

Abstract
Pseudomonas aeruginosa has two well-characterized quorum-sensing systems, Las and Rhl. These systems are composed of LuxR-type proteins, LasR and RhlR, and two acyl homoserine lactone (AHL) synthases, LasI and RhlI. LasI catalyzes the synthesis of N-(3-oxododecanoyl)homoserine lactone (3O-C12-HSL), whereas RhlI catalyzes the synthesis of N-butyryl-homoserine lactone. There is little known about the importance of AHLs in vivo and what effects these molecules have on eukaryotic cells. In order to understand the role of AHLs in vivo, we first tested the effects that deletions of the synthase genes in P. aeruginosa had on colonization of the lung. We demonstrate that in an adult mouse acute-pneumonia model, deletion of the lasI gene or both the lasI and rhlI genes greatly diminished the ability of P. aeruginosa to colonize the lung. To determine whether AHLs have a direct effect on the host, we examined the effects of 3O-C12-HSL injected into the skin of mice. In this model, 3O-C12-HSL stimulated a significant induction of mRNAs for the cytokines interleukin-1α (IL-1α) and IL-6 and the chemokines macrophage inflammatory protein 2 (MIP-2), monocyte chemotactic protein 1, MIP-1β, inducible protein 10, and T-cell activation gene 3. Additionally, dermal injections of 3O-C12-HSL also induced cyclooxygenase 2 (Cox-2) expression. The Cox-2 enzyme is important for the conversion of arachidonic acid to prostaglandins and is associated with edema, inflammatory infiltrate, fever, and pain. We also demonstrate that 3O-C12-HSL activates T cells to produce the inflammatory cytokine gamma interferon and therefore potentially promotes a Th1 environment. Induction of these inflammatory mediators in vivo is potentially responsible for the significant influx of white blood cells and subsequent tissue destruction associated with 3O-C12-HSL dermal injections. Therefore, the quorum-sensing systems of P. aeruginosa contribute to its pathogenesis both by regulating expression of virulence factors (exoenzymes and toxins) and by inducing inflammation.
Pseudomonas aeruginosa has the unique ability to adapt from its natural environment in the water and soil to induce a wide range of infections in humans. Many of the virulence factors needed to make this transition are regulated by cell-to-cell communication (quorum sensing) (4). This communication relies on the production of small diffusible signal molecules called acyl homoserine lactones (AHLs) that bind to and activate transcriptional regulators and induce gene transcription. P. aeruginosa has two well-studied quorum-sensing systems, Las and Rhl (6, 15). The Las system includes LasI, which is the synthase for the signal molecule N-(3-oxododecanoyl)homoserine lactone (3O-C12-HSL [also referred to as PAI-1 or OdDHL]), and LasR, which is a transcriptional regulator (19). The Rhl system includes RhlI, the synthase for the signal molecule N-butyryl-homoserine lactone (C4-HSL [also referred to as PAI-2 or BHL]), and the RhlR transcriptional regulator (20). While both AHLs diffuse into and out of the bacteria, 3O-C12-HSL is also actively pumped out by the MexAB-OprM efflux system (22). As the extracellular concentrations of AHLs increase, due to an increase in bacterial density, an intracellular threshold concentration is reached and 3O-C12-HSL and C4-HSL bind to and activate their cognate transcription factors LasR and RhlR. The LasR-3O-C12-HSL and RhlR-C4-HSL complexes induce the expression of multiple virulence genes as well as the production of additional quorum-sensing signals (7, 11, 17, 21, 23, 24, 29). Expression of quorum-sensing genes in P. aeruginosa not only is important for the production of virulence factors but also is essential for twitching motility (8) and the formation of differentiated biofilms (3). This biofilm differentiation is thought to protect the organisms from host defenses and provide enhanced resistance to antibiotics.
Based on these data, the expression of quorum-sensing genes may be a mechanism whereby P. aeruginosa gains a protective advantage, by making a concerted effort as a population to overcome host defenses and establish an infection. The role of quorum sensing in the pathogenesis of P. aeruginosa has been studied in several animal models, including an acute lung infection model utilizing neonatal mice and a chronic lung infection model in adult rats (18, 38). Despite the fact that P. aeruginosa causes acute infections in immunocompromised and intubated adult patients, no one has yet examined the role of quorum sensing in an acute lung infection model utilizing adult animals. In the models of infection thus far tested, it has been shown that in the absence of a complete las and or rhl quorum-sensing system, P. aeruginosa is significantly attenuated in its ability to colonize the host, induce inflammation, disseminate, and cause mortality (18, 26, 35, 38). Additional studies have examined the role of quorum sensing in human infections. For example, in the sputa from cystic fibrosis (CF) patients with chronic P. aeruginosa infections, levels of mRNA transcripts for lasR correlated with those of the lasR-regulated genes, lasA, lasB, and toxA (34). Additionally, the production of AHLs was also detected in P. aeruginosa-infected CF sputa (31). These studies indicate that during a vigorous P. aeruginosa infection in humans, the LasR-3O-C12-HSL complex is actively regulating the expression of virulence genes and therefore is enhancing the pathogenesis of the organism.
Several studies have suggested that the expression of 3O-C12-HSL during P. aeruginosa infections modulates the immune response and therefore may alter the pathogenesis of the organism. 3O-C12-HSL stimulation of in vitro cultures of human bronchial epithelial cells is a potent inducer of the neutrophil chemokine interleukin-8 (IL-8) (5, 32). A specific mitogen-activated protein kinase pathway regulates this induction of IL-8. This pathway leads to the induction of the transcription factor NF-κB and subsequent IL-8 production (32). Additional studies have shown that 3O-C12-HSL can also activate an immune response in lymphocytes (36). In this paper we examine the contribution of quorum sensing in an adult mouse acute lung infection model. We also identify key inflammatory mediators induced in vivo by 3O-C12-HSL. This is the first demonstration that 3O-C12-HSL is a potent inducer of inflammation in vivo.
MATERIALS AND METHODS
Mice.
