

Abstract
UhpA, a member of the NarL family of response regulators, activates transcription of the Escherichia coli uhpT gene for the sugar phosphate transporter UhpT in response to extracellular glucose-6-phosphate. UhpA binds with different affinities to adjacent regions in the uhpT promoter, termed the strong-binding (S) region from −80 to −50 and the weak-binding (W) region from −50 to −32. Transcription activation by UhpA is stimulated by the catabolite gene activator protein (CAP)-cyclic AMP complex and depends on the C-terminal domains of the RNA polymerase RpoA and RpoD subunits. Because single-base substitutions in the UhpA-binding region had little effect on promoter activity, nucleotide substitutions in successive 4-bp blocks throughout this region were examined for their effects on promoter activation and UhpA binding. Changes in three of four blocks within the W region substantially impaired the ability of UhpA to bind to this region, to drive expression of a uhpT-lacZ reporter, and to support UhpA-dependent in vitro transcription. These W region variant promoters were strongly stimulated by CAP. Changes in several parts of the S region impaired UhpA binding to both the S and W regions and decreased promoter activity in vivo and in vitro. Thus, binding of UhpA to the W region is crucial for UhpA-dependent activation and depends on occupancy of the S region. None of these substitutions eliminated promoter function. The orientation of UhpA-binding sites was assessed by the affinity cleavage method. The iron chelate FeBABE [iron (S)-1-(p-bromoacetamidobenzyl) EDTA] was covalently attached to engineered cysteine residues near the DNA-binding region in UhpA. Hydroxyl radicals generated by the iron chelate attached at position 187 resulted in DNA strand cleavages in two clusters of sites located in the middle of the S and W regions. These results are consistent with the binding of two dimers of UhpA. Each dimer binds to an inverted repeat of monomer-binding sites with the consensus sequence CCTGRR, where R is A or G, and each is separated by 6 bp. It is likely that members of the NarL family bind to dyad targets, in contrast to the binding of OmpR family response regulators to direct-repeat targets.
Many bacterial promoters are controlled by multiple transcription activators, which enhance transcription by affecting DNA topology or by interacting with each other or with the RNA polymerase (RNAP) holoenzyme. Specific surfaces of the RNAP components RpoD (σ70) or the N-terminal domain of RpoA are sites of functional contact by some transcription factors that bind near the −35 promoter region, such as FNR, PhoB, or λcI protein (25). The C-terminal domain of RpoA (α-CTD) is used for activation by other proteins which bind further upstream in the promoter. The catabolite gene activator protein CAP can interact with each of these three domains of RNAP, depending on the location of its binding site within the promoter (7). In addition, transcription activation can result from changes in DNA topology owing to protein-induced bending (13, 35, 36).
The Escherichia coli uhpT gene encodes the sugar phosphate transporter UhpT and is induced by extracellular glucose-6-phosphate (Glc6P) through action of the transcription activators UhpA and CAP (22). The regulon-specific regulator UhpA is a member of the NarL family of response regulator (RR) proteins of two-component regulatory systems (reviewed in reference 40). It is expected to have a two-domain structure very similar to that of the nitrate/nitrite-responsive regulator NarL (3), with an N-terminal phosphorylation module characteristic of RRs and a C-terminal DNA-binding/transcription activation domain containing a conserved and widely distributed helix-turn-helix motif (19). UhpA is essential for detectable uhpT expression in vivo and in vitro (33, 42). The activity of UhpA is controlled by its phosphorylation by the cognate membrane-bound sensor kinase complex composed of UhpB and UhpC (21). Phosphorylation of UhpA strongly increases but is not required for binding to the −80 to −32 region of the uhpT promoter (all promoter coordinates are relative to the transcription start site) (9, 10). Overexpression of UhpA allows phosphorylation-independent transcription activation. DNase footprinting and gel shift analyses indicated that UhpA binds to different portions of this promoter region with different affinities. UhpA occupies the −80 to −50 region of the promoter, called the strong-binding (S) region, more efficiently than the −50 to −32 region, called the weak-binding (W) region. UhpA was seen to protect five contiguous, helically phased DNA segments in this region from hydroxyl radical-mediated cleavage (10).
UhpA-dependent transcription of the uhpT promoter is strongly stimulated by the global regulator CAP-cyclic AMP (cAMP) complex, which binds to positions −117 to −90 (28, 33). The effects on in vivo and in vitro uhpT transcription caused by alanine substitutions in the C-terminal portions of RpoA and RpoD supported a bipartite model of transcription activation. The ability of UhpA to activate uhpT transcription depended on certain residues (K593, L598, and K599) in the C-terminal domain of RpoD (σ-CTD) (34). This requirement for σ-CTD was greatly reduced when CAP was present. In contrast, stimulation by CAP was strongly reduced when α-CTD was deleted or carried certain alanine substitutions affecting its DNA-binding ability (particularly residues R265, G296, and S299). In the bipartite activation model, UhpA bound to the downstream W region might allow recruitment and positioning of RNAP through interaction with σ-CTD. UhpA bound to the upstream S region could enhance binding of UhpA to the W region and interact directly with CAP and α-CTD.
To better understand the binding and action of UhpA, we analyzed a systematic set of base substitutions throughout the UhpA-binding region in the uhpT promoter for their effect on UhpA occupancy and transcription activation. The results indicate the cooperativity of binding and action of UhpA. The stoichiometry and orientation of bound UhpA molecules were assessed by an affinity cleavage technique. The helix-loop-helix-containing RRs of the OmpR family, OmpR and PhoB, bind in a direct or head-to-tail arrangement to recognition sites upstream or adjacent to the RNAP-binding region, where they interact with α-CTD or σ-CTD, respectively (16, 17, 32). In contrast, RR of the NarL family (including UhpA, NarL, NarP, BvgA, and DegU) may recognize multiple sites with dyad symmetry, suggesting that they bind as tail-to-tail dimers (2, 4, 11). For example, NarL recognizes a 7-2-7 dyad-repeat motif with half-site consensus of TACYNMT, where Y = C or T and M = A or C (14, 24). In this study, cysteine residues introduced into UhpA were derivatized with a DNA cleavage reagent, and the resulting pattern of hydroxyl radical-mediated DNA cleavage was consistent with the binding of two dimers of UhpA, with each dimer occupying one region in tail-tail manner.
MATERIALS AND METHODS
Bacterial strains and plasmids.
The bacterial strains, plasmids, and phages used in this study are listed in Table 1. Plasmid DNA isolation and recombinant DNA manipulation were carried out by standard methods (38) or the recommendations of the manufacturers of the reagents.
TABLE 1.
