

Abstract
Immunoglobulin mu binding protein 2 (IGHMBP2) is a DNA/RNA helicase with a putative role in transcriptional regulation and splicing. A recessive mutation of the Ighmbp2 gene in neuromuscular degeneration (nmd) mice causes progressive neurogenic atrophy of limb muscles. Affected mice show significant loss of motor neurons with large caliber axons and a moderate reduction of neurons with small caliber axons in the ventral nerve roots of the spinal cord. To investigate the role of Ighmbp2 in the pathogenesis of neuromuscular degeneration, we generated two independent lines of transgenic mice expressing the full-length Ighmbp2 cDNA specifically in neurons. Histopathological evaluation of L4 ventral nerve roots revealed that transgenic expression of the Ighmbp2 cDNA prevented primary motor neuron degeneration, while restoring the normal axonal morphology and density in nmd mice. A similar neuronal improvement is found in mutant mice carrying the CAST/EiJ-derived modifier of nmd (MnmC). Intriguingly, both the transgenic and modified nmd mice went on to develop a previously unobserved cardiac and skeletal myopathy. Necropsy of nmd mice in end-stage heart failure revealed a primary dilated cardiomyopathy with secondary respiratory failure that was confirmed by in vivo ECG and echocardiographic measures. Our results suggest that reduced levels of IGHMBP2 in nmd mice compromise the integrity and function not only of motor neurons, but also of skeletal and cardiac myocytes. These findings highlight the important role of IGHMBP2 in the maintenance and survival of these terminally differentiated cell types.
Introduction
The Ighmbp2nmd–2J neuromuscular degeneration mutation (hereafter referred to as nmd) arose spontaneously at The Jackson Laboratory and was identified by positional cloning and sequence analysis as a splice donor mutation, reducing functional IGHMBP2 expression to 20–25% of controls (1). Mice homozygous for the nmd mutation have life spans that range from 12 to 138 days and exhibit progressive and severe muscle wasting of the hind limbs, followed by the forelimbs, secondary to loss of motor neuron innervation.
Immunoglobulin mu binding protein-2 (IGHMBP2) is a member of the DEXDc DEAD-like superfamily of DNA/RNA helicases and is thought to act as a transcriptional activator or associate with pre-mRNA splicing complexes (2–4). Mutations in the human IGHMBP2 gene were recently identified in patients with spinal muscular atrophy with respiratory distress type 1 (SMARD1). This disease is genetically distinct from Chr. 5q13 spinal muscular atrophy (SMA) and chronic distal SMA that maps near SMARD1 at Chr. 11q13 (5–9). SMARD1 patient mutation analysis revealed missense, nonsense, splice donor, inframe deletion and frameshift mutations in the IGHMBP2 gene, presumably leading to loss of function (5, 7–9). Although SMARD1 involves life-threatening respiratory distress consequent to paralysis of the diaphragm, it is also accompanied by severe limb muscle atrophy due to progressive loss of muscle innervation, similar to the nmd mutant mouse (5–9). As a model for human SMARD1, the nmd mouse provides a novel experimental system to begin addressing the selective vulnerability of neuronal subtypes and identifying factors that can alter the onset of clinical disease (10).
We previously reported that a major genetic modifier of the nmd phenotype from CAST/EiJ mice (MnmC) is located on mouse Chr. 13 (1). Mice homozygous for both the nmd mutation and the MnmC modifier locus display a less progressive, if not arrested, neurogenic muscular atrophy. We show here that neuron-specific expression of the wild-type mouse Ighmbp2 cDNA also prevents motor neuron and axonal degeneration in nmd mice. However, despite their greatly improved mobility and muscle function, nmd mice carrying either the Ighmbp2 transgene or the MnmC modifier locus display a mild skeletal myopathy and succumb to congestive heart failure due to dilated cardiomyopathy (DCM). These data suggest that Ighmbp2 expression is required not only in motor neurons, but also in skeletal and cardiac myocytes.