Pathogen-free C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, Maine). DO11.10 αβ T-cell receptor transgenic mice were kindly supplied by J. Woodward (University of Kentucky). All mice were housed in accordance with National Institutes of Health guidelines. Mice were 6 to 8 weeks old at the start of the experiments.
Bacterial strains.
Nonmucoid P. aeruginosa strain PAO1 (obtained from Dennis Ohman, Virginia Commonwealth University), strain PAO-JP1 (the lasI deletion mutant), and strain PAO-JP2 (the lasI rhlI deletion mutant) were used in these studies (21).
Reagents.
3O-C12-HSL and C4-HSL were chemically synthesized as previously described (19, 20). These molecules are structurally and functionally identical to those obtained from P. aeruginosa cultures. Each lot of 3O-C12-HSL and C4-HSL was shown to be pure by high-pressure liquid chromatography. No detectable levels of endotoxin were found in preparations of AHLs by using a Limulus amebocyte lysate assay (Cape Cod Associates, Falmouth, Mass.). Nonacylated homoserine lactone was obtained from Sigma Chemical, St. Louis, Mo.
Adult mouse acute pneumonia model.
Bacteria were grown from frozen stocks overnight on Luria agar plates. A swab of bacteria was resuspended in Luria broth to give an optical density at 660 nm of 0.1 and grown at 37°C with shaking to an optical density at 660 nm of 0.5. Bacteria were pelleted and resuspended in phosphate-buffered saline (PBS), and dilutions were made to give the final concentrations of bacteria that were to be inoculated into each mouse. Mice were anesthetized with an intraperitoneal injection of 20 μl of Avertin (1% [wt/vol] 2,2,2-tribromoethanol and 1% [vol/vol] tert-amyl alcohol) per g, and 10 μl of bacteria was slowly dropped into each nostril, to give a total volume of 20 μl. After 24 h, mice were euthanized, their lungs were homogenized, and total lung CFU were determined by plating dilutions onto Pseudomonas isolation agar plates. A 250- to 300-mg portion of the liver was removed, homogenized in 0.5 ml of PBS, and plated on Pseudomonas isolation agar. Dissemination of the bacteria was determined by the presence of at least one colony in the liver.
Dermal inflammation.
Mice were anesthetized with Avertin as described above. Hair was shaved to expose the skin, and 50 μl of PBS or AHL dissolved in PBS was injected intradermally. After 24 h, mice were examined for inflammation at the site of injection. Skin samples were removed for histology and protein and RNA extraction.
Histology.
Skin samples from mice were fixed in 10% buffered formalin for at least 24 h. Samples were embedded in paraffin blocks, and 5-μm sections were cut. Samples were deparaffinized by heating for 1 h at 60°C and washing in xylene. Rehydrated samples were either stained with hematoxylin and eosin to determine the degree of inflammation or stained with anti-cyclooxygenase 2 (anti-Cox-2) antibody. Cox-2 immunostaining was performed as follows. Samples were incubated for 10 min with 3% H2O2 to quench any endogenous peroxidase activity. Between steps, samples were washed three times with PBS-0.05% Tween 20 for 5 min each. Antigen retrieval was performed by incubating samples at room temperature for 4 min with a proteinase K solution (Dako, Carpinteria, Calif.). Incubation of samples for 1 h at room temperature with 5% normal goat serum diluted in PBS-0.1% bovine serum albumin blocked any nonspecific antibody binding. Rabbit anti-mouse Cox-2 polyclonal antibody (Cayman Chemical, Ann Arbor, Mich.) was diluted 1:500 in PBS-0.1% bovine serum albumin. As a negative control for staining, an equal amount of the Cox-2 blocking peptide (Cayman Chemical) was added to antibody preparations and incubated for 1 h at room temperature. Antibody or antibody plus blocking peptide was added to samples and incubated overnight at 4°C. Sections were then stained with a 1:200 dilution of biotinylated goat anti-rabbit immunoglobulin G (Vector Laboratories, Burlingame, Calif.) for 1 h at room temperature. Bound antibodies were detected by the addition of a 1:1,000 dilution of streptavidin-horseradish peroxidase (HRP) for 1 h at room temperature. AEC (Zymed Laboratories, San Francisco, Calif.), which reacts with the HRP to form a red precipitate, was used to visualize antibody binding.
RNA isolation and RNase protection assays.
Skin samples were frozen in liquid nitrogen and ground to a fine powder with a mortar and pestle. TriReagent (Molecular Research Center, Cincinnati, Ohio) was added to samples, and RNA was extracted as per the manufacturer's procedures. Total RNA was quantified by spectrophotometry, and 4 μg was used for each assay. DNA templates for mouse cytokines and chemokines (PharMingen, San Diego, Calif.) were used to make 32P-labeled RNA probes. Probes were mixed with sample RNA, and a RibQuant kit (PharMingen) was used to perform RNase protection assays as per the manufacturer's procedures. Protected RNA fragments were resolved on a 5% polyacrylamide gel and quantified by densitometry. Densitometry was performed using image software from Eastman Kodak (Rochester, N.Y.). GAPDH (glyceraldehyde-3-phosphate dehydrogenase) controls were used to standardize the quantification of RNA samples.
T-cell isolation and stimulation.