Bacterial strains, plasmids, and phages used in this work
| Strain, plasmid, or phage | Relevant characteristics | Source or reference |
|---|---|---|
| Strains | ||
| MC4100 | ΔlacU169 araD139 deoC1 flbB5301 ptsF25 rbsR rpsL150 | 8 |
| RK1251 | MC4100 Δuhp(A15-T427) tna::Tn10 | R. Kadner |
| RK1280 | MC4100 recA tna::Tn10 | R. Kadner |
| BL21(DE3) | ompT hsdSB gal dcm (DE3) | Novagen |
| Plasmids | ||
| pRJK10 | ApruhpABCT | 41 |
| pUT1 (and derivatives) | ApruhpT, rrnB t1+2 terminator (derivatives carrying mutations M1 to M12) | 33 |
| This work | ||
| pRS415 | AprlacZ fusion plasmid | 39 |
| pRS415:PT (and derivatives) | uhpT promoter-lacZ operon fusion plasmid (derivatives carrying mutations M1 to M12) | This work |
| pET15b | Apr T7 promoter | Novagen |
| pET15b-uhpA+ (and derivatives) | ApruhpA, T7 promoter control (derivatives carrying mutations A154C, V155C, G181C, V182C, D185C, E187C, and A189C) | This work |
| Phages | ||
| λRZ5 | lacZ fusion vector | 37 |
| λRZ5-PTlac (and derivatives) | ApruhpT-lacZ fusion phage (derivatives carrying mutations M1 to M12) | 29 |
| This work |
Site-directed mutagenesis.
Mutations that change four successive residues in the uhpT promoter or that result in substitution of cysteine codons in the uhpA gene were introduced by PCR amplification with mutagenic primers (20). Sequences of mutagenic primers are available upon request. Plasmid pRJK10 carrying the wild-type uhpABCT locus (41) was used as a template, and standard PCR cycling conditions were followed. The amplimers carrying variant uhpT promoter regions were cloned by restriction fragment exchange for the 180-bp EcoRI-BamHI fragment of plasmid pUT1 (33), carrying the wild-type uhpT promoter from positions −140 to +40.
PCR products carrying fragments of the wild-type and mutated uhpA genes (variants D185C, E187C, and A189C) were cloned as 610-bp BspHI-BamHI fragments into the NcoI-BamHI-digested plasmid pET15b (Novagen, Inc.) to create plasmids expressing the uhpA genes under the control of the T7 late promoter and producing products with the same length as wild-type UhpA. The PCR products that encoded the A154C, V155C, G181C, and V182C variants were cloned into NdeI-BamHI-digested plasmid pET15b to generate UhpA proteins with an N-terminal six-His tag. All constructs were verified by DNA sequencing at the University of Virginia Biomolecular Resource Center.
β-Galactosidase assays.
The 180-bp EcoRI-BamHI fragments carrying the wild-type or variant uhpT promoters were cloned into EcoRI-BamHI-digested plasmid pRS415 (39) to generate transcriptional fusions to the lacZYA operon. Each fusion was transferred by homologous recombination to phage λRZ5 (37, 39), and recombinant phages were used to form single lysogens in strain RK1280. The β-galactosidase activities of at least six separate lysogens were measured in triplicate following growth of cells in A salts medium with glycerol as the carbon source and with and without induction by 250 μM Glc6P. Enzyme activity was determined from the continuous rate of o-nitrophenyl-β-d-galactopyranoside hydrolysis in a plate reader (Molecular Dynamics), as previously described (29).
DNase footprinting assays.
DNase I protection assays were performed as previously described (33), with 5′-32P-labeled DNA fragments carrying the wild-type or mutant uhpT promoter region from −140 to +40. The DNA fragments were generated by PCR in which one primer had been 5′ labeled by [γ-32P]ATP and polynucleotide kinase with plasmid pUT1 or its variants as a template. The amplimers were purified by electrophoresis on 10% polyacrylamide gels. Binding reactions were carried out in TXN buffer (40 mM Tris-HCl [pH 8.0], 50 mM KCl, 10 mM MgCl2, 10 mM dithiothreitol, 200 μM cAMP) with specified concentrations of UhpA and with DNA fragments at a final concentration of 1 nM (10,000 to 50,000 cpm). Quantification of radioactive fragments was obtained with a PhosphorImager and ImageQuant software (Molecular Dynamics) (1).
Transcription assays.
Transcription activity was determined with purified components by a single-round transcription assay, as previously described (33). DNA templates were added as supercoiled plasmid pUT1 or its derivatives at 1 nM final concentration. They were incubated at 37°C in TXN buffer with 220 nM UhpA in the absence or presence of 20 nM CAP. After 10 min, RNAP (30 nM final concentration) was added, followed after 2 min by the NTPs (ATP, CTP, and GTP at 200 μM, and [α-32P]UTP (2.5 Ci/nmol) at 40 μM) and heparin (50 μg/ml). After 2 min, transcription was stopped by addition of 5 μl of Stop solution (7 M urea, 0.1 M EDTA, 0.4% sodium dodecyl sulfate, 40 mM Tris-Cl [pH 8.0], 0.5% bromophenol blue, 0.5% xylene cyanol). Products were separated by electrophoresis on 5% polyacrylamide-7 M urea gels, and the amount of the 230-nucleotide uhpT transcript was determined by PhosphorImager. Values were normalized to the level of the RNA I transcript from the plasmid vector.
Proteins.
UhpA proteins without an epitope tag were purified as described previously (10), but without isopropyl-β-d-thiogalactopyranoside (IPTG) induction of the T7 RNA polymerase. The uninduced expression in strain BL21(DE3) was generally sufficient for overexpression of the proteins. In brief, cell lysates obtained by sonication were fractionated by polyethyleneimine precipitation, ammonium sulfate fractionation, and chromatography on DEAE and Sephacryl S-200 columns. This method typically yielded protein of 90 to 95% purity in the range of 0.2 to 1 mg per liter of culture. The N-terminal His-tagged versions of UhpA were expressed as described above and purified by Ni(II)-affinity chromatography to >95% purity by the protocol recommended by the manufacturer (Novagen). CAP protein was purified by cAMP affinity chromatography (44). E. coli RNA polymerase holoenzyme was purchased from Amersham-Pharmacia Biotech, Inc. Protein concentrations were determined by the dye-binding method of Bradford (6).
Conjugation of UhpA with FeBABE.
Purified wild-type and Cys-substitution variants of UhpA were dialyzed for 1 h at 4°C against conjugation buffer C (50 mM HEPES [pH 7.9], 50 mM KCl, 1 mM EDTA, 10% glycerol [vol/vol]) with Slide-A-Lyzer Mini Dialysis units (Pierce, Rockford, Ill.). Conjugation was performed by mixing 1 μl of 26 mM FeBABE [iron (S)-1-(p-bromoacetamidobenzyl) EDTA] in dimethyl sulfoxide (kindly provided by C. F. Meares, University of California, Davis) with 25 μl of 100 μM UhpA for 1 h at ambient temperature. The reaction was halted by the addition of 26 μl of 1 M Tris-HCl (pH 8.0) containing 10% glycerol. Free FeBABE was removed by dialysis against buffer C as described above, and the UhpA-FeBABE proteins were stored at −70°C.