Chronic heart failure and other cardiac dysfunctions stemming from diseases of the myocardium are morphologically and hemodynamically classified as dilated cardiomyopathy (the most common form in humans) and hypertrophic and/or restrictive cardiomyopathies (11, 12). Mechanisms leading to cardiomyopathy include impaired force generation due to deficiency of sarcomeric proteins (13), reduced force transmission due to reduction of cytoskeletal proteins (14), abnormal intracellular signaling or nuclear membrane instability (15, 16), mitochondrionopathy and deficits in cardiac energetics (17), and facilitated cell death (apoptosis and/or necrosis) (18–22). We show here that reduced expression of the DNA/RNA helicase IGHMBP2 in nmd mice leads to cardiomyocyte death and morphological and functional alteration of the myocardium. Our discovery of a progressive cardiomyopathy in the nmd mouse suggests that treatment of neurogenic atrophy in SMARD1 patients may reveal a similar role for IGHMBP2 in human cardiomyopathy.
Results
Tissue-specific Rescue of Neuromuscular Degeneration in Transgenic nmd Mice.
As the Ighmbp2 gene is ubiquitously expressed, we reasoned that motor neuron degeneration in nmd mice could be caused by a specific requirement for IGHMBP2 in neurons themselves, in cells that interact with motor neurons such as astrocytes or Schwann cells, or in the skeletal muscle targets of motor innervation. To address the role of Ighmbp2 in neurons, we generated transgenic nmd mice on the C57BL/6J (B6) background expressing a full-length Ighmbp2 cDNA under the control of the rat enolase 2, gamma, neuronal (Eno2) promoter (C57BL/6J-TgN(Eno2-Ighmbp2)), hereafter referred to as TgNI. A schematic representation of the transgenic construct is shown in Fig. 1A. RT-PCR analysis revealed that TgNI-derived Ighmbp2 mRNA expression was limited to the central nervous system including forebrain, cerebellum and spinal cord in two independent transgenic lines (Fig. 1B, and data not shown).
Figure 1.
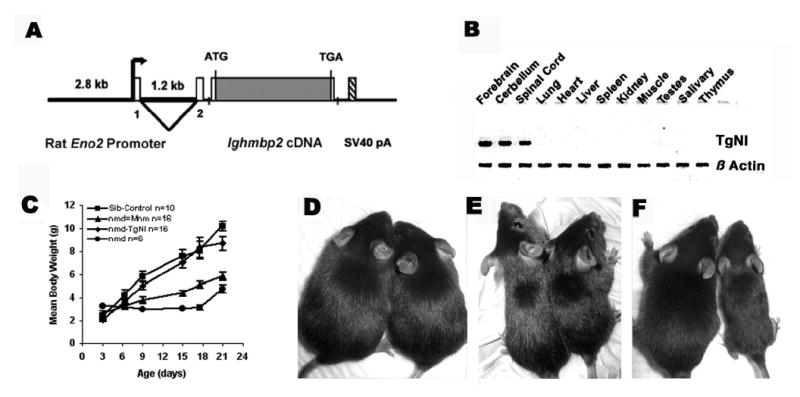
(A) Schematic illustration of the TgN(Eno2-Ighmbp2) transgenic construct. Tissue-specific expression of the Ighmbp2 cDNA was regulated by the rat neuron-specific enolase (Eno2) gene (34). (B) RT-PCR analysis of transgene expression in twelve adult tissues reveals that expression is limited to neuronal tissues compared to a ubiquitous β-actin control. (C) Neonatal-to-weaning growth curve and mean body weight of combined male and female nmd, modified nmd-MnmC and transgenic nmd mice compared to their unaffected littermate controls (+/+ or +/nmd). (D–F) Photographs of transgenic nmd, nmd-MnmC, and nmd mice (right) with their sib controls (left) at approximately 7 wk of age. (D) The transgenic nmd mouse is indistinguishable from its +/nmd sib control while the modified nmd-MnmC mouse (E) is visibly smaller than a +/nmd sib control. (F) A homozygous nmd mouse is significantly smaller than its +/nmd sib control due to progressive skeletal muscle atrophy.