Naive T cells were isolated from the splenocytes of the transgenic DO11.10 mice, which are on a BALB/c background. These cells express only the T-cell receptor for the ova peptide 323 to 339 (14). Cells were depleted of erythrocytes by ammonium chloride treatment, and then adherent cells were depleted for 2 h at 37°C. Cells were then passed over a T-cell enrichment column (R&D Systems, Minneapolis, Minn.) according to the manufacturer's instructions. The resulting purified cells were >99% T cells and were found to be >85% CD4-positive cells. All cells were found to be negative for the activation marker CD25 (data not shown). Antigen-presenting cells (APCs) were isolated from BALB/c splenocytes that were depleted of erythrocytes and irradiated at 2,500 rads (Gammacell 1000 cesium source; Nordion International, Ontario, Canada). Naive T cells at a concentration of 2.5 × 105 per ml were incubated with 2.5 × 106 irradiated APCs and 0.3 μM ova (peptide 323 to 339) in 24-well tissue culture plates for 3 days at 37°C in supplemented RPMI 1640 medium with 10% fetal bovine serum (Life Technologies, Grand Island, N.Y.). Cultures were then expanded threefold and incubated for an additional 3 days at 37°C, after which cells were washed with medium to remove any cytokine or 3O-C12-HSL previously added to cultures. Fresh medium with APCs and ova peptide was added, and cells were incubated for an additional 48 h. 3O-C12-HSL (5 μM) was added on days 0 and 3 of culture. As controls, the cytokine IL-12 (100 U), which is a potent inducer of T-cell gamma interferon (IFN-γ) (Th1 phenotype), or IL-4 (1,000 U), a potent inducer of T cells to produce IL-4 (Th2 phenotype), was added to separate cultures on days 0 and 3. Culture supernatants were harvested and analyzed in cytokine enzyme-linked immunosorbent assays (ELISAs).
Cytokine ELISAs.
The IFN-γ cytokine ELISA was performed as the manufacturer (Endogen, Woburn, Mass.) instructed, with the coating antibody (R4-6A2) at 1 μg/ml and the detecting antibody (XMG1.2) at 1 μg/ml. The limit of detection of the assay is 30 pg/ml. The IL-4 cytokine ELISA was performed as instructed by the manufacturer (Endogen), with the coating antibody (11B11) at 2 μg/ml and the detecting antibody (BVD6.24G2) at 1 μg/ml. The limit of detection of the assay is 20 pg/ml.
RT-PCR.
RNA was collected as described above. Reverse transcription-PCRs (RT-PCRs) for Cox-2 and Cox-1 were preformed as previously described (9).
Protein isolation and Western blotting.
Protein from skin samples was isolated from TriReagent extracts as per the manufacturer's instructions and quantified using a bicinchoninic acid kit (Pierce, Rockford, Ill.). Protein extracts (20 μg) were denatured by boiling for 5 min in a sodium dodecyl sulfate sample buffer. Samples were separated on sodium dodecyl sulfate-10% polyacrylamide gels and transferred to Hybond-C extra nitrocellulose membranes (Amersham, Piscataway, N.Y.). Membranes were blocked for 2 h at room temperature with 10% skim milk in PBS with 0.1% Tween 20. Primary anti-Cox-2 antibody (Cayman Chemical) was added at a 1:1,000 dilution in 5% skim milk in PBS-Tween and left overnight at 4°C. After each step the membranes were washed three times for 5 min each with PBS-Tween. Immunoreactive proteins were detected with a 1:2,000 dilution of anti-mouse immunoglobulin G-HRP (Santa Cruz Biotechnology, Santa Cruz, Calif.). Specific bands were visualized using an enhanced chemiluminescence kit (Amersham).
NF-κB nuclear mobilization.
Keratinocytes were isolated from the skin of C57BL/6 mice. Skin samples were treated with 0.5% trypsin at 37°C for 1 h. The epidermal layer was removed and treated with 0.05% DNase for 10 min at 37°C. Fetal bovine serum was added to give a final concentration of 5%, and the suspensions were gently passed through a 60-ml syringe and a 100-μm-pore-size nylon filter to dissociate cells. Cells were cultured in epithelial growth medium (Biowhittaker, Walkersville, Md.) in eight-well chamber slides (Nalge Nunc, Naperville, Ill.) at 104 cells/well. Cells were stimulated for 2 h with or without 100 μM 3O-C12-HSL in serum-free medium. Cells were then stained with anti-NF-κB p65 antibody at 2 μg/ml (Santa Cruz Biotechnology) as previously described (32).
RESULTS
Production of 3O-C12-HSL and C4-HSL is essential for colonization of the lung in adult mice.
The adult model of acute pneumonia, where anesthetized adult mice are nasally infected with P. aeruginosa, has been used to study the importance of various virulence factors in the pathogenesis of this organism (2). However, the role of quorum sensing in this model or any other acute lung infection models utilizing adult mice has not been examined. To determine if deletions in quorum-sensing genes would have an effect on P. aeruginosa pathogenesis in this model, we infected C57BL/6 mice with the wild-type P. aeruginosa strain PAO1 or the quorum-sensing mutant PAO-JP1 or PAO-JP2. PAO-JP1 contains a deletion in the lasI gene and produces no 3O-C12-HSL and only small amounts of C4-HSL; PAO-JP2 contains deletions in both the lasI and rhlI genes and therefore produces neither of the two major AHLs (21). After 24 h, mice were euthanized and total lung CFU were determined. At higher inocula of the wild-type strain PAO1 (1 × 106 and 1 × 107 CFU), the mice exhibited increasing signs of illness, such as lethargy and ruffled coats, while even at the highest inocula (∼2 × 107 CFU) of PAO-JP1 or PAO-JP2, mice appeared healthy. In all cases where equal numbers of CFU were inoculated into mice, a lower number of bacteria were isolated from the lungs of mice infected with either of the quorum-sensing mutants tested (Fig. 1). When mice were infected with 107 CFU, there was a 6-log-unit difference between the numbers of bacteria isolated from the lungs of PAO1- and PAO-JP1- or PAO-JP2-infected mice after 24 h. At this concentration it was observed that 100% of the mice infected with PAO1 had disseminated infections, evidenced by the presence of bacteria in the liver, while only 29% of the mice infected with PAO-JP2 and 33% of the mice infected with PAO-JP1 had bacteria in their livers. At an inoculum of 106 CFU, there was a 4-log-unit-smaller amount of bacteria isolated from PAO-JP1-infected mice and a 3-log-unit-smaller amount of bacteria isolated from the lungs of mice infected with PAO-JP2 compared to that isolated from mice infected with the wild type (Fig. 1). In addition, 85% of mice infected with 106 CFU of PAO1 were found to have disseminated infections, while none of the mice infected with 106 CFU of PAO-JP1 or PAO-JP2 had bacteria in their livers. These data demonstrate that the production of AHLs has a profound effect on the ability of P. aeruginosa to cause an acute lung infection and disseminate into the bloodstream in adult mice.
