

Abstract
Cigarette-induced endothelial dysfunction could be an early mediator of atherosclerosis. In this study, we explored the mechanisms of cigarette smoke extract (CSE)-induced human aortic endothelial cells (HAEC) apoptosis. We found that 10–65% of HAECs underwent apoptotic changes when HAECs were exposed to 0.001–0.02 cigarette equivalent unit of CSE for 4 h. CSE activated the caspases-3 and 8, the p38 MAP kinase and stress activated protein kinase/c-Jun N-terminal protein kinase (SAPK/JNK). Specific inhibitors of p38 MAP or SAPK/JNK reduced CSE-induced caspase activation. We further showed that eNOS pre-activation by l-arginine reduced endothelial apoptosis from 65% to 5%; and eNOS inhibition by N-ω-nitro-l-arginine methyl ester accentuated CSE-induced endothelial apoptosis. We suggest that appropriate endogenous NO production may be an important protective mechanism against smoking-induced endothelial damage.
Keywords: Caspase, Cigarette smoke extract, eNOS, Nitric oxide, Apoptosis
1. Introduction
Cigarette smoke exposure has been implicated as a major risk factor for atherosclerosis and other vascular diseases. The mechanisms are not, however, well understood [1]. Endothelium, as the first line of defense in the vessel wall [2], is the primary target of many atherogenic risk factors including hypercholesterolemia, inflammation, and cigarette smoking. Numerous studies have demonstrated that cigarette smokers have dysfunctional endothelium, which can potentially lead to initiation and progression of atherosclerosis, acute rupture of existing atherosclerotic lesions, and increased thrombosis [2–6].
Among various dysfunctional changes in endothelial cells, uncontrolled apoptosis plays a significant role in atherogenesis and thrombogenesis. Apoptosis is a programmed cell death by which cells maintain homeostasis through a balance between cell proliferation and cell death. Aberration of this balance contributes to the pathogenesis of many diseases. Apoptosis can be modulated by several signaling molecules including nitric oxide (NO). NO is a short lived free radical with a wide spectrum of physiological and pathological functions [7]. NO likely exerts its physiological function mainly through cGMP signaling pathways. Recently, various other pathways have been found relevant. NO interacts with the sulfhydryl groups of proteins and forms S-nitrosothiols (S-NO) via S-nitrosylation of cysteine, which result in functional modification of cellular proteins such as cathepsin B, aldolase and aldehyde dehydrogenase [8]. NO-mediated S-nitrosylation of the cysteine-containing proteins, including caspases and tissue trans-glutaminase, modulates apoptotic cell death [9,10]. NO contributes to the physiologic balance of apoptosis between pro-apoptotic and anti-apoptotic shift in the cells by modulating signaling molecules in apoptotic pathway [11]. NO may protect cells from free radical-mediated cytotoxicity by scavenging superoxide anions, reducing toxic ferryl species to ferrous ion, thereby blocking the hemoprotein-mediated Fenton-like reaction, and also by terminating the propagation of radical-mediated lipid peroxidation [12,13].
Recently, we and others have shown that cigarette smoke can induce excessive apoptosis in endothelial cells [14] and other cell types [15,16]. Our earlier studies showed that physiological NO may inhibit TNFα-induced endothelial apoptosis via the cGMP pathway [17]. We hypothesize that endothelial derived NO may have a protective role in cigarette smoke extract (CSE)-induced endothelial apoptosis. To explore the hypothesis, we used CSE, which has both water-soluble gas and particulate phase contents of cigarette smoking [16,18,19], to induce apoptotic changes in human aortic endothelial cells (HAECs). We found that CSE activated p38 MAP kinase and c-Jun N-terminal protein kinase/stress activated protein kinase (JNK/SAPK). Pre-activation of eNOS by l-arginine attenuated CSE-induced apoptosis and inhibited stress pathway. Pretreatment with N-ω-nitro-l-arginine methyl ester (l-NAME) further confirmed the involvement of NO and eNOS on CSE-induced apoptosis in HAECs.
2. Materials and methods
2.1. Cells and chemicals
HAECs were purchased from Cell Applications, Inc. (San Diego, CA). p38 MAP kinase inhibitor SB 202190 and JNK/SAPK inhibitor SP600125 were purchased from Calbiochem (San Diego, CA). HAECs were cultured in F-12K media (ATCC, Manassas, VA) containing bovine endothelial cell growth supplement (45 μg/mL), heparin (100 μg/ mL, Acros, NJ), penicillin (50 IU/L), streptomycin (50 μg/L, Cellgro, Herndon, VA) and 20% fetal calf serum (Invitrogen Corporation, Carlsbad, CA), in a 5% CO2/air atmosphere. Cells within 3–8 passages were used for the experiments at ~90% confluence.
2.2. Preparation of CSE
CSE was prepared as described previously [18–20]. Briefly, the smoke of a research cigarette (2R4F, from Tobacco Health Research, University of Kentucky, contains nicotine: 0.85 mg/cigarette and tar: 9.70 mg/cigarette) was bubbled into a flask containing 20 mL of pre-warmed F-12K complete medium. The ignited cigarette was completely consumed in puffs by 10 mL syringe suction at one end of the tube within 5 min. The aqueous smoke extract was filtered through 0.2 μm syringe filter and the pH of CSE was adjusted to 7.4. Since there is no standardized way to express the quantity of CSE, the concentration of CSE was calculated in arbitrary units as cigarette equivalents per milliliter of the medium. One cigarette smoked into 20 mL medium yields one unit cigarette (U). Therefore, one milliliter of prepared medium contains 0.05 U CSE.
2.3. Apoptosis detection
HAECs were cultured on gelatin-coated petri dishes (1 × 106 cells) and treated with CSE. At the end of the experimental period, floating and adherent cells were collected and centrifuged for 5 min at 1000 × g. The cells with apoptotic changes were measured by In Situ Cell Death Detection Kit, Fluorescein (Roche, Indianapolis, IN), which detects DNA fragmentation during apoptosis using a FACScan Becton Dickinson Instrument and the Cellquest software (Franklin Lakes, NJ). The degree of apoptosis was determined by the percentage of apoptotic cells over total cells. Endothelial apoptosis was also confirmed by fluorescence microscopy using Annexin-V-Fluos (Roche) labeling, which detects cell membrane changes during early apoptosis [21].