To assess whether expression of the Ighmbp2 transgene could rescue nmd motor neuron degeneration, transgenic carriers from lines 17 and 90 were independently crossed with B6-+/nmd mice to produce transgene-positive homozygous nmd/nmd mutant mice. Morphologic, histopathologic and functional data were compared between nmd mice with and without the transgene as well as nmd mice homozygous for our previously described CAST/EiJ-derived MnmC modifier locus (1). As shown in Figure 1C, growth rates for nmd mice, as measured by mean body weight, are indistinguishable from their control littermates’ from birth up to approximately 1 week of age. During the second and third weeks of life, however, the muscle atrophy of nmd mice results in a significantly reduced mean body weight, such that by 21 days of age the weights of nmd mice were less than half those of their control sibs (4.0 ± 0.33 g, n = 9 vs. 9.5 ± 0.43 g, n = 28, p < .0001). In contrast, nmd mice carrying either the line 17 or line 90 transgene had a growth rate that was not significantly different from their control littermates’ (+/+ or +/nmd) at any time point. The nmd mice carrying the MnmC modifier locus displayed an intermediate growth rate that led to weights significantly different from those of nmd-TgNI mice at 21 days of age, 5.8 ± 0.3 g (n = 33) vs. 9.9 ± 0.43 g (n = 34, p < 0.0001), respectively. Observed differences in body weight among the three groups of nmd mice were readily apparent at weaning (Fig. 1D–F), and were further exacerbated by the onset of dilated cardiomyopathy (described below) as early as 4–6 weeks of age. Both male and female transgenic nmd mice from lines 17 and 90 displayed equivalent growth rates, average life spans (54 days) and performance scores in functional tests (described below); therefore mice from these two independent lines were combined for all subsequent analyses.
To investigate the nature of the motor improvement in nmd-TgNI and nmd-MnmC mice, gross and microscopic evaluation of the ventral spinal nerve roots and hind limb skeletal muscles was conducted. Seven-week-old nmd mice display significant neuronal loss as evidenced by a gross reduction in the diameter of L4 ventral nerve root cross-sections (Fig. 2A and B). Similar reductions were observed in all lumbar ventral roots examined (L2–L6, not shown). In contrast, normal axonal morphology and nerve root diameters are observed in the ventral roots of aged-matched nmd-TgNI mice (Fig. 2D). Furthermore, a modest reduction in diameter of the ventral roots of 7 wk nmd-MnmC mice is observed, suggesting that the MnmC modifier does not affect as complete a rescue of motor neuron degeneration as does the Ighmbp2 transgene (Fig. 2C and D). Higher magnifications of nmd ventral root cross-sections reveal numerous dystrophic large and small caliber axons and a paucity of healthy large caliber axons compared to age-matched B6 controls (Fig. 2E and F). The effect of the transgene is evident from the absence of dystrophic axons and the restoration of normal axonal morphology and density in nmd-TgNI mice (Fig. 2H). Dystrophic axons are occasionally observed in nmd-MnmC ventral nerve roots (Fig. 2G).
Figure 2.