FIG. 1.

Quorum-sensing mutants PAO-JP1 and PAO-JP2 are attenuated in their ability to colonize mice in an adult mouse model of pneumonia. Data are expressed as the mean total CFU recovered from the lungs of mice 24 h after nasal infection. Inoculating doses varied two- to threefold between samples and experiments. Each bar is representative of results from at least four mice. Error bars indicate standard deviations.
3O-C12-HSL is a potent inducer of inflammation in the skin of mice.
The fact that a P. aeruginosa strain that is unable to produce AHLs was attenuated in its ability to colonize the lung does not directly address the effects that AHLs may have on the host. When AHLs were instilled into the lungs of mice, we found that 3O-C12-HSL induced inflammation, but these results were variable, possibly due to inconsistencies in the delivery process (data not shown). Hence, to begin to examine the effects of AHLs in vivo, we developed a dermal inflammation model where PBS or AHL was injected intradermally in mice and the injection site was examined 24 h later for inflammation. This model enabled us to reproducibly induce a localized effect that could be easily examined and excised. Hematoxylin and eosin staining of tissue samples from mice injected with PBS showed no signs of inflammation (Fig. 2). In contrast, samples from mice injected with 3O-C12-HSL showed a significant induction in inflammation. Hematoxylin and eosin staining revealed a significant infiltration of inflammatory cells, edema, and destruction of the epidermal layer (Fig. 2). With as little as 1 μM 3O-C12-HSL there was significant infiltration of inflammatory cells and tissue destruction. With increasing concentrations of 3O-C12-HSL, the inflammation (infiltrating cells and edema) was found deeper in the dermal and fatty layers of the skin, and the induced tissue destruction and amount of inflammatory infiltration were also more extensive. A higher magnification of the infiltrating population (insets in Fig. 2) shows that the cellular infiltrate is a mix of both polymorphonuclear and mononuclear cells. To determine if the structure of the AHL (i.e., length or presence of the acyl side chain) was important for the induction of inflammation, mice were injected with either C4-HSL or nonacylated homoserine lactone. When mice were injected with C4-HSL, a second AHL produced by P. aeruginosa, there was no inflammation induced with 1 or 10 μM C4-HSL and only a very modest induction with 100 μM C4-HSL. Dermal injections of 10 or 100 μM nonacylated homoserine lactone induced no inflammation (data not shown). These data indicate that the length and/or modifications of the acyl side chain are important for the induction of this inflammatory response.
FIG. 2.

3O-C12-HSL induces cellular infiltration and tissue destruction in mouse skin. Skin sections were stained with hematoxylin and eosin 24 h after dermal injection of PBS or 3O-C12-HSL. Insets show a ×800 magnification of the cells associated with the inflamed area. Arrows indicate both polymorphonuclear and mononuclear cells.
3O-C12-HSL induces production of chemokines and cytokines.
To determine how 3O-C12-HSL was inducing inflammation in the skin of mice, tissue samples were examined for the presence of inflammatory chemokines and cytokines. RNA was extracted from skin samples of mice injected with PBS or 10 μM 3O-C12-HSL, and a multiprobe RNase protection assay was used to determine the chemokines and cytokines being induced. With standardization to GAPDH controls, a five- to sixfold induction was found with the chemokines macrophage inflammatory protein-1β (MIP-1β), MIP-2, and monocyte chemotactic protein-1 (MCP-1). A three- to fourfold increase in mRNA levels was found for the chemokines inducible protein 10 (IP-10) and T-cell activation gene 3 (TCA-3) (Fig. 3A). MIP-2 is produced by activated epithelial cells, fibroblasts, macrophages, and T cells and is a potent chemotactic factor for neutrophils. Induction of MIP-2 correlates with the histology data, which show a significant infiltration of polymorphonuclear cells to the site of 3O-C12-HSL injections. MIP-1β and MCP-1 are mainly chemotactic for monocytes and T cells, and induction of these mRNAs is consistent with the migration of mononuclear cells observed in 3O-C12-HSL stimulated skin samples (Fig 2). IP-10 is able to induce the chemotaxis of naive T cells, B lymphocytes, and NK cells, while TCA-3 has been shown to be chemotactic for neutrophils, monocytes, and T cells.
FIG. 3.

3O-C12-HSL is a potent inducer of proinflammatory cytokines and chemokines in vivo. RNA was extracted from the skin of mice injected with PBS or 10 μM 3O-C12-HSL and was used in RNase protection assays to identify induction of inflammatory chemokines (A) or cytokines (B). Induction was quantified by densitometry and standardized to internal GAPDH controls. Graphs show the mean fold induction compared to PBS controls. Each bar is the mean from samples from four separate mice. Error bars indicate standard deviations.
A three- to fourfold induction in the mRNA levels for proinflammatory cytokines IL-1α and IL-6 was found with 3O-C12-HSL stimulation in vivo (Fig. 3B). IL-6 is a potent activator of lymphocytes and results in an increase in antibody production and induction of an acute-phase response. IL-1α activates macrophages to produce additional inflammatory cytokines and stimulates Cox and prostaglandin production from multiple cell types. These data demonstrate that 3O-C12-HSL is a potent inducer of both inflammatory cytokine and chemokine genes in vivo.
3O-C12-HSL induces an inflammatory Th1 phenotype in T cells.