2.4. Caspase-3 activity
Endothelial cell lysates were used for cellular caspase-3 activity measurements using a Caspase-3 Cellular Activity Assay Kit Plus (Cat#-AK-703, Biomol, Plymouth Meeting, PA). The kit measures protease activity of caspase-3 by the cleavage of a tetrapeptide substrate (Ac-DEVD-pNA), which is monitored colorimetrically by increased absorption at 405 nm. The caspase-3 activity was expressed as pmol/ min/μg of total protein.
2.5. Western-blot analysis for eNOS protein and apoptosis related proteins
HAECs were seeded in six-well plates and grew to 90% confluency. On the next day, the cells were treated with different doses of CSE (0–0.025 U) for 24 h. Each well of cells was lysed into 200 μl of lysis buffer containing 20 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerol phosphate, 1 mM Na3VO4, 10 μg/ml of each protease inhibitors aprotinin, leupeptin and pepstatin, and 1 mM phenylmethylsulfonyl fluoride. Twenty-five micrograms of protein was loaded to each lane and separated by 8% SDS–polyacrylamide gel and transferred to nitrocellulose membrane. The membrane was blocked with 5% non-fat powdered milk in TBST (50 mM Tris, pH 7.5, 150 mM NaCl, and 0.05% Tween 20), and then incubated with the polyclonal anti-phospho-eNOS (Ser1177) and anti-eNOS antibody (Cell Signaling Technology, Beverly, MA. 1:1000 dilution) overnight at 4 °C. The membrane was washed with PBST and incubated with anti-rabbit antibody horseradish peroxidase conjugate (Amersham Biosciences, Piscataway, NJ). Actin was detected using anti-human β-actin antibody in the same membrane after the primary detecting antibody was stripped as described previously [22]. Bands were visualized with ECL (Amersham Biosciences).
To detect apoptosis related protein, HAECs were treated with 0.02 U CSE for 4 h. Total proteins were isolated as described above. Twenty-five micrograms of protein was separated by 12% SDS–PAGE. The membrane was incubated with the primary antibodies separately, including polyclonal cleaved caspase-3, monoclonal cleaved caspase-8, polyclonal p38 MAP kinase, polyclonal phospho-p38 MAP kinase, polyclonal SAPK/JNK and polyclonal phospho-SAPK/JNK (Cell Signaling Technology). After incubation with primary antibodies, membranes will be washed three times before incubated with secondary anti-rabbit or anti-mouse horseradish peroxidase-labeled antibody (Amersham Biosciences), and processed for ECL detection.
2.6. RNA isolation and real-time RT-PCR for eNOS mRNA analysis
HAECs were treated with CSE as described above. Total RNA was isolated using TRIzol reagent (Gibco-BRL Life Technologies). One microgram of total RNA was used for the synthesis of cDNA by iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA). Real-time PCR was performed using iQ SYBR Green Supermix Kit (BioRad) following the manufacturer’s instructions. All quantitative gene expression by real-time RT-PCR was evaluated using the comparative threshold cycle (CT) method and was normalized against the endogenous control β-actin. The PCR primer sequences are: eNOS forward: AGGAACCTGTGTGACCCTCA, eNOS reverse: CGAG-GTGGTCCGGGTATCC. The primers for β-actin are: forward: CTGGAACGGTGAAGGTGACA, reverse: AAGGGACTTCCTG-TAACAATGCA. All samples were run in duplicate and repeated three times.
2.7. Determination of nitrite and nitrate (NOx) levels in the culture medium
At the end of each experimental period, the culture media were collected and the amount of NO metabolites (NOx) was determined by a modified Griess reaction [23]. Briefly, the nitrate in the culture media was first converted to nitrite by the action of NADPH-dependent nitrate reductase. The Griess reaction was initiated by the addition of 1% (w/v) sulfanilamide and 0.1% (w/v) N-[1-naphthyl]-ethylenediamine. The absorbance of the reaction mixture was read at 540 nm and the NOx levels were expressed as μM/μg of total cellular protein where the medium was collected for NOx levels.
2.8. S-nitrosothiols level
S-NO content was measured using Saville–Griess assay as described previously [24–26]. After treatment, HAECs were washed with PBS once, then lysed in 300 μl of Griess lysis buffer containing 50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 50 mM KCl, 1% NP40, 1 mM phenylmethysulfonyl fluoride, 1 mM bathocuproinedisulfonic acid, 1 mM diethylenetriaminepenta-acetic acid, and 10 mM N-ethylmaleimide. 80 μg of cell lysate was incubated with 1% sulfanilamide and 0.1% of N-[1-naphthyl]-ethylenediamine in the presence or absence of 3.75 mM of p-chloromercuribenzosulfonic acid at room temperature for 20 min. S-NO contents were measured absorbance at 540 nm. The amount was calculated using GSNO concentration as a standard.
2.9. Measurement of eNOS activity
After treatment, the cells were washed with ice-cold PBS and lysed in protein lysis buffer for 1 h on ice. Protein concentration was measured by the Bradford method and the cell lysates were used for the estimation of total eNOS activity as described previously [27]. The final result was expressed as cpm/μg of protein.
2.10. Statistical analysis
Data were expressed as means ± S.E.M. and analyzed by analysis of variance (ANOVA) for comparison among multiple groups. P values of <0.05 were considered significant. In the ANOVA analyses, we used the Dunnett’s post hoc test for P values when we compared the changes to the levels of no treatment (CSE = 0) as shown in Fig. 1; we used the Bonferroni’s correction for P values when we compared between group differences as shown in Figs. 6 and 7.
Fig. 1.