DNA cleavage by UhpA-FeBABE.
Wild-type or Cys-substituted UhpA-FeBABE (1.3 μM) was incubated with DNA fragments labeled with 32P at either 5′ end (1 nM; 50,000 cpm) in buffer C (20-μl final volume) for 10 min at 37°C. DNA cleavage was initiated by addition of ascorbate and H2O2 (5 mM final concentration of each). After 5 min at 37°C, the reaction was stopped by ethanol precipitation of the DNA. DNA was washed with 70% ethanol, dried under vacuum, and dissolved in loading buffer. DNase footprinting of the same samples was carried out by addition of DNase I instead of ascorbate and H2O2. Products of both assays were resolved by electrophoresis on 5% polyacrylamide sequencing gels (38).
RESULTS
Effect of mutations in the UhpA-binding region on uhpT-lacZ expression.
Previous mutagenesis of the uhpT promoter showed that base substitutions at 12 sites within the 50-bp UhpA-binding region caused little defect in uhpT-lacZ expression (27). For a systematic test of the contribution of the residues in the −80 to −33 region, we changed every residue in blocks of 4 successive bp. Directed mutagenesis by PCR techniques generated 12 variants, called M1 through M12, in which each set of 4 bp was changed to their most extreme alternatives: i.e., a purine was converted to the pyrimidine that pairs with the other purine. For example, in the M12 mutant, the wild-type sequence CAGG at −36 to −33 was changed to ACTT. The presence of the expected sequences was verified by DNA sequencing. To assay promoter activity in vivo, the variant UhpA-binding regions flanked by the entire wild-type uhpT promoter sequences (residues −140 to +40) were cloned into the promoter-reporter plasmid pRS415 (39) to form transcriptional fusions to the lac operon. These uhpT-lacZ fusions were transferred by homologous recombination into phage λRZ5, and single-copy lysogens were obtained. The variant promoters were also cloned into plasmid pSR, which contains a strong Rho-independent transcription terminator downstream of the promoter (23) for assay of in vitro transcription. The pSR plasmids were also used as templates in PCRs to generate end-labeled DNA fragments for DNase footprinting and affinity cleavage reactions. Figure 1 shows the relevant portion of the uhpT promoter and the sequence changes in each mutation.
FIG. 1.

Sequence and mutational changes in the UhpA-binding region of the uhpT promoter. Above the nucleotide sequence of the UhpA-binding region are shown the base substitutions in each of the 12 promoter variants. Coordinates are relative to the transcription start site. The extents of the S and W UhpA-binding regions are indicated.
β-Galactosidase production by single-copy λRZ5 uhpT-lacZ lysogens carrying the wild-type and variant UhpA-binding regions were assayed in Glc6P-induced cells. In all cases, expression of β-galactosidase was fully dependent on the presence of UhpA and induction by Glc6P (data not shown). Many promoter variants showed greatly decreased Glc6P-induced uhpT-lacZ expression, relative to the wild-type promoter (Fig. 2). Variants M2, M3, and M5 in the S region and M9, M11, and M12 in the W region were strongly affected and exhibited a 7- to 15-fold decrease in activity. Substitutions M1, M4, M6, and M7 had a more modest effect, causing a two- to fourfold decrease in activity. Substitutions M8 and M10 had only a marginal effect, with at most a twofold decrease. Thus, certain positions in both the S and W regions are important for promoter activity, but none of the 4-bp changes resulted in complete loss of activity, as occurs upon deletion of the uhpA gene (<10 units).
FIG. 2.

Expression of β-galactosidase from uhpT-lacZ fusions carrying substitutions in UhpA-binding region. Units of activity are Δ optical density at 415 nm (OD415) × min−1 × OD630−1 and were measured as described in Materials and Methods. Cultures were grown with 250 μM Glc6P; β-galactosidase activities in the absence of Glc6P were <20 units. Values are averaged from triplicate determinations with at least four isolates of each strain, with standard errors indicated.
In vitro transcription activity of variant promoters.
Promoter activity of the uhpT variants was measured with single-round transcription assays with purified RNAP and UhpA in the absence or presence of CAP, as described previously (33). Synthesis of the uhpT-specific transcript was detected by PhosphorImager (a representative example is shown in Fig. 3), quantified by ImageQuant analysis, and normalized to the level of the vector RNA I transcript (average values of replicate experiments are shown in Fig. 4). Transcription from all uhpT promoters required addition of UhpA (data not shown), which was used here at 220 nM (see below). In the absence of CAP, the variant promoters showed different levels of transcription. The W region variants M11 and M12, which carried changes closest to the RNAP-binding region, were highly defective for UhpA-dependent transcription and showed a >20-fold decrease relative to the wild-type promoter, similar to their effect on uhpT-lacZ expression. UhpA-stimulated activities of variants M1, M2, M3, M4, and M5 in the S region and of M9 in the W region were decreased to 30 to 50% of the level of the wild-type promoter. Variants M6, M7, M8, and M10 were not impaired and showed activities similar to or greater than those of the wild-type promoter. Thus, only the sequences from −40 to −33 altered in variants M11 and M12 contributed strongly to transcription activated by UhpA alone. These sequences may be involved in binding of UhpA, RNAP, or both proteins. Changes in the downstream part of the S region allowed full expression, whereas changes in the upstream part partially decreased stimulation by UhpA. These results indicate that bases in the S region contribute to but are not required for UhpA action.
FIG. 3.

Effect of promoter substitutions on in vitro transcription of the uhpT promoter. Single-round transcription reactions were carried out as described in Materials and Methods with 30 nM RNAP and 220 nM UhpA in the absence (−) and presence (+) of 20 nM CAP, as indicated. Radiolabeled products were resolved by electrophoresis and visualized by PhosphorImager. WT, wild type.
FIG. 4.

Quantification of uhpT transcript produced in single-round transcription assay with UhpA-binding region variants. Product formation by 30 nM RNAP plus 220 nM UhpA is indicated by open bars; product formed with the additional presence of 20 nM CAP is indicated by solid bars. The amount of transcript is in arbitrary units from PhosphorImager and ImageQuant analysis of data similar to that presented in Fig. 3. All values are normalized to the amount of the RNA I transcript and are presented relative to the amount of transcript formed by the wild-type (WT) uhpT promoter in the presence of CAP. Results are averaged from three experiments, with standard error ranges indicated.