Activated CD4+ T cells can be classified into two subgroups, Th1 and Th2. These groups are characterized by the cytokine profiles produced from the different populations (13). Th1 cells typically produce the cytokine IFN-γ, while Th2 cells produce the cytokines IL-4 and IL-13. The production of IFN-γ by Th1 cells leads to the activation of macrophages, while IL-4 from Th2 cells is important for the activation of B cells (1). Having demonstrated that 3O-C12-HSL is a potent inducer of chemokines that are known to stimulate the migration of both activated and naive T cells, we wanted to determine what effects, if any, 3O-C12-HSL would have on these cells. When naive T cells were activated with antigen and APCs and cultured with 5 μM 3O-C12-HSL, there was a significant induction in the production of the inflammatory cytokine IFN-γ (Fig. 4). The levels of IFN-γ produced with 3O-C12-HSL stimulation, although not as substantial, were similar to those induced by the cytokine IL-12, a potent stimulator of IFN-γ production in T cells. 3O-C12-HSL was not able to stimulate T cells to produce IL-4, a cytokine produced by Th2 cells. These data indicate that 3O-C12-HSL is a potent activator of T cells and is able to induce the inflammatory Th1 phenotype.
FIG. 4.

3O-C12-HSL stimulation of T cells induces Th1-type cytokines but not Th2. Purified T cells were activated with ova peptide and APCs and stimulated with IL-12 (100 U), IL-4 (1,000 U), or 3O-C12-HSL (5 μM). Data are expressed as the percentage of cytokine produced in comparison to the control (cultures containing only T cells, ova peptide, and APCs). Error bars indicate standard deviations.
3O-C12-HSL induces production of Cox-2.
Having demonstrated that 3O-C12-HSL is a potent inducer of inflammatory chemokines and cytokines; we wanted to determine if it was also inducing additional inflammatory mediators in vivo. Cox enzymes are essential in the conversion of arachidonic acid to prostaglandins. Prostaglandins are involved in both normal and pathophysiological processes, such as inflammation, edema, platelet aggregation, fever, and hyperalgesia. There are two known isoforms of Cox enzymes, Cox-1 and Cox-2. Cox-1 is constitutively expressed in most cells and is thought to be important for normal physiological functions. However, Cox-2 is usually expressed at low basal levels and is rapidly induced with inflammatory stimulation. Cox-2 is classified as an immediate-early gene product due to the rapid induction of mRNA after stimulation (33). Using RT-PCR to screen for Cox mRNA, it was determined that skin samples from mice injected with PBS expressed no mRNA for Cox-2 but did have the constitutively expressed Cox-1 mRNA (Fig. 5A). In contrast, samples from mice injected with 10 μM 3O-C12-HSL contained high levels of both Cox-1 and Cox-2 mRNAs (Fig. 5A). Protein extracts from the skin of mice injected with PBS showed low basal levels of Cox-2 protein by Western blotting. When mice were injected with 10 or 100 μM 3O-C12-HSL, there was a significant induction in Cox-2 protein (9- and 17-fold, respectively, over that of PBS samples) (Fig. 5B). Using immunohistochemistry to stain for Cox-2 in the tissue, it was found that injections of PBS did not induce expression of Cox-2, but 10 μM 3O-C12-HSL stimulated a significant induction (Fig. 6). This staining showed that Cox-2 localized to the cells associated with the inflammatory infiltrate as well as isolated cells, such as fibroblasts, lower in the dermis. These data demonstrate that 3O-C12-HSL is a potent inducer of Cox-2 expression in vivo.
FIG. 5.

3O-C12-HSL induces Cox-2 mRNA and protein in the skin of mice. RNA and protein were extracted from the skin of mice injected with PBS or 3O-C12-HSL. (A) Agarose gel showing ethidium bromide-stained RT-PCR products specific for Cox-1 and Cox-2. Lanes 1 and 2, samples from two separate mice injected with PBS; lanes 3 and 4, samples from two mice injected with 10 μM 3O-C12-HSL. (B) Proteins from the skin of mice injected with PBS or 3O-C12-HSL were evaluated in a Western blot specific for Cox-2. This is a representative blot from four separate experiments.
FIG. 6.

Cox-2 expression is induced in skin after 3O-C12-HSL stimulation. Mice were given dermal injections of PBS or 10 μM 3O-C12-HSL, and after 24 h skin sections were stained with an anti-Cox-2 antibody. As a control for background staining, a blocking peptide (BP) was added to the antibody prior to incubation with samples. The red precipitate in the sections shows the expression of Cox-2 in the samples. Samples were counterstained with hematoxylin. Arrows indicate areas of staining.
3O-C12-HSL stimulates NF-κB mobilization in mouse keratinocytes.
Our laboratory previously demonstrated that 3O-C12-HSL is a potent activator of the transcription factor NF-κB. 3O-C12-HSL induction of NF-κB was found to regulate the production of the chemokine IL-8 (32). The chemokines MIP-2, MIP-1β, IP-10, and MCP-1, which are induced by 3O-C12-HSL in vivo, have NF-κB binding sites in their promoter regions (12, 16, 25, 37). To determine if 3O-C12-HSL is activating inflammation in the skin through the induction of NF-κB, keratinocytes from mouse skin were purified and stained with an anti-NF-κB antibody to visualize the localization of NF-κB in the cell. Without any stimulation NF-κB remained sequestered in an inactive form in the cytoplasm. It was only when cells were stimulated with 3O-C12-HSL that NF-κB was able to translocate into the nucleus, where it could then potentially induce the transcription of multiple genes (Fig. 7). These data support the hypothesis that the inflammation stimulated by 3O-C12-HSL is mediated through the induction of NF-κB.
FIG. 7.
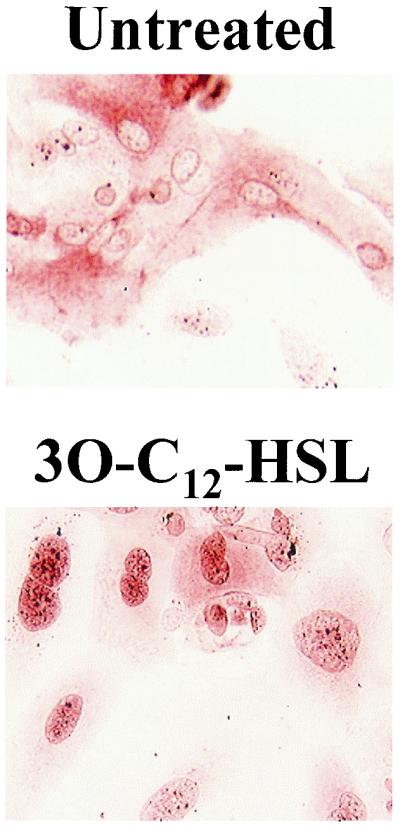
3O-C12-HSL stimulates nuclear mobilization of NF-κB in mouse keratinocytes. Keratinocytes were isolated from the skin of C57BL/6 mice and were cultured with or without 50 μM 3O-C12-HSL. Cells were stained with an anti-NF-κB p65 antibody. The red precipitate indicates the localization of NF-κB staining.