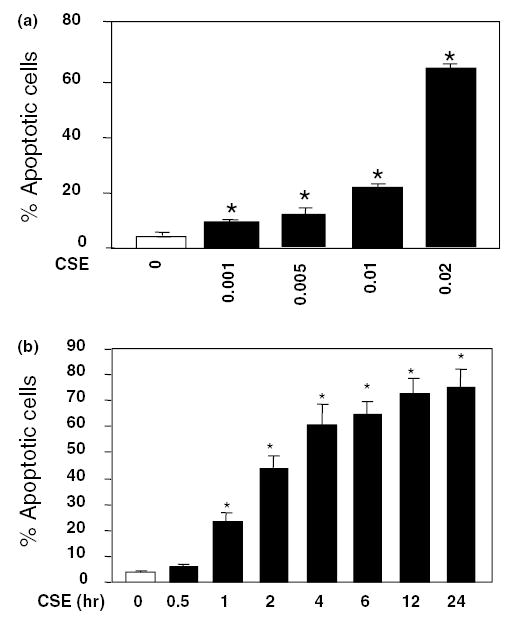
CSE induces apoptosis in HAECs. (a) Dose-dependent effects of CSE on endothelial apoptosis. Cells were treated with 0–0.02 U of CSE for 4 h and percentages of apoptotic cells (means + S.E.M., N = 3) were determined by FACS using TUNEL labeling. (b) Time course of HAEC apoptosis induced by 0.02 CSE from 0 to 24 h. The apoptotic rate increased rapidly during the first 4 h, and continued the increase till 24 h. Data are expressed as means + S.E.M. at each time point from three individual experiments. The among group differences in the percent of apoptotic cells were statistically significant (F = 762, P < 0.001). *P < 0.01 by the post hoc Dunnett’s test, when compared with control group in which endothelial cells were treated with the standard medium only.
Fig. 6.
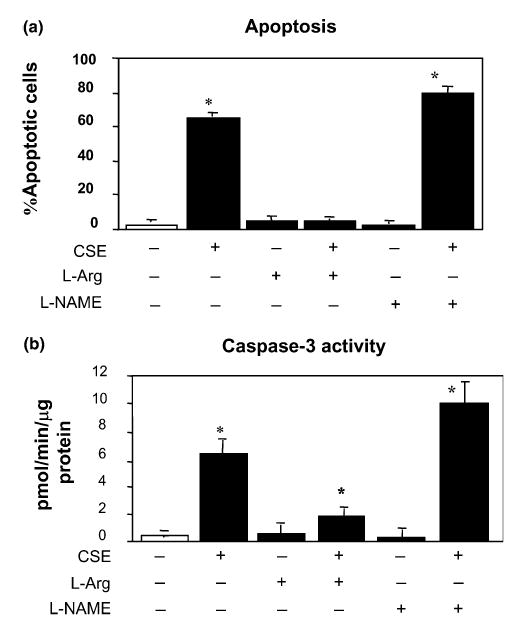
l-Arginine blocks the CSE-induced apoptosis in endothelial cells. HAECs were incubated with 200 μM of l-arginine (columns 3 and 4), or 400 μM of l-NAME (columns 5 and 6) for 30 min followed by 0.02 U CSE exposure for 4 h (columns 2, 4 and 6). l-Arginine or l-NAME pretreatment altered CSE-induced endothelial apoptosis (a) and caspase-3 activity (b). Data are presented as means + S.E.M. (N = 3) at each treatment condition. There was a significant among group difference by ANOVA test (P < 0.01). CSE treatment increased cell death significantly (P < 0.001 comparing to controls). However, l-arginine pretreatment reduced the cell death to the level not different from the control cells (P = NS). Yet, l-arginine treatment alone did not alter the rate of cell death. Although l-NAME treatment alone also did not change the cell death, l-NAME pre-conditioning followed by CSE treatment significantly increased the percentage of cell death comparing to CSE treatment alone (P < 0.05). Changes in caspase-3 activity were similar to the percentage of cell death except that the caspase-3 activity did not return to the control level in cells preconditioned with l-arginine and treated with CSE. *The post hoc Bonferroni correction was applied to the P values for between group comparisons.
Fig. 7.
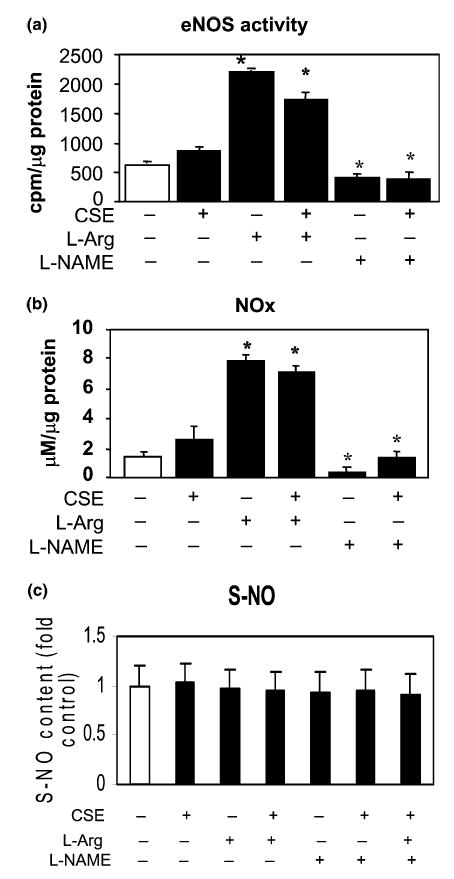
Changes in eNOS activity and levels of NOx and S-NO. Pre-incubating HAECs with l-arginine for 30 min significantly increased the eNOS activity (means + S.E.M., N = 3, P < 0.01, a) and NOx production (P < 0.01, b), but not S-NO levels (P = NS, c). Inhibition of eNOS by l-NAME pretreatment resulted in reduced eNOS activity (P < 0.01, a) and NOx production (P < 0.01, b) comparing to control cells, but not the S-NO level (P = NS, c). CSE treatment for 4 h changed very little in eNOS activity (P = NS, a) and NOx levels (P = NS, b), and no change in S-NO (c). CSE treatment also did not induce further changes in eNOS activity (a) and NOx levels (b) in cells pretreated with l-arginine, which were still significantly higher than those in controls (P < 0.01 for both). In cells pretreated with l-NAME, which reduced eNOS activity and NOx levels, further CSE treatment made no change to eNOS activity (a) but increased the culture medium NOx levels (b), which became comparable to the control cells. Data are presented as means + S.E.M. at each treatment condition from three individual experiments. Among group difference was analyzed using ANOVA test (P < 0.01 for all). *Bonferroni correction was applied to all P values for between group comparisons.
