacking of eukaryotic functions by intracellular bacterial pathogens. Int. Microbiol. 2004;7:181–191. [PubMed] [Google Scholar]
- 8.Schoen C., Stritzker J., Goebel W., Pilgrim S. Bacteria as DNA vaccine carriers for genetic immunization. Int. J. Med. Microbiol. 2004;294:319–335. doi: 10.1016/j.ijmm.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 9.Brockstedt D.G., Bahjat K.S., Giedlin M.A., Liu W., Leong M., Luckett W., Gao Y., Schnupf P., Kapadia D., Castro G., et al. Killed but metabolically active microbes: a new vaccine paradigm for eliciting effector T-cell responses and protective immunity. Nature Med. 2005;11:853–860. doi: 10.1038/nm1276. [DOI] [PubMed] [Google Scholar]
- 10.Glaser P., Frangeul L., Buchrieser C., Rusniok C., Amend A., Baquero F., Berche P., Bloecker H., Brandt P., Chakraborty T., et al. Comparative genomics of Listeria species. Science. 2001;294:849–852. doi: 10.1126/science.1063447. [DOI] [PubMed] [Google Scholar]
- 11.Milohanic E., Glaser P., Coppee J.Y., Frangeul L., Vega Y., Vazquez-Boland J.A., Kunst F., Cossart P., Buchrieser C. Transcriptome analysis of Listeria monocytogenes identifies three groups of genes differently regulated by PrfA. Mol. Microbiol. 2003;47:1613–1625. doi: 10.1046/j.1365-2958.2003.03413.x. [DOI] [PubMed] [Google Scholar]
- 12.Arous S., Buchrieser C., Folio P., Glaser P., Namane A., Hebraud M., Hechard Y. Global analysis of gene expression in an rpoN mutant of Listeria monocytogenes. Microbiology. 2004;150:1581–1590. doi: 10.1099/mic.0.26860-0. [DOI] [PubMed] [Google Scholar]
- 13.Helloin E., Jansch L., Phan-Thanh L. Carbon starvation survival of Listeria monocytogenes in planktonic state and in biofilm: a proteomic study. Proteomics. 2003;3:2052–2064. doi: 10.1002/pmic.200300538. [DOI] [PubMed] [Google Scholar]
- 14.Weeks M.E., James D.C., Robinson G.K., Smales C.M. Global changes in gene expression observed at the transition from growth to stationary phase in Listeria monocytogenes ScottA batch culture. Proteomics. 2004;4:123–135. doi: 10.1002/pmic.200300527. [DOI] [PubMed] [Google Scholar]
- 15.Folio P., Chavant P., Chafsey I., Belkorchia A., Chambon C., Hebraud M. Two-dimensional electrophoresis database of Listeria monocytogenes EGDe proteome and proteomic analysis of mid-log and stationary growth phase cells. Proteomics. 2004;4:3187–3201. doi: 10.1002/pmic.200300841. [DOI] [PubMed] [Google Scholar]
- 16.Trost M., Wehmhoner D., Karst U., Dieterich G., Wehland J., Jansch L. Comparative proteome analysis of secretory proteins from pathogenic and nonpathogenic Listeria species. Proteomics. 2005;5:1544–1557. doi: 10.1002/pmic.200401024. [DOI] [PubMed] [Google Scholar]
- 17.Calvo E., Pucciarelli M.G., Bierne H., Cossart P., Albar J.P., Garcia-del Portillo F. Analysis of the Listeria cell wall proteome by two-dimensional nanoliquid chromatography coupled to mass spectrometry. Proteomics. 2005;5:433–443. doi: 10.1002/pmic.200400936. [DOI] [PubMed] [Google Scholar]
- 18.Wehmhoner D., Dieterich G., Fischer E., Baumgartner M., Wehland J., Jansch L. ‘LaneSpector’, a tool for membrane proteome profiling based on sodium dodecyl sulfate-polyacrylamide gel electrophoresis/liquid chromatography-tandem mass spectrometry analysis: application to Listeria monocytogenes membrane proteins. Electrophoresis. 2005;26:2450–2460. doi: 10.1002/elps.200410348. [DOI] [PubMed] [Google Scholar]
- 19.Stajich J.E., Block D., Boulez K., Brenner S.E., Chervitz S.A., Dagdigian C., Fuellen G., Gilbert J.G., Korf I., Lapp H., et al. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002;12:1611–1618. doi: 10.1101/gr.361602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schaumburg J., Diekmann O., Hagendorff P., Bergmann S., Rohde M., Hammerschmidt S., Jansch L., Wehland J., Karst U. The cell wall subproteome of Listeria monocytogenes. Proteomics. 2004;4:2991–3006. doi: 10.1002/pmic.200400928. [DOI] [PubMed] [Google Scholar]
- 22.Dieterich G., Karst U., Wehland J., Jansch L. MineBlast: a literature presentation service supporting protein annotation by data mining of BLAST results. Bioinformatics. 2005;21:3450–3451. doi: 10.1093/bioinformatics/bti528. [DOI] [PubMed] [Google Scholar]
- 23.Arai M., Ikeda M., Shimizu T. Comprehensive analysis of transmembrane topologies in prokaryotic genomes. Gene. 2003;304:77–86. doi: 10.1016/s0378-1119(02)01181-2. [DOI] [PubMed] [Google Scholar]
- 24.Claros M.G., von Heijne G. TopPred II: an improved software for membrane protein structure predictions. Comput. Appl. Biosci. 1994;10:685–686. doi: 10.1093/bioinformatics/10.6.685. [DOI] [PubMed] [Google Scholar]
- 25.van Helden J. Regulatory sequence analysis tools. Nucleic Acids Res. 2003;31:3593–3596. doi: 10.1093/nar/gkg567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nelson K.E., Fouts D.E., Mongodin E.F., Ravel J., DeBoy R.T., Kolonay J.F., Rasko D.A., Angiuoli S.V., Gill S.R., Paulsen I.T., et al. Whole genome comparisons of serotype 4b and 1/2a strains of the food-borne pathogen Listeria monocytogenes reveal new insights into the core genome components of this species. Nucleic Acids Res. 2004;32:2386–2395. doi: 10.1093/nar/gkh562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stoeckert C.J., Jr Functional genomics databases on the web. Cell. Microbiol. 2005;7:1053–1059. doi: 10.1111/j.1462-5822.2005.00553.x. [DOI] [PubMed] [Google Scholar]