DISCUSSION
Quorum sensing is a mechanism whereby bacteria are able to sense the environment and, as a population, regulate the expression of various genes. In several gram-negative and gram-positive bacteria this mechanism is used to regulate expression of genes important for the virulence of the organism. Therefore, the bacteria in a joint effort are able to subvert the host defenses and cause infection. In P. aeruginosa, quorum sensing regulates expression of numerous virulence genes that are thought to be important for the pathogenesis of the organism. Several models of P. aeruginosa infection have been used to examine the role of quorum sensing in the pathogenesis of the organism. In all cases a deletion of one or more of the components in the quorum-sensing systems led to reduction in the infection elicited by P. aeruginosa (18, 26, 35, 38). In adult humans with deficiencies in their normal immune functions (e.g., human immunodeficiency virus, cancer, and ventilator patients), P. aeruginosa frequently causes acute lung infections; therefore, it was important that these earlier animal studies examining quorum-sensing mutants be extended to include an adult model of acute lung infection. In this report we demonstrate that in an adult mouse acute-pneumonia model, PAO-JP2 and PAO-JP1 are attenuated in their ability to colonize the lung compared to wild-type PAO1 (Fig. 1). Studies evaluating this model showed that anesthetized adult mice were able to rapidly aspirate into the lungs 67 to 100% of the bacteria applied to the nostrils (2). Therefore, we found that this model was very sensitive and allowed us to see differences between wild-type PAO1 and quorum-sensing mutants with relatively low numbers of bacteria (∼2 × 106 CFU). It has also been shown that PAO1 has a 50% lethal dose of 3 × 107 CFU in this model (2). Although we did not calculate the 50% lethal dose with our strains, an inoculum of 2 × 108 CFU of PAO-JP2 did not kill mice after 24 h (data not shown). These data, along with those from other animal studies, emphasize the importance of complete quorum-sensing systems and the production of AHLs for the pathogenesis of P. aeruginosa.
There are several studies demonstrating that 3O-C12-HSL interacts in vitro with human and mouse cells and stimulates an immune response (5, 27, 32, 36). The data reported here are the first demonstration that 3O-C12-HSL directly stimulates an inflammatory response in vivo. Although 10 to 100 μM 3O-C12-HSL was needed to stimulate cells in vitro, we have shown that a concentration of 1 μM was sufficient to induce significant inflammation in the skin of mice (Fig. 2). This concentration is well below what can be found in planktonic cultures of P. aeruginosa (≈5 to 10 μM). A closer examination of the induced inflammation revealed that 3O-C12-HSL was stimulating the production of mRNAs for multiple inflammatory chemokines and cytokines (Fig. 3). During chronic P. aeruginosa infections in CF patients, it is the exuberant induction of the chemokine IL-8 and the subsequent infiltration of neutrophils that are thought to be the major source of tissue destruction associated with this infection (28). Therefore, the demonstration that 3O-C12-HSL significantly induces the neutrophil chemokine MIP-2 in vivo gives additional evidence that 3O-C12-HSL is a potent activator of neutrophil migration during P. aeruginosa infections. Also, the demonstration that 3O-C12-HSL induces multiple chemokines known to stimulate the migration of neutrophils, monocytes, and T cells to the site of infection indicates that 3O-C12-HSL stimulates a broad range of inflammation that includes several types of cells involved in inflammation.
We have also demonstrated that 3O-C12-HSL is a potent inducer of IFN-γ production in T cells (Fig. 4). Production of IFN-γ induces an inflammatory phenotype by activating macrophages, which leads to enhanced bacterial killing and the production of inflammatory cytokines. It was previously shown that 3O-C12-HSL inhibits the production of IL-12 from activated monocytes (36). Since IL-12 is a potent stimulator of T cells to produce IFN-γ, it was hypothesized that its inhibition by 3O-C12-HSL would lead to a decrease in T cells with the Th1 phenotype and therefore induce a Th2 phenotype. Our data demonstrate that 3O-C12-HSL directly interacts with T cells to produce IFN-γ in the absence of IL-12 production (Fig. 4). Therefore, we propose that 3O-C12-HSL is able to modulate the immune response during P. aeruginosa infection by stimulating the inflammatory Th1 phenotype and not a Th2 response as previously hypothesized.
We also demonstrate that 3O-C12-HSL is a potent inducer of Cox-2. Expression of Cox-2 is associated with the induction of inflammation, fever, and pain. One of the major prostaglandins produced by Cox-2 is prostaglandin E2. In burn wounds, it has been shown that infection with Pseudomonas led to a significant induction in Cox-2 expression and prostaglandin E2 production. This induction was directly associated with the ability of the organism to disseminate and cause a septic infection (10, 30). In this same model, PAO-JP2, which produces neither of the two major AHLs, had a greatly reduced ability to spread at the site of infection and, more importantly, to disseminate throughout the body and cause septicemia and death (26). Thus, the ability of wild-type P. aeruginosa to disseminate and cause septicemia may be associated with the production of 3O-C12-HSL and its ability to induce Cox-2 expression. Therefore, new Cox-2-selective inhibitory drugs currently in use may have significant pharmacological effects in the control of Pseudomonas infections by inhibiting 3O-C12-HSL-induced inflammation.