3. Results
3.1. CSE dose-dependently induced endothelial apoptosis
First, we investigated dose and time-dependent induction of HAEC apoptosis by CSE. Cells were treated with CSE from 0 to 0.02 U for 4 h. As shown in Fig. 1(a), CSE dose-dependently induced endothelial apoptosis from 3% to 65% when CSE doses were increased from 0 to 0.02 U for 4 h of the treatment. Although induction of apoptosis was evident at 0.001 U CSE, the maximal effect was observed at 0.02 U. We also tested a higher CSE dose (0.05 U). At this dosage, majority of the cells died during early hours (1–2 h) of the treatment. CSE at the dose of 0.02 U also induced endothelial apoptosis in a time-dependent manner (Fig. 1(b)). The CSE-induced apoptosis started at 30 min and the percentage of apoptotic cells increased substantially within first 4 h; and the increase in cell death continued up to 75% at 24 h.
3.2. CSE reduced eNOS expression and protein phosphorylation
To investigate if CSE has an effect on eNOS expression, HAECs were treated with different doses of CSE for 24 h. Total eNOS protein was detected using anti-human eNOS antibody by Western blotting. As shown in Fig. 2(a), eNOS protein levels were decreased with the CSE treatment in a dose-dependent manner. The similar pattern of reduction was also found for eNOS mRNA levels (Fig. 2(b)). On the other hand, however, the phospho-eNOS (Ser1177) appeared to be increased (Fig. 2(a)).
Fig. 2.
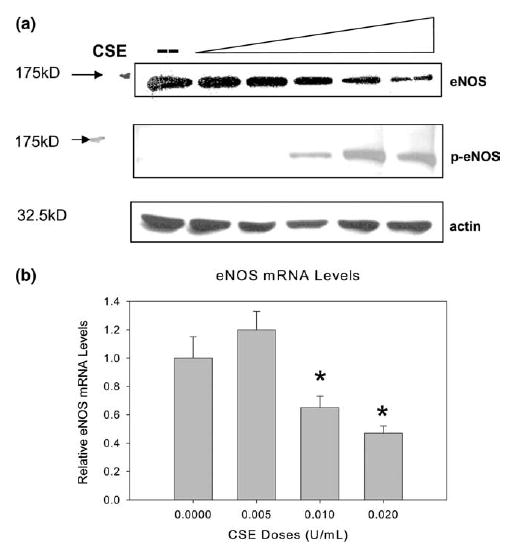
CSE causes dose-dependent change in eNOS protein and mRNA level. (a) Western blot of the eNOS protein levels in HAECs treated with CSE at doses of 0, 0.005, 0.001, 0.015, 0.02, and 0.025 U (lanes 1–6) for 24 h. Twenty-five micrograms of cellular protein was separated by 8% SDS–PAGE and blotted with anti-eNOS antibody and anti-phospho eNOS (Ser1177). Experiments were repeated three times and the representative electrophoresis is shown in the figure. (b) HAECs treated with CSE showed dose-dependent changes in eNOS mRNA level (means + S.E.M., N = 3) measured by quantitative real-time RT-PCR and expressed as relative units comparing to control cells. HAECs were treated with indicated doses of CSE for 24 h. House keeping gene β-actin mRNA was used as internal control. Among group difference was statistically significant (P < 0.01) by ANOVA test; *P < 0.01 by the post hoc Dunnet’s test when compared with control cells treated with the standard medium only.
3.3. Effects of CSE on caspase-3 and caspase-8
Caspase-3, one of the executing proteases for apoptosis, is normally present in an inactive pro-enzyme form. It can be activated by proteolytic processing of its inactive zymogen into activated p17 and p19 subunits. We examined the caspase-3 activation by CSE in HAECs. As shown in Fig. 3(a), caspase-3 activity increased substantially after CSE treatment from 0 to 0.02 U. This caspase-3 activation pattern coincided with the pattern of the increased endothelial apoptosis by CSE treatment (Fig. 1(a)). The increased activity of caspase-3 in 0.02 CSE treated cells was further confirmed by a large increase in cleaved caspase-3 active subunit p17 in Western-blot analysis (Fig. 3(b)).
Fig. 3.
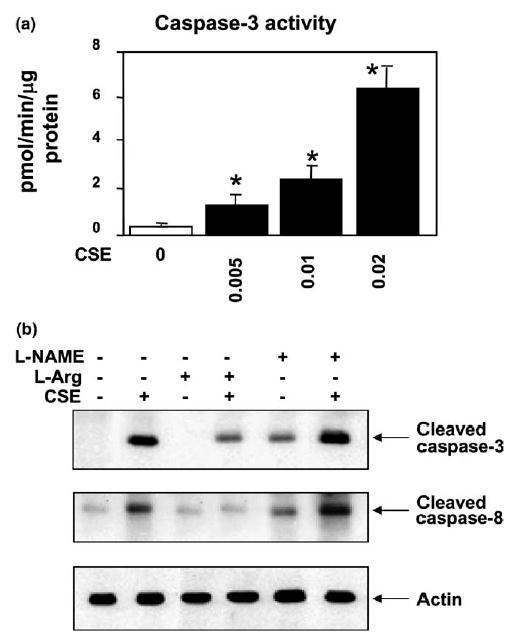
Effects of CSE on caspase-3 and -8 activations in HAECs. (a) Caspase-3 activity (means + S.E.M., N = 3) was increased with increasing CSE concentrations (F = 40.3, P = 0.01), and the caspase-3 activities treated with three different doses of CSE (0.005, 0.01 and 0.02 U) were significantly higher (*P < 0.01) than the control cells treated with standard medium only by the post hoc Dunnett’s test. (b) HAECs were treated with (lane 2) or without (lane 1) CSE (0.02 U) for 4 h. Treatment conditions are shown on the top of the figure. Cell lysates were separated on 12% SDS–PAGE. The membrane was immunoblotted with either anti-cleaved caspase-3 antibody (top panel of b) or anti-cleaved caspase-8 antibody (middle panel of b). CSE activated the cleavage of caspases-3 and 8 at 4 h. In pretreatment experiments, cells were incubated with 200 μM of l-arginine (lanes 3 and 4), or 400 μM of l-NAME (lanes 5 and 6) for 30 min followed by 0.02 U CSE exposure for 4 h (lanes 4 and 6). Pre-activation of eNOS by l-arginine attenuated the CSE-induced caspases-3 and 8 activations whereas inhibition of eNOS by l-NAME increased the CSE-induced caspase activation. Western blotting with anti-actin antibody was carried out to assess for equal loading (lower panel). Experiments were repeated three times and the representative electrophoresis is shown in the figure.
