


Abstract
Transterm has now been publicly available for >10 years. Major changes have been made since its last description in this database issue in 2002. The current database provides data for key regions of mRNA sequences, a curated database of mRNA motifs and tools to allow users to investigate their own motifs or mRNA sequences. The key mRNA regions database is derived computationally from Genbank. It contains 3′ and 5′ flanking regions, the initiation and termination signal context and coding sequence for annotated CDS features from Genbank and RefSeq. The database is non-redundant, enabling summary files and statistics to be prepared for each species. Advances include providing extended search facilities, the database may now be searched by BLAST in addition to regular expressions (patterns) allowing users to search for motifs such as known miRNA sequences, and the inclusion of RefSeq data. The database contains >40 motifs or structural patterns important for translational control. In this release, patterns from UTRsite and Rfam are also incorporated with cross-referencing. Users may search their sequence data with Transterm or user-defined patterns. The system is accessible at http://uther.otago.ac.nz/Transterm.html.
INTRODUCTION
The fate of a large number of mRNAs is determined by motifs or structures encoded within them. These motifs are often located in the 3′-untranslated region (3′-UTR) or 5′-UTR but may be located in coding regions. Non-coding regions have been the focus of much research, reviewed in (1–3), and are implicated in the regulation of gene expression by microRNAs (4).
RELEVANT MRNA REGIONS EXTRACTED FROM GENBANK AND REFSEQ
The 5′-UTR, CDS and 3′-UTRs were extracted from all CDS entries that have a termination codon in Genbank (5) and were analysed using our previously described methods (6) and references therein. As most CDS do not have known and annotated 3′ or 5′ ends, we extract 1000 bases prior to the initiation codon, or 3000 bases after the termination codon for sequences from eukaryote species and 200 prior and 600 after for bacterial sequences. Entries are truncated at the next annotated feature if it overlaps (e.g. next CDS in bacteria). This results in files that will include the 3′- and 5′-UTRs, but may extend beyond them. A small proportion of long UTRs will be truncated by this method. Our analysis of 17 048 non-redundant human RefSeq mRNAs shows only 3% were >3000 bases in length. This gives a redundant set, e.g. for human 3′-UTRs 94 791 due to the redundancy in Genbank. A non-redundant set is derived (e.g. 33 332 sequences for humans) according to our published methods (6). These non-redundant datasets are analysed by species to give summary files, e.g. the frequency of bases around the termination codon for these 33 332 genes analysed by several means (*.termnrttmatrix, *.termnrttbit, *.termnrttchi, *.termnrttcvs, files; see also Figure 1 legend) (6). As expected, these show a bias toward A and G in the position immediately after the termination codon. Purines in this position have previously shown to enhance termination (7). These summary files represent the most commonly used codons or initiation and termination contexts for each species.
Figure 1.
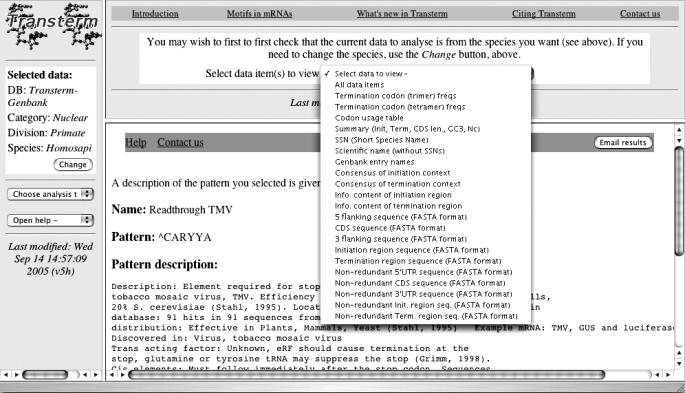
Data available for each species. Shown is a selection of the type of pre-processed data to view in progress, with the results of a pattern description search from a previous action in the low frame (see also Table 1). The file contents for each type of data have been described previously (15). These include redundant and non-redundant 3′- and 5′-flanks, CDS, initiation and termination contexts; consensuses and information content of the initiation and termination contexts; codon usage; list of entries making up the dataset; scientific and short names of the species; an overall summary file.
PATTERN/MOTIF DESCRIPTIONS
The Transterm database also contains descriptions of experimentally defined motifs from mRNAs. These are derived from the literature, or other databases [UTRdb (8) and Rfam (9)], reviewed, updated and integrated into the Transterm database. An example of a Transterm motif description is shown in Table 1. The element described promotes read-through of a termination codon, hindering termination in ∼5% of ribosome passes. The entry contains the pattern, a description of its function as well as key references and cross-references to other databases (in this case Recode, 10). An interesting feature of this pattern is that it contains a C in the position immediately after the stop codon, this is both less frequent and efficient in eukaryotic termination (7). These files represent features important for particular mRNAs.
Table 1.