Our data demonstrate that 3O-C12-HSL not only is essential in the regulation of multiple virulence factors but also acts as a virulence factor itself by stimulating an inflammatory response in the host. In immunocompetent individuals the induction of inflammation by 3O-C12-HSL in vivo would be beneficial for helping to clear the infection. However, in patients who have defects in their normal defense mechanisms, the stimulation of an inflammatory response may be less beneficial. Since AHLs are cell density-regulated molecules, Pseudomonas may have already established a prominent infection prior to the production of significant amounts of 3O-C12-HSL. Therefore, the induction of a potent inflammatory response may be nonproductive in its ability to clear the infection and would instead lead to significant tissue destruction. In CF patients who have chronic Pseudomonas infections, there is stimulation of a significant neutrophil infiltration into the lungs, but the infection is not cleared. Instead, there is extensive tissue destruction due to the overzealous activation of the immune response. Therefore, regulation of the inflammation induced by 3O-C12-HSL may be essential in reducing the amount of tissue destruction found during such chronic P. aeruginosa infections. Having an understanding of the effects of various components of P. aeruginosa on the host is crucial in determining the optimal treatment regimens. Our data show that the inhibition of the production of 3O-C12-HSL or its activation of inflammation would be excellent targets for therapeutic development.
Acknowledgments
This work was supported by Public Health Service grants AI33713, HL07216, HL56002, DE11390, ES01247, and T32 HL66988 and by Cystic Fibrosis Foundation grant 00Z0.
REFERENCES
- 1.Allen, J. E., and R. M. Maizels. 1997. Th1-Th2: reliable paradigm or dangerous dogma? Immunol. Today 18:387-392. [DOI] [PubMed] [Google Scholar]
- 2.Allewelt, M., F. T. Coleman, M. Grout, G. P. Priebe, and G. B. Pier. 2000. Acquisition of expression of the Pseudomonas aeruginosa ExoU cytotoxin leads to increased bacterial virulence in a murine model of acute pneumonia and systemic spread. Infect. Immun. 68:3998-4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davies, D. G., M. R. Parsek, J. P. Pearson, B. H. Iglewski, J. W. Costerton, and E. P. Greenberg. 1998. The involvement of cell-to-cell signals in the development of a bacterial biofilm. Science 280:295-298. [DOI] [PubMed] [Google Scholar]
- 4.de Kievit, T. R., and B. H. Iglewski. 2000. Bacterial quorum sensing in pathogenic relationships. Infect. Immun. 68:4839-4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DiMango, E., H. J. Zar, R. Bryan, and A. Prince. 1995. Diverse Pseudomonas aeruginosa gene products stimulate respiratory epithelial cells to produce interleukin-8. J. Clin. Invest. 96:2204-2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gambello, M. J., and B. H. Iglewski. 1991. Cloning and characterization of the Pseudomonas aeruginosa lasR gene, a transcriptional activator of elastase expression. J. Bacteriol. 173:3000-3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gambello, M. J., S. Kaye, and B. H. Iglewski. 1993. LasR of Pseudomonas aeruginosa is a transcriptional activator of the alkaline protease gene (apr) and an enhancer of exotoxin A expression. Infect. Immun. 61:1180-1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glessner, A., R. S. Smith, B. H. Iglewski, and J. B. Robinson. 1999. Roles of Pseudomonas aeruginosa las and rhl quorum-sensing systems in control of twitching motility. J. Bacteriol. 181:1623-1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graf, B. A., D. A. Nazarenko, M. A. Borrello, L. J. Roberts, J. D. Morrow, J. Palis, and R. P. Phipps. 1999. Biphenotypic B/macrophage cells express COX-1 and up-regulate COX-2 expression and prostaglandin E2 production in response to pro-inflammatory signals. Eur J. Immunol. 29:3793-3803. [DOI] [PubMed] [Google Scholar]
- 10.Hahn, E. L., H. H. Tai, L. K. He, and R. L. Gamelli. 1999. Burn injury with infection alters prostaglandin E2 synthesis and metabolism. J. Trauma 47:1052-1057. [DOI] [PubMed] [Google Scholar]
- 11.Latifi, A., M. Foglino, K. Tanaka, P. Williams, and A. Lazdunski. 1996. A hierarchical quorum-sensing cascade in Pseudomonas aeruginosa links the transcriptional activators LasR and RhIR (VsmR) to expression of the stationary-phase sigma factor RpoS. Mol. Microbiol. 21:1137-1146. [DOI] [PubMed] [Google Scholar]
- 12.Mori, N., A. Ueda, R. Geleziunas, A. Wada, T. Hirayama, T. Yoshimura, and N. Yamamoto. 2001. Induction of monocyte chemoattractant protein 1 by Helicobacter pylori involves NF-κB. Infect. Immun. 69:1280-1286. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Mosmann, T. R., H. Cherwinski, M. W. Bond, M. A. Giedlin, and R. L. Coffman. 1986. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 136:2348-2357. [PubMed] [Google Scholar]
- 14.Murphy, K. M., A. B. Heimberger, and D. Y. Loh. 1990. Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science 250:1720-1723. [DOI] [PubMed] [Google Scholar]
- 15.Ochsner, U. A., and J. Reiser. 1995. Autoinducer-mediated regulation of rhamnolipid biosurfactant synthesis in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 92:6424-6428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohmori, Y., and T. A. Hamilton. 1995. The interferon-stimulated response element and a kappa B site mediate synergistic induction of murine IP-10 gene transcription by IFN-gamma and TNF-alpha. J. Immunol. 154:5235-5244. [PubMed] [Google Scholar]
- 17.Passador, L., J. M. Cook, M. J. Gambello, L. Rust, and B. H. Iglewski. 1993. Expression of Pseudomonas aeruginosa virulence genes requires cell-to-cell communication. Science 260:1127-1130. [DOI] [PubMed] [Google Scholar]