The addition of 20 nM CAP along with 220 nM UhpA stimulated transcription from the wild-type promoter by about threefold under these conditions (Fig. 3 and 4) (33). Each promoter variant was stimulated by CAP (Fig. 3). Notably, variants M11 and M12, which had the lowest activity in the absence of CAP, showed the highest degree of stimulation by CAP. Variants M1, M2, and M4 near the CAP-binding region also exhibited about fivefold stimulation by CAP. The M3, M5, M6, M9, M11, and M12 variants had lower than wild-type activity. The lack of consistent correlation between the in vitro and in vivo assays may reflect the use of saturating concentrations of the transcription factors, the absence of competing promoters, and the different ionic conditions. The activities and the extent of CAP stimulation of the remaining variants were close to the wild-type level. Thus, no mutations in the UhpA-binding region prevented CAP-dependent stimulation.
Promoter mutations affect efficiency of UhpA action.
The changes in DNA sequence could affect various steps of the interaction of UhpA with the promoter or RNAP. The effects of UhpA concentration on transcription from the wild-type and the M4 variant promoters were compared in a single-round transcription assay (Fig. 5). The M4 promoter showed about 35% of wild-type activity in the in vivo and in vitro assays. As described previously (33), activation by UhpA shows apparent cooperative behavior, and the amount of transcript synthesized was fit to the following equation for sigmoidal rate dependence:
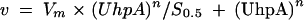 |
FIG. 5.

Dependence of in vitro uhpT transcription on concentration of UhpA. Single-round transcription assays were carried out in the presence of 30 nM RNAP and indicated concentrations of UhpA, as described in Materials and Methods. Template DNAs were plasmid pUT1 derivatives carrying the wild-type sequence (squares) or the M4 variant (circles). The amount of uhpT-specific transcript was quantified by PhosphorImager and ImageQuant analysis, with background subtraction of regions above and below the band corresponding to the specific product. The curve is a fit to the sigmoid rate equation by the DeltaGraph program.
where Vm is maximal velocity and S0.5 is the concentration of UhpA for half-maximal velocity.
The presence of 220 nM UhpA, as was used in the experiment in Fig. 3, resulted in near-maximal rates of transcription at both promoters. Vm calculated by the curve-fitting program for the M4 mutant promoter was 54% of the wild-type value, in fair agreement with the previous assays. The Hill coefficients (n) for the wild-type and M4 promoters were 4.2 and 6.3, respectively, which is consistent with the model that at least four molecules of monomeric UhpA bind in a cooperative manner during the assembly of the transcription initiation complex. This difference in Hill coefficients may not be significant. The UhpA concentrations for half-maximal activity at the wild-type and M4 promoters were 92 and 150 nM, respectively. Similar dependence on UhpA concentration was observed with the variants M1 and M9, which were examined across a more limited range of UhpA concentrations (data not shown). Thus, sequence changes in the S region had a moderate effect on the maximal level of transcription, the concentration dependence for UhpA action, and possibly the cooperativity of UhpA action.
UhpA binding to variant promoters.
The effect of the promoter mutations on UhpA binding was determined by using DNase I footprinting assays. Consistent with previous findings (9, 33), UhpA conferred extensive protection from DNase cleavage in the S region from −74 to −50 on the top strand and from −78 to −51 on the bottom strand (Fig. 6 and data not shown). Weaker protection occurred in the W region from −50 to −33 on the top strand and −48 to −34 on the bottom strand. UhpA induced the appearance of DNase-hypersensitive cleavage sites at −58 on the top strand and at −80, −74, −63, and −50 on the bottom strand. Only qualitative conclusions regarding DNA occupancy by UhpA are presented here because of technical problems which interfered with reliable quantification. There are only a few DNase I cleavage sites in the A+T-rich S region. Several of the sites at which cleavage by DNase was blocked by UhpA were close to sites where cleavage was enhanced by UhpA. Third, the mutational changes often resulted in the appearance of new sites of DNase cutting.
FIG. 6.

Effect of base substitutions on UhpA binding to uhpT promoter. DNase I protection assay was carried out on the uhpT promoter variants in the 180-bp DNA fragment extending from −140 to +40 and in the absence or presence of 800 nM UhpA. Nucleotide coordinates are listed on the left and were based on the position of A/G cleavage reactions (data not shown). The positions of the S and W regions are shown on the right. WT, wild type.
Protection against DNase I cleavage by saturating levels of UhpA (800 nM) in replicate experiments showed that the DNase I-susceptible sites in the S region were strongly protected by UhpA in variants M1, M2, and M6 through M12 (Fig. 6). Occupancy of the S region was markedly decreased in mutants M3 and M5 and was partially decreased in the M4 variant. In all cases in which UhpA protected the S region from cleavage, UhpA also elicited the appearance of the DNase-hypersensitive sites. Thus, the S region sequences changed in variants M3 to M5 (positions −72 and −61) are important for binding of UhpA. The S region variants M3 to M5, which decreased UhpA binding, also decreased binding to the W region. Thus, UhpA occupancy of the W region depends on occupancy of the S region. In contrast, the W region variants M9, M11, and M12 exhibited decreased occupancy of the W region, but normal occupancy of the S region. Variants M6 to M8 at −60 to −49 and M10 showed no major defect in UhpA binding.
New DNase I cleavage sites appeared at the positions altered by the base substitutions (Fig. 6). These sites were protected by UhpA to a similar degree as were the flanking wild-type sequences. This protection of nonnative sequences inserted within the target site indicates that occupancy by UhpA of a specific region is not highly dependent on the sequence of that region, as long as the flanking sequences are intact, consistent with a cooperative binding process.
Sites for attachment of affinity cleavage reagent.
The location and orientation of the UhpA-binding regions were investigated by affinity cleavage reactions, in which a DNA-cleaving moiety was attached near the DNA-binding region of UhpA (12). Cysteine residues were introduced at nine positions of the C-terminal domain of UhpA adjacent to the region of homology to the DNA-binding helix-turn-helix motif of NarL (3). UhpA contains four cysteines in the N-terminal phosphorylation module. Substitution of these native Cys residues to Ser resulted in substantial loss of protein stability and activity in DNA binding and transcription assays (data not shown). Hence, these Cys residues were retained.