Caspase-3 can be activated by several upstream caspases, one of which is caspase-8. We next examined whether CSE activated caspase-8 in HAECs using Western blotting in which an active caspase-8 would show cleaved forms of p18 and p10. Cells were stimulated with or without CSE (0.02 U) for 4 h and cell lysates were immunoblotted with anti-caspase-8 antibody. As shown in Fig. 3(b), there was a clear increase in the presence of cleaved active caspase-8. An active caspase-8 is able to cleave and activate downstream caspases, including caspase-1 and caspase-3.
3.4. p38 MAP kinase and JNK/SAPK activation in CSE-induced HAEC apoptosis
We further examined the signaling events involved in CSE-induced apoptosis in HAECs. Caspases can be activated by several upstream signaling molecules. To understand the pathways involved in the activation of caspases in CSE-induced apoptosis, we investigated the activation of p38 MAP kinase in CSE-treated HAECs. The p38 MAP kinase can be activated by the phosphorylation and p38 MAP kinase activation has been shown to trigger the apoptotic pathway. HAECs were incubated with or without CSE (0.02 U) and cell lysates were immunoblotted with anti-phospho p38 MAP kinase antibody. We found that p38 MAP kinase was substantially activated by the CSE treatment (Fig. 4).
Fig. 4.
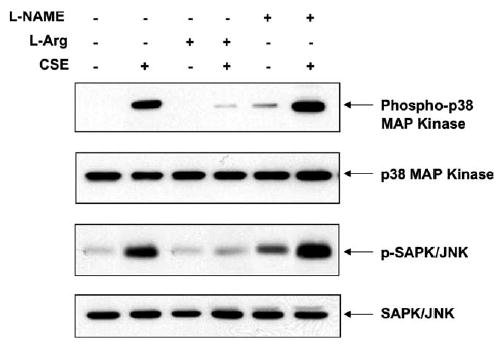
Activation of p38 MAP kinase and SAPK/JNK by CSE treatment. CSE induced the activation of p38 MAP kinase and SAPK/ JNK in HAECs. Treatments are listed on the top of the figure in the same condition as described in Fig. 3. A representative blot from three individual experiments is shown. Cell lysates were separated on a 10% SDS–PAGE. The membrane was immunoblotted with either anti-phospho p38 MAP kinase or anti-phospho SAPK/JNK antibodies. Pre-activation of eNOS by l-arginine prevented the CSE-induced kinase activation whereas inhibition of eNOS by l-NAME increased the CSE-induced activation. Total p38 MAP kinase and total SAPK/ JNK were unchanged.
A variety of environmental stressors can activate SAPK/JNK by phosphorylation. Since CSE is known to induce oxidative stress [28], we explored whether SAPK/JNK was also activated by CSE in HAECs. Control and CSE treated cell lysates were immunoblotted with anti-phospho-JNK/SAPK antibody. As shown in Fig. 4, CSE (0.02 U) induced the SAPK/JNK phosphorylation at 4 h when compared to control.
To further confirm the involvement of p38 MAP kinase and SAPK/JNK pathways in CSE-induced apoptosis, the HAECs were pretreated with 10 μM of SB 202190 – a p38 MAP kinase inhibitor and/or SP600125 – a SAPK/JNK inhibitor for 2 h, which was followed by exposure to CSE for 4 h. As shown in Fig. 5, p38 MAP kinase inhibitor partially inhibited CSE-induced caspase-3 activation in HAECs. The inhibition of SAPK/JNK by SP600125 also partially blocked the caspase-3 cleavage induced by CSE. When both kinase inhibitors were used together, there was a near complete inhibition of CSE-activated caspase-3 and a dramatic reduction in CSE-induced endothelial apoptosis. This clearly indicates that CSE-induced apoptosis requires the activation of both p38 MAP kinase and SAPK/JNK pathways in HAECs.
Fig. 5.
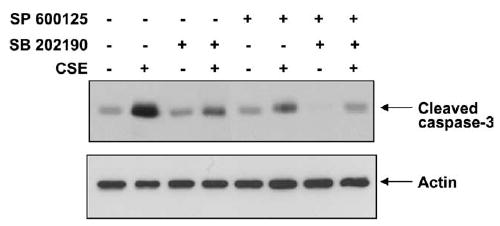
Effects of p38 MAP kinase and/or SAPK/JNK inhibition on caspase-3 activation. Cells were pretreated with 10 μM of SB 202190, a p38 MAP kinase inhibitor, and/or SP600125, a JNK/SAPK inhibitor for 2 h as summarized on the top of the figure. This was followed by CSE (0.02 U) exposure for 4 h. Cell lysates were separated on 12% SDS–PAGE. The membrane was immunoblotted with anti-cleaved caspase-3 antibody. Inhibition of either p38 MAP kinase (lanes 3 and 4) or JNK/SAPK (lanes 5 and 6) resulted in partial reduction in CSE-induced caspase-3 activation. Furthermore, active caspase-3 returned to the control level when both p38 MAP kinase and SAPK/JNK were inhibited at the same time (lanes 7 and 8). Western blot with anti-actin was used as a loading control. Experiments were repeated three times and representative electrophoresis is shown in the figure.
3.5. Pre-activation of eNOS protected against CSE-induced HAEC apoptosis
We found that CSE treatment decreased eNOS protein and mRNA level (Fig. 2), and increased caspase-3 activity and apoptosis (Figs. 1(a) and 3(a)). We hypothesized that the attenuated eNOS activity by the high-dose CSE (0.02 U) could compromise the endogenous anti-apoptotic mechanism so that it is indirectly responsible for CSE-induced HAEC apoptosis. To prove this hypothesis, we pretreated endothelial cells with l-arginine – a known substrate of eNOS, at the dose of 200 μM for 30 min prior to CSE exposure for 4 h. As shown in Fig. 6, l-arginine pretreatment significantly decreased CSE-induced apoptosis to a level (4.8 ± 0.2%; P < 0.001) similar to controls (3.0 ± 0.03%). Pretreatment with d-arginine, on the other hand, had no effect on CSE-induced apoptosis (data not shown). Pretreatment with l-NAME (400 μM) – a non-specific NOS inhibitor, further increased CSE-induced apoptosis (81.9 ± 1.1%) when compared to CSE alone treated cells (65.2 ± 0.2%, p < 0.05, Fig. 6(a)). Treatment by l-arginine or l-NAME alone or together did not inhibit or accelerate the naturally occurring apoptotic process during in vitro cell culture (4.4 ± 0.5% and 1.6 ± 0.4%, respectively, Fig. 6(a)). The protective effect of l-arginine was also observed under light microscopy for the number of adherent cells (data not shown).