An example of a pattern entry; the upper portion of this can be seen in Figure 1
| Readthrough TMV | |
| Pattern | CARYYA |
| Description | Element required for stop codon read-through in the plant virus tobacco mosaic virus, TMV. The motif ‘stop codon CARYYA’ was defined by mutagenesis studies in plants (2). The efficiency is ∼5% in plants, 1–3% in mammalian cells and 20% in Saccharomyces cerevisiae (1). A recent compilation of 91 unique viral sequences showed that CARYYA motifs were the most effective (3–4% in mammalian cells), with other 18 bases read-through contexts causing 0.75–2.25% read-through (5). |
| Location | 5′ end of 3′-UTR |
| Indicative hits in database | 91 in 27 796 non-viral eukaryotic 3′-UTRs |
| Confirmed phylogenetic distribution | Effective in plants, mammals, yeast |
| Example mRNA | TMV genomic RNA |
| Discovered in | Tobacco mosaic virus |
| Trans acting factor | eRF should facilitate termination at the stop (6), glutamine or tyrosine tRNAs may suppress the stop (4) |
| Cis elements | Must follow immediately after the stop codon. Sequences, particularly CAA prior to stop may be important (3). |
| Signal is sufficient in vivo in a heterologous message? | Yes (1,5) |
| Structural classification | Sequence |
| Related TransTerm entry | Readthrough elements |
| Related entries in other databases | ‘Codon redefinition’ entries (eg ID 289) in the recode database (recode.genetics.utah.edu). |
| Bibliography | (1) Stahl,G., Bidou,L., Rousset,J.P. and Cassan,M. (1995) Versatile vectors to study recoding: conservation of rules between yeast and mammalian cells. Nucleic Acids Res., 23, 1557–1560 |
| (2) Skuzeski,J.M., Nichols,L.M., Gesteland,R.F. and Atkins,J.F. (1991) The signal for a leaky UAG stop codon in several plant viruses includes the two downstream codons. J Mol. Biol., 218, 365–373 | |
| (3) Bonetti,B., Fu,L.W., Moon,J. and Bedwell,D.M. (1995) The efficiency of translation termination is determined by a synergistic interplay between upstream and downstream sequences in Saccharomyces cerevisiae. J. Mol. Biol., 251, 334–345 | |
| (4) Grimm,M., Nass,A., Schull,C. and Beier,H. (1998) Nucleotide sequences and functional characterization of two tobacco UAG suppressor tRNA(Gln) isoacceptors and their genes. Plant Mol. Biol., 38, 689–697 | |
| (5) Harrell,L., Melcher,U. and Atkins, J.F. (2002) Predominance of six different hexanucleotide recoding signals 3′ of read-through stop codons. Nucleic Acids Res. 30, 2011–2017 | |
| (6) Brown,C.M., Quigley,F.R. and Miller,W.A. (1995) Three eukaryotic release factor one (eRF1) homologs from Arabidopsis thaliana Columbia (Accession Nos. U40217, U40218, X69374, X69375). Plant Physiol., 110, 336 | |
| (7) Chapman,B and Brown,C.M. (2004) Translation termination in A. thaliana: characterization of three versions of release factor 1. Gene, 341, 219–225 | |
| Entry Added | 20/2/98 |
| Last Modified | 2/10/2005 |
ACCESS TO THE DATABASE
Processed sequence data and the programs used to make them can be obtained from the website. The interface has been redesigned for this release. Subsets of the database can be searched for putative motifs using regular expressions and matrices using the program scan_for_matches (10) or BLAST (11). Subsets may be user-chosen regions of a gene (5′- or 3′-UTR, CDS, translation start and stop context) for specified Genbank divisions or species (patterns only).
User-defined pattern searches can include a wide range of elements including simple sequences, gaps, reverse complemented sequences, palindromes, mismatches, n mismatches in a pattern, range of gap sizes, weight patterns and repeats. The on-line Help Browser that is part of Transterm contains detailed notes under help on ‘Motif patterns (scan-for-matches)’.
We have added the facility to search using longer query sequences with BLAST using empirically altered defaults to make it suitable for finding motifs. This approach will be useful to users with sequences of ∼50–100 bases, which they expect contains a conserved motif. The motif must have retained at least seven identical bases, but elsewhere in the motif sequence, it may have undergone insertions, deletions and substitutions that are common in UTRs. For such long motifs regular expression-based algorithms are usually impractical, as they would need to include a high tolerance for mismatches, insertions and deletions, which makes them inefficient.
The additional BLAST parameters given, presented in the ‘Other advanced options’ section of the BLAST search form, are ‘-W 7 -G 2 -E 1 -q -2 -r 2 -e 100 -S 1’. These, in order, with the default value for blastn in square brackets, are W, initial (seed) word size [11]; G, gap opening penalty [5]; E, gap extension penalty [2]; q, nucleotide mismatch score [−3]; r, score for a nucleotide match [1]; e, threshold expectation value for keeping an alignment [10] and S, search only the top strand. These parameters are suitable for matching small motifs, which may contain gaps and substitutions, and may occur fairly frequently.