Cysteine residues were introduced into UhpA or a variant carrying an N-terminal six-His extension, at positions 154, 155, 181, 182, 183, 185, 189, and 191 (Table 2). Each variant protein was tested for its ability to complement a ΔuhpA strain for expression of a uhpT-lacZ fusion. UhpA-Cys proteins that were active for complementation were expressed and purified. The S183C and R191C variants gave low protein yields and were not studied further. The remaining seven variants were tested for their binding to the uhpT promoter fragment by the DNase protection assay. Cys substitutions at positions 155, 181, and 182 interfered with DNA binding, particularly to the W region. The remaining four variant proteins showed near-normal occupancy of the S and W regions and partial-to-full activity for in vitro transcription and activation of uhpT-lacZ expression. These four variant proteins and wild-type UhpA were derivatized with the sulfhydryl-reactive DNA cleavage agent FeBABE (kindly provided by C. F. Meares, University of California, Davis) (15) and tested for their ability to cause DNA strand cleavage at the uhpT promoter. The chelated iron atom catalyzes formation of hydroxyl radicals by the Fenton reaction upon addition of H2O2 plus ascorbate. The radicals form within 20 Å of the derivatized cysteine residue, and their diffusion is limited by the presence of glycerol as a quenching agent. In this assay, only the UhpA E187C substitution displayed substantially greater DNA cleavage in the S and W regions than did FeBABE-derivatized wild-type UhpA protein. The D185C variant stimulated cutting in the W region, but not the S region (data not shown).
TABLE 2.
Effect of cysteine substitutions in the C-terminal domain of UhpA on DNA binding, transcription activity, and site-specific cleavage
| UhpA varianta | Relative protein yieldb | DNA bindingc
|
Transcription activity
|
FeBABE cleavagef | |||
|---|---|---|---|---|---|---|---|
| S | W | LacZd | In vitroe
|
||||
| −CAP | +CAP | ||||||
| Wild type | 100 | + | + | 100 | 83 | 100 | — |
| H-WT | 100 | + | + | 300 | — | ||
| H-A154C | 100 | + | + | 100 | — | ||
| H-V155C | 66 | ± | − | 37 | |||
| H-G181C | 24 | + | − | 53 | |||
| H-V182C | 33 | ± | − | 38 | |||
| S183C | <15 | 63 | |||||
| D185C | 43 | + | + | 51 | 15 | 52 | W |
| E187C | 100 | + | + | 70 | 17 | 50 | S, W |
| A189C | 42 | + | + | 179 | 23 | 92 | — |
| R191C | <10 | 47 | |||||
UhpA proteins carried the indicated cysteine substitutions. Proteins whose name begins with “H” were synthesized with an N-terminal His tag; all others had the same terminal sequences as the wild-type protein.
The amount of protein, estimated by densitometry of Coomassie blue-stained gels following standard protein purification and resolution by sodium dodecyl sulfate-acrylamide gel electrophoresis, is expressed relative to that of the wild-type protein.
Binding of the purified proteins to the uhpT promoter-containing DNA fragment was determined by DNase footprinting assay. Results were expressed as comparable to the wild-type protein for protection of the S and W regions (+), weaker protection than the wild-type (±), or no detectable protection (−).
Wild-type and cysteine-substitution variants of UhpA protein were expressed from plasmids in cells carrying the uhpT-lacZ fusion. Levels of Glc6P-induced β-galactosidase activities are expressed relative to the wild-type protein.
uhpT-specific transcription driven by 30 nM RNAP-220 nM UhpA variant, in the presence or absence of 20 nM CAP. The amount of labeled RNA product is expressed as the percent uhpT-specific transcript produced by the wild-type UhpA protein.
DNA cleavage of the S or W regions following incubation of DNA with FeBABE-modified UhpA proteins in the presence of hydrogen peroxide and ascorbate. Enhanced cleavage in the S or W regions is indicated. —, no enhanced cleavage.
DNA cleavage by UhpA.
The 5′-labeled DNA fragments complexed with UhpA derivatives were tested by the DNase protection assay (Fig. 7A and B, lanes 3 to 5). Native UhpA and the FeBABE-derivatized wild-type and E187C variant proteins protected the same regions of the uhpT promoter against DNase I cleavage, suggesting that the presence of the adduct did not interfere with DNA occupancy. However, the extent of derivatization by FeBABE was not determined due to the presence of the four native Cys residues, and thus the DNase footprint may reflect unmodified protein.
FIG. 7.

Affinity cleavage reactions at the uhpT promoter. The uhpT promoter fragment carrying residues −140 to +40 were 5′ end labeled on the top (A) or bottom (B) strand. All proteins were treated with FeBABE. In panels A and B, lane 1 shows the product of the affinity cleavage reaction following addition of hydrogen peroxide and ascorbate with wild-type UhpA-FeBABE; lane 2 shows the affinity cleavage products with UhpA E187C-FeBABE; lane 3 shows the DNase I cleavage products in the absence of UhpA proteins; lanes 4 and 5 show DNase protection reactions in the presence of wild-type UhpA-FeBABE and UhpA E187C-FeBABE, respectively. All proteins were present at 1.3 μM. The nucleotide coordinates are shown on the left, and the locations of the S and W regions are indicated on the right. (C) Quantification of the cleavage products from the reactions with UhpA E187C-FeBABE (lanes 2) on the top and bottom strands. The lengths of the bars extending from each base represent the amount of each corresponding cleavage product, as determined by PhosphorImager and ImageQuant analysis.
The FeBABE-derivatized wild-type UhpA caused a low level of DNA cutting, which was interspersed with regions of protection (Fig. 7A and B, lane 1). The low level of DNA cutting was presumably the result of hydroxyl radicals generated by iron atoms of FeBABE coupled to the Cys residues in the phosphorylation module. The pattern of protection was similar to that seen when hydroxyl radicals generated in solution were blocked by bound UhpA (10). The FeBABE-UhpA E187C protein catalyzed increased DNA cleavage at several sites (lanes 2). The strongest cleavage occurred at a cluster of symmetric sites at positions −66 to −61 on the top strand and positions −69 to −64 on the bottom strand. The pattern of offset of the cleavage sites on the two strands indicated that the iron atom was located over the DNA minor groove (12).
The FeBABE-UhpA E187C protein also elicited cleavage in the W region, centered at −41 (positions −43 to −39) on the top strand and at −45 (positions −47 to −43) on the bottom strand (Fig. 7). These cleavages had the same offset pattern on the two strands as in the S region. Cleavage by the UhpA E187C protein in the W region was less intensive than in the S region, consistent with the lower occupancy seen in the DNase I protection assay. The same sites of affinity cleavage were seen in the M5 and M12 variant promoters, although the relative extents of DNA cleavages in the S and W regions were different (data not shown). The observed presence of a single locus of cleavage sites in the middle of each region is most consistent with the conclusion that UhpA binds to each region as a dyad or tail-to-tail protein dimer.