l-Arginine pretreatment resulted in increased eNOS activity and NOx level in HAECs, whereas l-NAME pre-incubation inhibited the eNOS activity and decreased the NOx level in CSE-treated HAECs as shown in Figs. 7(a) and (b). The S-NO levels in the cell lysates, however, were not changed by the l-arginine and l-NAME pre-incubation (Fig. 7(c)). Pre-activation of eNOS by l-arginine inhibited the CSE-induced activation of p38 MAP kinase and JNK/SAPK, whereas l-NAME pretreatment increased CSE-induced p38 MAP kinase and SAPK/JNK activation (Fig. 4). Furthermore, pre-activation of eNOS by l-arginine reduced CSE-induced caspase-3 and -8 activations whereas inhibition of eNOS by l-NAME treatment resulted in an increase of active caspase levels (Fig. 3). The measured caspase-3 activities (Fig. 6(b)) in the pretreatment experiments were well correlated with the levels of active caspase-3 by Western blot (Fig. 3).
4. Discussion
There is no doubt that cigarette smoking causes endothelial dysfunction and is associated with vascular diseases. However, knowledge of the underlying molecular mechanisms of pathological changes is limited. Previously, we have shown that smoking-induced endothelial apoptosis could be one of the potential mechanisms [14]. In the current study, we observed a dose- and time-dependent increase in endothelial apoptosis after exposing to CSE. The excessive apoptosis induced by CSE could contribute to smoke-induced endothelial injury, which may then lead to atherogenesis. The pro-apoptotic effects by cigarette smoking have also been demonstrated in other cell types such as epithelial cells in gastric mucosa [16], which could be relevant to smoking-induced carcinogenesis.
Our investigation in the current study has revealed two new pathways that have not been reported previously in relation to smoking-induced cell death. We have shown that activation of p38 MAP kinase and SAPK/JNK may partially mediate CSE-induced apoptosis. We have also demonstrated that endogenous NO production may have a protective effect against CSE-induced apoptosis in endothelial cells.
NO is an essential signaling molecule for endothelial integrity and growth [2,29,30]. A moderate basal NO production can protect endothelial cells from damaging effects of risk factors such as TNFα [17]. We hypothesize that if intrinsic protective mechanisms could be pre-activated by moderate NO production in endothelial cells cultured in vitro, these cells could be better prepared to the ensuing CSE assault. Our experiments indeed demonstrated such protective role by the pre-activation of eNOS against the CSE-induced apoptotic changes. We show that NO inhibits the caspases-3 and 8 activations in l-arginine treated cells and thereby inhibits the CSE-induced apoptosis, which is consistent with early findings [31–33]. Inhibition of eNOS by l-NAME has eliminated the eNOS-mediated protective mechanisms by augmenting CSE-induced caspase activation. Inhibition of NO synthesis resulted in an increased caspase activity that has been shown in macrophages [34] and bovine aortic endothelial cells [35]. It should be further acknowledged, however, that the protective effect by l-arginine could also be mediated through non-eNOS-dependent pathways, since l-arginine could be substrates for other metabolic reactions. This requires further investigation.
NO can regulate apoptosis via two distinct mechanisms: cGMP-dependent and cGMP-independent pathway [36]. Excessive free radicals produced by cigarette smoking could be one of the mechanisms responsible for the damaging effects. Therefore, we could speculate that NO exposure could induce modest but significant increase in cytosolic non-heme iron [37]. Increased cytosolic iron is known to upregulate the synthesis of ferritin, which may be an important protective response against oxidative injury by sequestering iron and thus preventing the formation of hydroxyl radical via the Fenton reaction [38]. This will be an interesting pathway to investigate further.
Our experiments have shown that p38 MAP kinase and SAPK/JNK pathways may be involved in the activation of apoptotic process induced by CSE exposure. Activated caspases-3 and 8 by CSE are accompanied by the increased p38 MAP kinase and SAPK/JNK activation. Inhibition of p38 MAP kinase and SAPK/JNK by specific inhibitors resulted in reduced caspase-3 activation, hence less apoptotic endothelial cells. The l-arginine pretreatment has further resulted in decreased activations of p38 MAP kinase and JNK/SAPK, and decreased active forms of the caspase-3 and 8. While the precise upstream molecules activated by the eNOS pre-activation are not clear, our findings are consistent with those reported by others. For example, NO protects B lymphocytes from antigen-induced apoptosis [39], and delays spontaneous apoptosis in cultured ovarian follicles [40] and eosinophils [41]. The physiological amount of eNOS-derived NO in endothelial cells is found sufficient to suppress caspase-3-like proteases [36]. Recently, Li and Liang [42] show that the l-arginine inhibits hepatocyte apoptosis via the inhibition of caspase-3 activity via increased NO synthesis. It is well known that JNK activity is required for stress-induced apoptosis [43]. As reported by So et al. [44], NO may inactivate the JNK by a cGMP-independent S-nitrosylation pathway and rescues the cells from the apoptotic death. The S-nitrosylation of thioredoxin (Trx) in the position Cys69 [25], can directly inhibit apoptosis signal-regulating kinase 1 (ASK1), which serves as the MAP kinase kinase kinase and leads to an activation of JNK and p38 MAP-kinases [25,45]. However, four-hour treatment by CSE with or without pre-conditioning of l-arginine or l-NAME did not appear to change the S-NO levels in the total protein extracts of the endothelial cells.
In summary, we have shown that CSE can induce endothelial apoptosis in a dose- and time-dependent manner. This apoptosis may be mediated by the activation of p38 MAP kinase, JNK/SAPK and caspases. Furthermore, pre-activation of eNOS by l-arginine offers significant protection against CSE-induced endothelial apoptosis. Our findings suggest that appropriate endogenous NO production may be an important protective mechanism against smoking-induced endothelial damage.
Acknowledgments
The work is supported by a grant from NIH (R01-HL066054). Dr. X.L. Wang is an AHA Established Investigator.