COMPARISON WITH OTHER TRANSLATIONAL CONTROL DATABASES
Databases of mRNA sequences
Transterm sequence files are provided for all CDS sequences in Genbank, making it the most comprehensive of the databases available of UTRs. UTRdb and UTRsite focus on those eukaryotic UTRs that are well annotated in the sequence databases (e.g. complete mRNAs rather than genomic sequences).
Databases that include translational control elements
Several specialized databases that include translational control elements are available and referenced on our website. Examples include ARED, a database of putative AU rich element containing mRNAs (12), the Recode database of recoding data (13) and the Rfam database of RNA families (9). Elements/motifs described in these databases and relevant to mRNA biology have been included in Transterm where it was possible to create an accurate pattern file and they complement the Transterm data.
Alternative approaches to identifying regulatory motifs in mRNAs include phylogenetic footprinting (14). The Ancient Conserved UnTranslated Sequence (ACUTS) database is available, but has not been recently updated. However, it contains descriptions of several hundred phylogenetically conserved elements in 3′- and 5′-UTRs (14). On the Transterm website access is also provided to search the conserved 5′- and 3′-UTRs from ACUTS.
FURTHER INFORMATION
Extensive help is available on the website. This includes an outline of approaches to finding motifs in mRNAs that may affect gene expression and links to other resources that facilitate such investigations.
Acknowledgments
The work was supported by a NZ Marsden fund grant to C.M.B., and NZ Health Research Council grant to W.P.T., Elisabeth Poole and C.M.B. Funding to pay the Open Access publication charges for this article was provided by the Health Research Council of New Zealand.
Conflict of interest statement. None declared.
REFERENCES
- 1.Mazumder B., Seshadri V., Fox P.L. Translational control by the 3′-UTR: the ends specify the means. Trends Biochem. Sci. 2003;28:91–98. doi: 10.1016/S0968-0004(03)00002-1. [DOI] [PubMed] [Google Scholar]
- 2.Waggoner S.A., Liebhaber S.A. Regulation of alpha-globin mRNA stability. Exp. Biol. Med. 2003;228:387–395. doi: 10.1177/153537020322800409. [DOI] [PubMed] [Google Scholar]
- 3.Kuersten S., Goodwin E.B. The power of the 3′ UTR: translational control and development. Nat. Rev. Genet. 2003;4:626–637. doi: 10.1038/nrg1125. [DOI] [PubMed] [Google Scholar]
- 4.Pasquinelli A.E. MicroRNAs: deviants no longer. Trends Genet. 2002;18:171–173. doi: 10.1016/s0168-9525(01)02624-5. [DOI] [PubMed] [Google Scholar]
- 5.Benson D.A., Karsch-Mizrachi I., Lipman D.J., Ostell J., Wheeler D.L. GenBank. Nucleic Acids Res. 2003;31:23–27. doi: 10.1093/nar/gkg057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacobs G.H., Rackham O., Stockwell P.A., Tate W., Brown C.M. Transterm: a database of mRNAs and translational control elements. Nucleic Acids Res. 2002;30:310–311. doi: 10.1093/nar/30.1.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCaughan K.K., Brown C.M., Dalphin M.E., Berry M.J., Tate W.P. Translational termination efficiency in mammals is influenced by the base following the stop codon. Proc. Natl Acad. Sci. USA. 1995;92:5431–5435. doi: 10.1073/pnas.92.12.5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pesole G., Liuni S., Grillo G., Licciulli F., Mignone F., Gissi C., Saccone C. UTRdb and UTRsite: specialized databases of sequences and functional elements of 5′ and 3′ untranslated regions of eukaryotic mRNAs. Update 2002. Nucleic Acids Res. 2002;30:335–340. doi: 10.1093/nar/30.1.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Griffiths-Jones S., Bateman A., Marshall M., Khanna A., Eddy S.R. Rfam: an RNA family database. Nucleic Acids Res. 2003;31:439–441. doi: 10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dsouza M., Larsen N., Overbeek R. Searching for patterns in genomic data. Trends Genet. 1997;13:497–498. doi: 10.1016/s0168-9525(97)01347-4. [DOI] [PubMed] [Google Scholar]
- 11.Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bakheet T., Frevel M., Williams B.R., Greer W., Khabar K.S. ARED: human AU-rich element-containing mRNA database reveals an unexpectedly diverse functional repertoire of encoded proteins. Nucleic Acids Res. 2001;29:246–254. doi: 10.1093/nar/29.1.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baranov P.V., Gurvich O.L., Hammer A.W., Gesteland R.F., Atkins J.F. RECODE 2003. Nucleic Acids Res. 2003;31:87–89. doi: 10.1093/nar/gkg024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duret L., Bucher P. Searching for regulatory elements in human noncoding sequences. Curr. Opin. Struct. Biol. 1997;7:399–406. doi: 10.1016/s0959-440x(97)80058-9. [DOI] [PubMed] [Google Scholar]
- 15.Jacobs G.H., Stockwell P.A., Schrieber M.J., Tate W.P., Brown C.M. Transterm: a database of messenger RNA components and signals. Nucleic Acids Res. 2000;28:293–295. doi: 10.1093/nar/28.1.293. [DOI] [PMC free article] [PubMed] [Google Scholar]