DISCUSSION
This study provides information regarding the recognition sequences for UhpA binding, the number and orientation of bound UhpA molecules, the contribution of UhpA-binding sites to transcription activation, and the existence of cooperativity in UhpA binding. The DNA-binding and activation domain of UhpA is well conserved and widely distributed in several families of transcriptional regulators, but it has not been extensively analyzed. The structure of the related protein NarL provides a framework to model transcription activation functions, although NarL and UhpA differ in the effect of phosphorylation on function. The DNA targets of RR proteins are often difficult to define compared to ligand-regulated high-affinity DNA-binding proteins. The NtrC family response regulators bind to enhancer-like sequences, but DNA binding is not affected by protein phosphorylation. OmpR binds to multiple sites in the ompF and ompC promoters, whose affinities are quite different and are affected by the presence of flanking sites (18). Affinity cleavage reactions suggest that OmpR binds to DNA targets in direct repeats (17). The OmpR family member PhoB binds to direct repeat target sites in the promoter (32). NarL binds to 16-bp regions arranged in a 7-2-7 manner, where the heptad half sites (TACYNMT) are arranged as inverted repeats separated by a 2-bp spacer (14, 24). The NarL-binding sites are often spread over long distances in its target promoters and contribute in different degrees to promoter activity. Indeed, NarL is proposed to act at the nir promoter to block the action of transcriptional silencers, rather than as a typical activator protein (43). UhpA action occurs through its binding to positions −80 to −32, flanked by the CAP-binding site one turn upstream and the RNAP-binding site immediately downstream. The five sequences of the uhpT promoter that UhpA protects from hydroxyl radicals (10) fit the consensus sequence AAAYY in the S region (where Y = C or T) and ARRYY in the W region (where R = A or G). However, the S and W regions lack much sequence similarity. The S region contains 61% A+T and extensive dyad symmetry, in which 24 of the 31 residues form a palindrome centered at position −64. In contrast, the 20-bp W region is only 36% A+T and is less symmetrical, with only an inverted repeat of the sequence GCCTG at each end. Definition of the UhpA recognition sequence required knowledge of the stoichiometry and orientation of the binding sites.
Mutational analysis was used to try to identify sequences important for UhpA binding; findings are summarized in Fig. 8. Our previous experience (27) was that single-base changes in the UhpA-binding region had little effect on promoter activity, but insertion or deletion of even a single base was detrimental (9). These results indicated the need for more drastic base substitutions to overcome the cooperative binding. Hence, we constructed a systematic and unbiased set of 12 mutations that introduced the most extreme replacement for blocks of four residues. The effects of the promoter mutations on in vivo uhpT-lacZ expression and on in vitro transcription were generally similar, but not identical, as might be expected from the different conditions used. Both assays showed that none of the mutations caused a complete loss of expression as occurs in the absence of UhpA protein. All promoter variants except M8 and M10 showed markedly decreased levels of β-galactosidase expression.
FIG. 8.

Identification of the UhpA-binding sequences. The nucleotide sequence is indicated in double-stranded format. The location of the variants is indicated above the sequence, and the regions of affinity cleavage, UhpA-induced DNase hypersensitivity, and hydroxyl radical protection are summarized below. The sequences of the deduced UhpA-binding sites are aligned in the 5′-to-3′ direction at the bottom.
The cooperative binding of UhpA molecules is suggested by the sigmoid dependence of DNA binding and transcription activation on the concentration of UhpA. The Hill coefficients for UhpA binding were ca. 2 at the S region and ca. 3 at the W region (9), and the coefficient for transcription activation was in the range of 4 to 6, which is consistent with the proposal that at least four monomeric UhpA molecules must bind simultaneously for efficient transcription (10). No mutations affected binding to just half of a region, suggesting that both regions can stably bind only a UhpA dimer. The S region variants M3 to M5 showed decreased occupancy of both regions, indicating that occupancy of the S region enhances occupancy of the W region.
Changes in the S region in the M1 through M5 variants resulted in decreased promoter activity. The M3 and M5 substitutions caused the greatest impairment of expression and strongly interfered with UhpA occupancy of both binding regions. The M4 variant showed a lesser degree of impairment in UhpA occupancy, and the M1 and M2 variants had little effect on UhpA occupancy, despite the substantial decrease in promoter activity. We suggest that the M3 to M5 mutations directly impair UhpA binding, but the flanking mutations interfere with bending of the bound complex into a topology needed for optimal promoter activity. A possible indication of the formation of different nucleoprotein structures in the S and W regions was that UhpA-dependent DNase I-hypersensitive sites were only seen in the S region. The W region substitutions M9, M11, and M12 were strongly impaired for UhpA binding and for transcription driven by UhpA and RNAP, suggesting that UhpA occupancy of the W region was crucial for activation by UhpA in the absence of CAP. Promoter activity of the W region variants was stimulated by CAP to a much greater degree than were other substitutions, suggesting that CAP can activate transcription as long as UhpA molecules are bound to the S region.
The DNA affinity cleavage technique provides a powerful tool to investigate the orientation of protein binding to DNA (for examples, see references 17, 30, and 31). Limitations of this technique include the need to find residues at which the cleavage reagent can be attached near the DNA-binding surface and oriented so that hydroxyl radicals produce localized cleavage, but without serious impact on DNA binding or function. We tested nine positions flanking the DNA-binding region, but only one, UhpA E187C, fulfilled all these criteria. Since the extent of labeling at position 187 and the other four cysteine residues in UhpA was not known, the effect of derivatization by FeBABE on DNA binding could not be determined.
The strongest cleavage specific for UhpA E187C occurred in the center of the S region, where symmetric cleavage sites were flanked by regions of protection. This cleavage pattern is most consistent with the binding of two UhpA molecules to inverted repeats on either side of the cleavage region. The M3 and M5 substitutions, which had the strongest effect on UhpA binding, flank the cleaved region and overlap the protected region (Fig. 8). The weaker affinity cleavage in the middle of the W region showed the same pattern as the S region and bore the same relationship to the M9 and M11 substitutions, which impaired UhpA binding to the W region, as occurred in the S region. Additional weak cleavages at both ends of the S region were also produced by FeBABE-derivatized wild-type UhpA. Since this cutting did not depend on the iron atom attached near the DNA-binding surface, it may indicate that UhpA binding distorts the DNA and increases the effectiveness of hydroxyl radical attack. These two sites are located near the ends of oligo(A) tracts, where DNA bending is expected.