References
- 1.Holbrook JH, Grundy SM, Hennekens CH, Kannel WB, Strong JP. Cigarette smoking and cardiovascular diseases. A statement for health professionals by a task force appointed by the steering committee of the American Heart Association. Circulation. 1984;70:1114A–1117A. [PubMed] [Google Scholar]
- 2.Moncada S. Nitric oxide in the vasculature: physiology and pathophysiology. Ann NY Acad Sci. 1997;811:60–67. doi: 10.1111/j.1749-6632.1997.tb51989.x. discussion 67–9. [DOI] [PubMed] [Google Scholar]
- 3.Nadler JL, Velasco JS, Horton R. Cigarette smoking inhibits prostacyclin formation. Lancet. 1983;1:1248–1250. doi: 10.1016/s0140-6736(83)92698-3. [DOI] [PubMed] [Google Scholar]
- 4.Powell JT, Higman DJ. Smoking, nitric oxide and the endothelium. Br J Surg. 1994;81:785–787. doi: 10.1002/bjs.1800810602. [DOI] [PubMed] [Google Scholar]
- 5.Kiowski W, Linder L, Stoschitzky K, Pfisterer M, Burckhardt D, Burkart F, Buhler FR. Diminished vascular response to inhibition of endothelium-derived nitric oxide and enhanced vasoconstriction to exogenously administered endothelin-1 in clinically healthy smokers. Circulation. 1994;90:27–34. doi: 10.1161/01.cir.90.1.27. [DOI] [PubMed] [Google Scholar]
- 6.Kalra VK, Ying Y, Deemer K, Natarajan R, Nadler JL, Coates TD. Mechanism of cigarette smoke condensate induced adhesion of human monocytes to cultured endothelial cells. J Cell Physiol. 1994;160:154–162. doi: 10.1002/jcp.1041600118. [DOI] [PubMed] [Google Scholar]
- 7.Beckman JS, Koppenol WH. Nitric oxide, super-oxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 8.Simon DI, Mullins ME, Jia L, Gaston B, Singel DJ, Stamler JS. Polynitrosylated proteins: characterization, bioactivity, and functional consequences. Proc Natl Acad Sci USA. 1996;93:4736–4741. doi: 10.1073/pnas.93.10.4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haendeler J, Weiland U, Zeiher AM, Dimmeler S. Effects of redox-related congeners of NO on apoptosis and caspase-3 activity. Nitric Oxide. 1997;1:282–293. doi: 10.1006/niox.1997.0134. [DOI] [PubMed] [Google Scholar]
- 10.Haendeler J, Zeiher AM, Dimmeler S. Nitric oxide and apoptosis. Vitam Horm. 1999;57:49–77. doi: 10.1016/s0083-6729(08)60640-8. [DOI] [PubMed] [Google Scholar]
- 11.Melino G, Bernassola F, Knight RA, Corasaniti MT, Nistico G, Finazzi-Agro A. S-nitrosylation regulates apoptosis. Nature. 1997;388:432–433. doi: 10.1038/41237. [DOI] [PubMed] [Google Scholar]
- 12.Kanner J, Harel S, Granit R. Nitric oxide as an antioxidant. Arch Biochem Biophys. 1991;289:130–136. doi: 10.1016/0003-9861(91)90452-o. [DOI] [PubMed] [Google Scholar]
- 13.Rubbo H, Radi R, Trujillo M, Telleri R, Kalyanaraman B, Barnes S, Kirk M, Freeman BA. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation. Formation of novel nitrogen-containing oxidized lipid derivatives. J Biol Chem. 1994;269:26066–26075. [PubMed] [Google Scholar]
- 14.Wang J, Wilcken DE, Wang XL. Cigarette smoke activates caspase-3 to induce apoptosis of human umbilical venous endothelial cells. Mol Genet Metab. 2001;72:82–88. doi: 10.1006/mgme.2000.3115. [DOI] [PubMed] [Google Scholar]
- 15.Vayssier M, Banzet N, Francois D, Bellmann K, Polla BS. Tobacco smoke induces both apoptosis and necrosis in mammalian cells: differential effects of HSP70. Am J Physiol. 1998;275:L771–L779. doi: 10.1152/ajplung.1998.275.4.L771. [DOI] [PubMed] [Google Scholar]
- 16.Wang HY, Ma L, Li Y, Cho CH. The role of nitric oxide on cigarette smoke-induced programmed cell death in the gastric mucosa. Scand J Gastroenterol. 2001;36:235–240. doi: 10.1080/003655201750074438. [DOI] [PubMed] [Google Scholar]
- 17.Shen YH, Wang XL, Wilcken DE. Nitric oxide induces and inhibits apoptosis through different pathways. FEBS Lett. 1998;433:125–131. doi: 10.1016/s0014-5793(98)00844-8. [DOI] [PubMed] [Google Scholar]
- 18.Ambalavanan N, Carlo WF, Bulger A, Shi J, Philips JB., 3rd Effect of cigarette smoke extract on neonatal porcine vascular smooth muscle cells. Toxicol Appl Pharmacol. 2001;170:130–136. doi: 10.1006/taap.2000.9094. [DOI] [PubMed] [Google Scholar]
- 19.Su Y, Han W, Giraldo C, De Li Y, Block ER. Effect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cells. Am J Respir Cell Mol Biol. 1998;19:819–825. doi: 10.1165/ajrcmb.19.5.3091. [DOI] [PubMed] [Google Scholar]
- 20.Wang J, Wilcken DEL, Wang XL. Cigarette smoking induces apoptosis of human umbilical venous endothelial cells: relevance of p53 expression. Mol Genet Metab. 2001;72:82–88. doi: 10.1006/mgme.2000.3115. [DOI] [PubMed] [Google Scholar]
- 21.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 22.Shen YH, et al. Human cytomegalovirus causes endothelial injury through the ataxia telangiectasia mutant and p53 DNA damage signaling pathways. Circ Res. 2004;94:1310–1317. doi: 10.1161/01.RES.0000129180.13992.43. Epub 2004 Apr 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J, Brown MA, Tam SH, Chan MC, Whitworth JA. Effects of diet on measurement of nitric oxide metabolites. Clin Exp Pharmacol Physiol. 1997;24:418–420. doi: 10.1111/j.1440-1681.1997.tb01212.x. [DOI] [PubMed] [Google Scholar]