- 18.Pearson, J. P., M. Feldman, B. H. Iglewski, and A. Prince. 2000. Pseudomonas aeruginosa cell-to-cell signaling is required for virulence in a model of acute pulmonary infection. Infect. Immun. 68:4331-4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pearson, J. P., K. M. Gray, L. Passador, K. D. Tucker, A. Eberhard, B. H. Iglewski, and E. P. Greenberg. 1994. Structure of the autoinducer required for expression of Pseudomonas aeruginosa virulence genes. Proc. Natl. Acad. Sci. USA 91:197-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pearson, J. P., L. Passador, B. H. Iglewski, and E. P. Greenberg. 1995. A second N-acylhomoserine lactone signal produced by Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 92:1490-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pearson, J. P., E. C. Pesci, and B. H. Iglewski. 1997. Roles of Pseudomonas aeruginosa las and rhl quorum-sensing systems in control of elastase and rhamnolipid biosynthesis genes. J. Bacteriol. 179:5756-5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pearson, J. P., C. Van Delden, and B. H. Iglewski. 1999. Active efflux and diffusion are involved in transport of Pseudomonas aeruginosa cell-to-cell signals. J. Bacteriol. 181:1203-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pesci, E. C., J. B. Milbank, J. P. Pearson, S. McKnight, A. S. Kende, E. P. Greenberg, and B. H. Iglewski. 1999. Quinolone signaling in the cell-to-cell communication system of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 96:11229-11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pesci, E. C., J. P. Pearson, P. C. Seed, and B. H. Iglewski. 1997. Regulation of las and rhl quorum sensing in Pseudomonas aeruginosa. J. Bacteriol. 179:3127-3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rezzonico, R., V. Imbert, R. Chicheportiche, and J. M. Dayer. 2001. Ligation of CD11b and CD11c beta(2) integrins by antibodies or soluble CD23 induces macrophage inflammatory protein 1alpha (MIP-1alpha) and MIP-1beta production in primary human monocytes through a pathway dependent on nuclear factor-kappaB. Blood 97:2932-2940. [DOI] [PubMed] [Google Scholar]
- 26.Rumbaugh, K. P., J. A. Griswold, B. H. Iglewski, and A. N. Hamood. 1999. Contribution of quorum sensing to the virulence of Pseudomonas aeruginosa in burn wound infections. Infect. Immun. 67:5854-5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saleh, A., C. Figarella, W. Kammouni, S. Marchand-Pinatel, A. Lazdunski, A. Tubul, P. Brun, and M. D. Merten. 1999. Pseudomonas aeruginosa quorum-sensing signal molecule N-(3-oxododecanoyl)-l-homoserine lactone inhibits expression of P2Y receptors in cystic fibrosis tracheal gland cells. Infect. Immun. 67:5076-5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schuster, A., A. Haarmann, and V. Wahn. 1995. Cytokines in neutrophil-dominated airway inflammation in patients with cystic fibrosis. Eur. Arch. Otorhinolaryngol. Suppl. 1:S59-S60. [DOI] [PubMed]
- 29.Seed, P. C., L. Passador, and B. H. Iglewski. 1995. Activation of the Pseudomonas aeruginosa lasI gene by LasR and the Pseudomonas autoinducer PAI: an autoinduction regulatory hierarchy. J. Bacteriol. 177:654-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shoup, M., L. K. He, H. Liu, R. Shankar, and R. Gamelli. 1998. Cyclooxygenase-2 inhibitor NS-398 improves survival and restores leukocyte counts in burn infection. J. Trauma 45:215-220. [DOI] [PubMed] [Google Scholar]
- 31.Singh, P. K., A. L. Schaefer, M. R. Parsek, T. O. Moninger, M. J. Welsh, and E. P. Greenberg. 2000. Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature 407:762-764. [DOI] [PubMed] [Google Scholar]
- 32.Smith, R. S., E. R. Fedyk, T. A. Springer, N. Mukaida, B. H. Iglewski, and R. P. Phipps. 2001. IL-8 production in human lung fibroblasts and epithelial cells activated by the Pseudomonas autoinducer N-3-oxododecanoyl homoserine lactone is transcriptionally regulated by NF-kappaB and activator protein-2. J. Immunol. 167:366-374. [DOI] [PubMed] [Google Scholar]
- 33.Smith, W. L., R. M. Garavito, and D. L. DeWitt. 1996. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J. Biol. Chem. 271:33157-33160. [DOI] [PubMed] [Google Scholar]
- 34.Storey, D. G., E. E. Ujack, H. R. Rabin, and I. Mitchell. 1998. Pseudomonas aeruginosa lasR transcription correlates with the transcription of lasA, lasB, and toxA in chronic lung infections associated with cystic fibrosis. Infect. Immun. 66:2521-2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang, H. B., E. DiMango, R. Bryan, M. Gambello, B. H. Iglewski, J. B. Goldberg, and A. Prince. 1996. Contribution of specific Pseudomonas aeruginosa virulence factors to pathogenesis of pneumonia in a neonatal mouse model of infection. Infect. Immun. 64:37-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Telford, G., D. Wheeler, P. Williams, P. T. Tomkins, P. Appleby, H. Sewell, G. S. Stewart, B. W. Bycroft, and D. I. Pritchard. 1998. The Pseudomonas aeruginosa quorum-sensing signal molecule N-(3-oxododecanoyl)-l-homoserine lactone has immunomodulatory activity. Infect. Immun. 66:36-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Widmer, U., K. R. Manogue, A. Cerami, and B. Sherry. 1993. Genomic cloning and promoter analysis of macrophage inflammatory protein (MIP)-2, MIP-1 alpha, and MIP-1 beta, members of the chemokine superfamily of proinflammatory cytokines. J. Immunol. 150:4996-5012. [PubMed] [Google Scholar]
- 38.Wu, H., Z. Song, M. Givskov, G. Doring, D. Worlitzsch, K. Mathee, J. Rygaard, and N. Hoiby. 2001. Pseudomonas aeruginosa mutations in lasI and rhlI quorum sensing systems result in milder chronic lung infection. Microbiology 147:1105-1113. [DOI] [PubMed] [Google Scholar]