The mutational analysis and affinity cleavage results allow deduction of the UhpA-binding consensus (Fig. 8). The W region contains the palindromic sequence 5′-GCCTGGA-N6-CTCAGGC symmetrically flanking the affinity cleavage sites. The corresponding position of the S region contains the related sequence 5′-AACTAAG-N6-TCCAGGT. The downstream SR site, 5′-ACCTGG, is closely related to both W region half-sites, whereas the upstream SL sequence, 5′-AACTAA, is less conserved. Both S region half-sites are preceded by the identical sequence 5′-GGCAA and are embedded in helically phased tracts of four A residues. Similar placement in regions of DNA bending occurs in the binding site for PhoB in the pst promoter (26). The four UhpA half-sites coincide with the regions protected against hydroxyl radical cleavage and with the sites at which mutations most strongly affected promoter activity. Based on these criteria, the consensus recognition sequence is deduced as RcCTgRR, in a 7-6-7 arrangement, where R = A or G, and lowercase letters indicate the preferred residues. The central six positions are variable, but their A-rich character in the S region may contribute to UhpA binding by promoting DNA bending. Recognition of this 16-bp inverted repeat is equivalent to the recognition by NarL of its 7-2-7 target. The cooperative but low-affinity binding of UhpA to its DNA targets complicates further mutational verification of this binding consensus. The basis for the difference in affinity of the S and W regions is not apparent and may depend on nucleoprotein geometry.
The identification of the UhpA-binding sites by our studies is consistent with the recently determined structure of the C-terminal domain of NarL complexed to its DNA target (A. E. Maris, M. R. Sawaya, M. Kaczor-Grzewkowiak, M. R. Jarvis, M. L. Kopka, R. P. Gunsalus, and R. E. Dickerson, submitted for publication), which shows binding of NarL protein as a dimer in inverted orientation with the majority of protein contacts to the DNA backbone rather than specific base contacts. Our results provide an interesting comparison to the binding of BvgA, the regulator of virulence gene expression in Bordetella pertussis, to high- and low-affinity binding sites in the fha promoter (5). Binding to the low-affinity region displays cooperativity with BvgA molecules at the high-affinity site, but binding to the low-affinity site shows little sequence specificity, suggesting that its occupancy is driven mainly by protein contacts with the BvgA molecules at the high-affinity sites. UhpA binding to the W sites shows cooperative dependence on occupancy of the S region, but also specific recognition of sequences in the W region. Taken together, these results suggest that UhpA contributes to transcription activation in several ways. Occupancy of the S region enhances binding of a UhpA dimer to the W region. Occupancy of the W region is needed for transcription activated by UhpA alone. The presence of CAP partly overcomes the decreased promoter activity of W region variants or RpoD mutants, but we have not found a defect in CAP activation due to impaired binding of UhpA. These results provide a basis for further studies of the interaction of UhpA with the other transcription components.
Acknowledgments
We appreciate insightful discussion with Ann Maris and Rob Gunsalus regarding the structure of the NarL-DNA complex. We are most grateful to Claude Meares for providing FeBABE and protocols for its use.
This work was supported by research grant GM38681 from the National Institute of General Medical Sciences.
REFERENCES
- 1.Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.). 1995. Current protocols in molecular biology, vol. 1. John Wiley & Sons, New York, N.Y.
- 2.Baikalov, I., I. Schröder, M. Kaczor-Grzeskowiak, D. Cascio, R. P. Gunsalus, and R. E. Dickerson. 1998. NarL dimerization? Suggestive evidence from a new crystal form. Biochemistry 37:3665-3676. [DOI] [PubMed] [Google Scholar]
- 3.Baikalov, I., I. Schröder, M. Kaczor-Grzeskowiak, K. Grzeskowiak, R. P. Gunsalus, and R. E. Dickerson. 1996. Structure of the Escherichia coli response regulator NarL. Biochemistry 35:11053-11061. [DOI] [PubMed] [Google Scholar]
- 4.Boucher, P. E., K. Murakami, A. Ishihama, and S. Stibitz. 1997. Nature of DNA binding and RNA polymerase interaction of the Bordetella pertussis BvgA transcriptional activator at the fha promoter. J. Bacteriol. 179:1755-1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boucher, P. E., M. S. Yang, D. M. Schmidt, and S. Stibitz. 2001. Genetic and biochemical analyses of BvgA interaction with the secondary binding region of the fha promoter of Bordetella pertussis. J. Bacteriol. 183:536-544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 7.Busby, S., and R. H. Ebright. 1999. Transcription activation by catabolite activator protein (CAP). J. Mol. Biol. 293:199-213. [DOI] [PubMed] [Google Scholar]
- 8.Casadaban, M. J. 1976. Transposition and fusion of the lac genes to selected promoters in E. coli using bacteriophage transposons. J. Mol. Biol. 104:541-555. [DOI] [PubMed] [Google Scholar]
- 9.Chen, Q., and R. J. Kadner. 2000. Effect of altered spacing between uhpT promoter elements on transcription activation. J. Bacteriol. 182:4430-4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dahl, J. L., B. Y. Wei, and R. J. Kadner. 1997. Protein phosphorylation affects binding of the Escherichia coli transcription activator UhpA to the uhpT promoter. J. Biol. Chem. 272:1910-1919. [DOI] [PubMed] [Google Scholar]
- 11.Darwin, A. J., K. L. Tyson, S. J. W. Busby, and V. Stewart. 1997. Differential regulation by the homologous response regulators NarL and NarP of Escherichia coli K-12 depends on DNA binding site arrangement. Mol. Microbiol. 25:583-595. [DOI] [PubMed] [Google Scholar]
- 12.Dervan, P. B. 1991. Characterization of protein-DNA complexes by affinity cleaving. Methods Enzymol. 208:497-515. [DOI] [PubMed] [Google Scholar]
- 13.Déthiollaz, S., P. Eichenberger, and J. Geiselmann. 1996. Influence of DNA geometry on transcriptional activation in Escherichia coli. EMBO J. 15:5449-5458. [PMC free article] [PubMed] [Google Scholar]
- 14.Dong, X. R., S. F. Li, and J. A. DeMoss. 1992. Upstream sequence elements required for NarL-mediated activation of transcription from the narGHJI promoter of Escherichia coli. J. Biol. Chem. 267:14122-14128. [PubMed] [Google Scholar]
- 15.Greiner, D. P., R. Miyake, J. K. Moran, A. D. Jones, T. Negishi, A. Ishihama, and C. F. Meares. 1997. Synthesis of the protein cutting reagent iron (S)-1-(p-bromoacetamidobenzyl)ethylenediaminetetraacetate and conjugation to cysteine side chains. Bioconjug. Chem. 8:44-48. [DOI] [PubMed] [Google Scholar]
- 16.Harlocker, S. L., L. Bergstrom, and M. Inouye. 1995. Tandem binding of six OmpR proteins to the ompF upstream regulatory sequence of Escherichia coli. J. Biol. Chem. 270:26849-26856. [DOI] [PubMed] [Google Scholar]
- 17.Harrison-McMonagle, P., N. Denissova, E. Martinez-Hackert, R. H. Ebright, and A. M. Stock. 1999. Orientation of OmpR monomers within an OmpR:DNA complex determined by DNA affinity cleaving. J. Mol. Biol. 285:555-566. [DOI] [PubMed] [Google Scholar]
- 18.Head, C. G., A. Tardy, and L. J. Kenney. 1998. Relative binding affinities of OmpR and OmpR-phosphate at the ompF and ompC regulatory sites. J. Mol. Biol. 281:857-870. [DOI] [PubMed] [Google Scholar]
- 19.Henikoff, S., J. C. Wallace, and J. P. Brown. 1990. Finding protein similarities with nucleotide sequence databases. Methods Enzymol. 183:111-132. [DOI] [PubMed] [Google Scholar]
- 20.Herlitze, S., and M. Koenen. 1990. A general and rapid mutagenesis method using polymerase chain reaction. Gene 91:143-147. [DOI] [PubMed] [Google Scholar]
- 21.Island, M. D., and R. J. Kadner. 1993. Interplay between the membrane-associated UhpB and UhpC regulatory proteins. J. Bacteriol. 175:5028-5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kadner, R. J. 1995. Expression of the Uhp sugar-phosphate transport system of Escherichia coli, p. 263-274. In J. A. Hoch and T. J. Silhavy (ed.), Two-component signal transduction. ASM Press, Washington, D.C.