- 24.Hoffmann J, Haendeler J, Zeiher AM, Dimmeler S. TNFalpha and oxLDL reduce protein S-nitrosylation in endothelial cells. J Biol Chem. 2001;276:41383–41387. doi: 10.1074/jbc.M107566200. Epub 2001 Aug 27. [DOI] [PubMed] [Google Scholar]
- 25.Haendeler J, Hoffmann J, Tischler V, Berk BC, Zeiher AM, Dimmeler S. Redox regulatory and anti-apoptotic functions of thioredoxin depend on S-nitrosylation at cysteine 69. Nat Cell Biol. 2002;4:743–749. doi: 10.1038/ncb851. [DOI] [PubMed] [Google Scholar]
- 26.Eu JP, Liu L, Zeng M, Stamler JS. An apoptotic model for nitrosative stress. Biochemistry. 2000;39:1040–1047. doi: 10.1021/bi992046e. [DOI] [PubMed] [Google Scholar]
- 27.Wang XL, Sim AS, Wang MX, Murrell GA, Trudinger B, Wang J. Genotype dependent and cigarette specific effects on endothelial nitric oxide synthase gene expression and enzyme activity. FEBS Lett. 2000;471:45–50. doi: 10.1016/s0014-5793(00)01356-9. [DOI] [PubMed] [Google Scholar]
- 28.Carnevali S, et al. Cigarette smoke extract induces oxidative stress and apoptosis in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2003;284:L955–L963. doi: 10.1152/ajplung.00466.2001. Epub 2003 Jan 24. [DOI] [PubMed] [Google Scholar]
- 29.Rudic RD, Shesely EG, Maeda N, Smithies O, Segal SS, Sessa WC. Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. J Clin Invest. 1998;101:731–736. doi: 10.1172/JCI1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bredt DS, Snyder SH. Nitric oxide: a physiologic messenger molecule. Annu Rev Biochem. 1994;63:175–195. doi: 10.1146/annurev.bi.63.070194.001135. [DOI] [PubMed] [Google Scholar]
- 31.Kwon YG, Min JK, Kim KM, Lee DJ, Billiar TR, Kim YM. Sphingosine 1-phosphate protects human umbilical vein endothelial cells from serum-deprived apoptosis by nitric oxide production. J Biol Chem. 2001;276:10627–10633. doi: 10.1074/jbc.M011449200. [DOI] [PubMed] [Google Scholar]
- 32.Li J, Billiar TR, Talanian RV, Kim YM. Nitric oxide reversibly inhibits seven members of the caspase family via S-nitrosylation. Biochem Biophys Res Commun. 1997;240:419–424. doi: 10.1006/bbrc.1997.7672. [DOI] [PubMed] [Google Scholar]
- 33.Kim YM, Chung HT, Simmons RL, Billiar TR. Cellular non-heme iron content is a determinant of nitric oxide-mediated apoptosis, necrosis, and caspase inhibition. J Biol Chem. 2000;275:10954–10961. doi: 10.1074/jbc.275.15.10954. [DOI] [PubMed] [Google Scholar]
- 34.Kim YM, Talanian RV, Li J, Billiar TR. Nitric oxide prevents IL-1beta and IFN-gamma-inducing factor (IL-18) release from macrophages by inhibiting caspase-1 (IL-1beta-converting enzyme) J Immunol. 1998;161:4122–4128. [PubMed] [Google Scholar]
- 35.Matsunaga T, Kotamraju S, Kalivendi SV, Dhanasekaran A, Joseph J, Kalyanaraman B. Ceramide-induced intracellular oxidant formation, iron signaling, and apoptosis in endothelial cells: protective role of endogenous nitric oxide. J Biol Chem. 2004 doi: 10.1074/jbc.M400977200. [DOI] [PubMed] [Google Scholar]
- 36.Kim YM, Talanian RV, Billiar TR. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J Biol Chem. 1997;272:31138–31148. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- 37.Kim YM, Bergonia HA, Muller C, Pitt BR, Watkins WD, Lancaster JR., Jr Loss and degradation of enzyme-bound heme induced by cellular nitric oxide synthesis. J Biol Chem. 1995;270:5710–5713. doi: 10.1074/jbc.270.11.5710. [DOI] [PubMed] [Google Scholar]
- 38.Halliwell B, Gutteridge JM. Oxygen free radicals and iron in relation to biology and medicine: some problems and concepts. Arch Biochem Biophys. 1986;246:501–514. doi: 10.1016/0003-9861(86)90305-x. [DOI] [PubMed] [Google Scholar]
- 39.Genaro AM, Hortelano S, Alvarez A, Martinez C, Bosca L. Splenic B lymphocyte programmed cell death is prevented by nitric oxide release through mechanisms involving sustained Bcl-2 levels. J Clin Invest. 1995;95:1884–1890. doi: 10.1172/JCI117869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chun SY, Eisenhauer KM, Kubo M, Hsueh AJ. Interleukin-1 beta suppresses apoptosis in rat ovarian follicles by increasing nitric oxide production. Endocrinology. 1995;136:3120–3127. doi: 10.1210/endo.136.7.7540548. [DOI] [PubMed] [Google Scholar]
- 41.Beauvais F, Michel L, Dubertret L. The nitric oxide donors, azide and hydroxylamine, inhibit the programmed cell death of cytokine-deprived human eosinophils. FEBS Lett. 1995;361:229–232. doi: 10.1016/0014-5793(95)00188-f. [DOI] [PubMed] [Google Scholar]
- 42.Li SQ, Liang LJ. Protective mechanism of l-arginine against liver ischemic-reperfusion injury in rats. Hepatobiliary Pancreat Dis Int. 2003;2:549–552. [PubMed] [Google Scholar]
- 43.Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 44.So HS, Park RK, Kim MS, Lee SR, Jung BH, Chung SY, Jun CD, Chung HT. Nitric oxide inhibits c-Jun N-terminal kinase 2 (JNK2) via S-nitrosylation. Biochem Biophys Res Commun. 1998;247:809–813. doi: 10.1006/bbrc.1998.8788. [DOI] [PubMed] [Google Scholar]
- 45.Sumbayev VV. S-nitrosylation of thioredoxin mediates activation of apoptosis signal-regulating kinase 1. Arch Biochem Biophys. 2003;415:133–136. doi: 10.1016/s0003-9861(03)00199-1. [DOI] [PubMed] [Google Scholar]