- 23.Kolb, A., D. Kotlarz, S. Kusano, and A. Ishihama. 1995. Selectivity of the Escherichia coli RNA polymerase E sigma 38 for overlapping promoters and ability to support CRP activation. Nucleic Acids Res. 23:819-826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li, S.-F., and J. A. DeMoss. 1988. Location of sequences in the nar promoter of Escherichia coli required for regulation by Fnr and NarL. J. Biol. Chem. 263:13700-13705. [PubMed] [Google Scholar]
- 25.Lonetto, M. A., V. Rhodius, K. Lamberg, P. Kiley, S. Busby, and C. Gross. 1998. Identification of a contact site for different transcription activators in region 4 of the Escherichia coli RNA polymerase σ70 subunit. J. Mol. Biol. 284:1353-1365. [DOI] [PubMed] [Google Scholar]
- 26.Makino, K., M. Amemura, T. Kawamoto, S. Kimura, H. Shinagawa, A. Nakata, and M. Suzuki. 1996. DNA binding of PhoB and its interaction with RNA polymerase. J. Mol. Biol. 259:15-26. [DOI] [PubMed] [Google Scholar]
- 27.Merkel, T. J. 1992. Ph.D. thesis. University of Virginia, Charlottesville.
- 28.Merkel, T. J., J. L. Dahl, R. H. Ebright, and R. J. Kadner. 1995. Transcription activation at the Escherichia coli uhpT promoter by the catabolite gene activator protein. J. Bacteriol. 177:1712-1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Merkel, T. J., D. M. Nelson, C. L. Brauer, and R. J. Kadner. 1992. Promoter elements required for positive control of transcription of the Escherichia coli uhpT gene. J. Bacteriol. 174:2763-2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyake, R., K. Murakami, J. T. Owens, D. P. Greiner, O. N. Ozoline, A. Ishihama, and C. F. Meares. 1998. Dimeric association of Escherichia coli RNA polymerase α subunits, studied by cleavage of single-cysteine α subunits conjugated to iron-(S)-1-[p-(bromoacetamido)benzyl]ethylenediaminetetraacetate. Biochemistry 37:1344-1349. [DOI] [PubMed] [Google Scholar]
- 31.Murakami, K., J. T. Owens, T. A. Belyaeva, C. F. Meares, S. J. W. Busby, and A. Ishihama. 1997. Positioning of two alpha subunit carboxy-terminal domains of RNA polymerase at promoters by two transcription factors. Proc. Natl. Acad. Sci. USA 94:11274-11278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okamura, H., S. Hanaoka, A. Nagadoi, K. Makino, and Y. Nishimura. 2000. Structural comparison of the PhoB and OmpR DNA-binding/transactivation domains and the arrangement of PhoB molecules on the phosphate box. J. Mol. Biol. 295:1225-1236. [DOI] [PubMed] [Google Scholar]
- 33.Olekhnovich, I. N., J. L. Dahl, and R. J. Kadner. 1999. Separate contributions of UhpA and CAP to activation of transcription of the uhpT promoter of Escherichia coli. J. Mol. Biol. 292:973-986. [DOI] [PubMed] [Google Scholar]
- 34.Olekhnovich, I. N., and R. J. Kadner. 1999. RNA polymerase α and σ70 subunits participate in transcription of the Escherichia coli uhpT promoter. J. Bacteriol. 181:7266-7273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parekh, B. S., and G. W. Hatfield. 1996. Transcriptional activation by protein-induced DNA bending: evidence for a DNA structural transmission model. Proc. Natl. Acad. Sci. USA 93:1173-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perez-Martin, J., and V. de Lorenzo. 1997. Clues and consequences of DNA bending in transcription. Annu. Rev. Microbiol. 51:593-628. [DOI] [PubMed] [Google Scholar]
- 37.Roland, K. L., C. Liu, and C. L. Turnbough. 1988. Role of the ribosome in suppressing transcriptional termination at the pyrB1 attenuator of Escherichia coli K-12. Proc. Natl. Acad. Sci. USA 85:7149-7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 39.Simons, R. W., F. Houman, and N. Kleckner. 1987. Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene 53:85-96. [DOI] [PubMed] [Google Scholar]
- 40.Stock, A. M., V. L. Robinson, and, P. N. Goudreau. 2000. Two-component signal transduction. Annu. Rev. Biochem. 69:183-215. [DOI] [PubMed] [Google Scholar]
- 41.Weston, L. A., and R. J. Kadner. 1987. Identification of the Uhp polypeptides and evidence for their role in exogenous induction of the sugar-phosphate transport system of Escherichia coli. J. Bacteriol. 169:3546-3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weston, L. A., and R. J. Kadner. 1988. Role of uhp genes in expression of the Escherichia coli sugar-phosphate transport system. J. Bacteriol. 170:3375-3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu, H., K. L. Tyson, J. A. Cole, and S. J. Busby. 1998. Regulation of transcription initiation at the Escherichia coli nir operon promoter: a new mechanism to account for co-dependence on two transcription factors. Mol. Microbiol. 27:493-505. [DOI] [PubMed] [Google Scholar]
- 44.Zhang, X., A. Gunasekera, Y. Ebright, and R. H. Ebright. 1991. Derivatives of CAP having no solvent-accessible cysteine residues, or having a unique solvent-accessible cysteine residue at amino acid 2 of the helix-turn-helix motif. J. Biomol. Struct. Dyn. 9:463-473. [DOI] [PubMed] [Google Scholar]