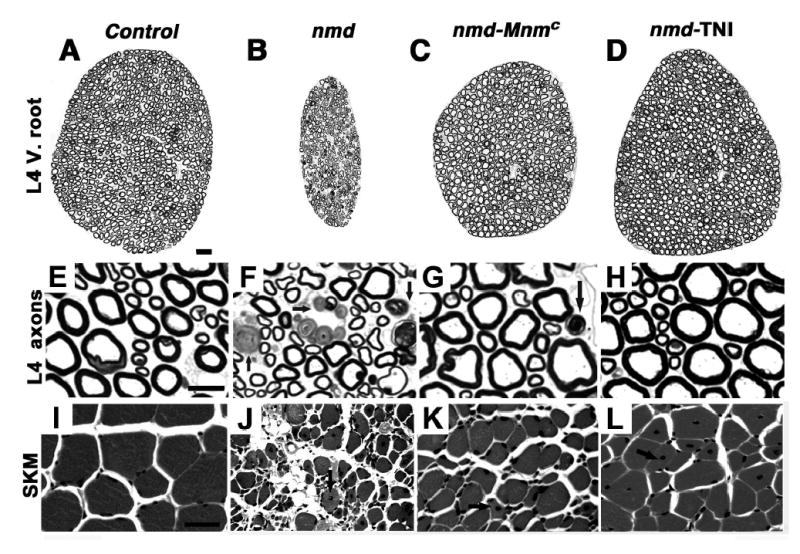
Histologic evaluation of spinal cord L4 ventral roots and hind limb muscles in 6–8 week old mutant nmd, nmd-MnmC, nmd-TgNI and age-matched C57BL/6J control mice (Scale bars: A to D, 20 μm, E to H, 10 μm, I to L, 25 μm). (A–D) Note the marked reduction in the overall diameter of the L4 ventral nerve root from an nmd mouse (B) compared to a control (A) ventral root. Significant improvement is observed in modified nmd-MnmC mice (C), and the presence of the transgene in nmd-TgNI mice (D) leads to restoration of L4 ventral nerve root diameter. Abundant dystrophic axons (arrow) are observed at higher magnification in the nmd L4 ventral root (F), unlike the modified nmd-MnmC mouse (G), which showed fewer dystrophic axons. In the transgenic nmd-TgNI mouse (H) both the morphology and density of the L4 axons were indistinguishable from controls (E). Panels I – L shows cross-sections of quadriceps skeletal muscles stained with H & E. Severe morphological changes of the myofibers including variation in fiber size and central nuclei (arrow) are evident in nmd muscle (J). In contrast, despite motor neuron rescue and less fiber size variation, modified nmd-MnmC (K) and transgenic nmd-TgNI (L) mice display evidence of an underlying myopathy (arrows).
Transgenic nmd-TgNI mice display a grossly normal hind limb muscle phenotype in contrast to their nmd littermates who exhibit severe muscle wasting and marked contracture of the hind limbs (Fig 1D and F). However, unlike the rescue of motor neuron degeneration observed in nmd-TgNI mice, histopathologic evaluation of hind limb skeletal muscle revealed mild myopathic changes, including degenerating and regenerating myocytes with centralized nuclei (Fig. 2I and L). In contrast, non-transgenic nmd mice display severe, diffuse myocyte degeneration and regeneration with extensive variability in fiber size, centralized nuclei and interstitial fibrosis and fatty infiltration (Fig. 2J). Modified nmd-MnmC muscles exhibit fewer changes with mild atrophy and moderate numbers of degenerating/regenerating myocytes that correlate with the observed reduction in ventral nerve root diameter (Fig. 2K). These observed morphological changes in the skeletal muscles of transgenic and MnmC-modified nmd mice suggest the presence of an underlying skeletal myopathy that was masked by the severe neurogenic muscular atrophy in nmd mutant mice.
A quantitative assessment of motor neuron rescue is reflected in the total and differential axonal diameter counts between nmd, nmd-TgNI and nmd-MnmC mice. As shown in Figure 3A, there is no significant difference in the total number of L4 ventral root motor axons between transgenic nmd and B6 control mice (1084 ± 26 vs. 1089 ± 25.5, p > 0.97). In contrast, greater than half (56%) of L4 motor axons are lost in non-transgenic nmd mice. In the presence of the MnmC modifier, this loss was significantly reduced – preserving 74% of the control number of axons in L4 ventral nerve roots (Fig. 3A). Interestingly, unlike in human SMARD1 patients, we found no evidence for axonal loss in the phrenic nerve innervating the diaphragm muscles of nmd mice (data not shown). Morphometric analysis revealed that the highest proportion of axonal loss in nmd mice occurred in large (102 ± 17 vs. 501 ± 16 axons in B6 controls, p < 0.001), followed by small (183 ± 17 vs. 328 ± 5 axons, p = 0.0014) and medium (208 ± 5 vs. 274 ± 14 axons, p = 0.046) caliber myelinated axons (Fig. 3A). Though less severe, a similar pattern of loss was observed in modified nmd-MnmC, with significant reductions in large (392 ± 19 axons, p = 0.026), small (233 ± 50 axons, p = 0.026) and medium (210 ± 24 axons, p = 0.001) caliber axons.
Figure 3.
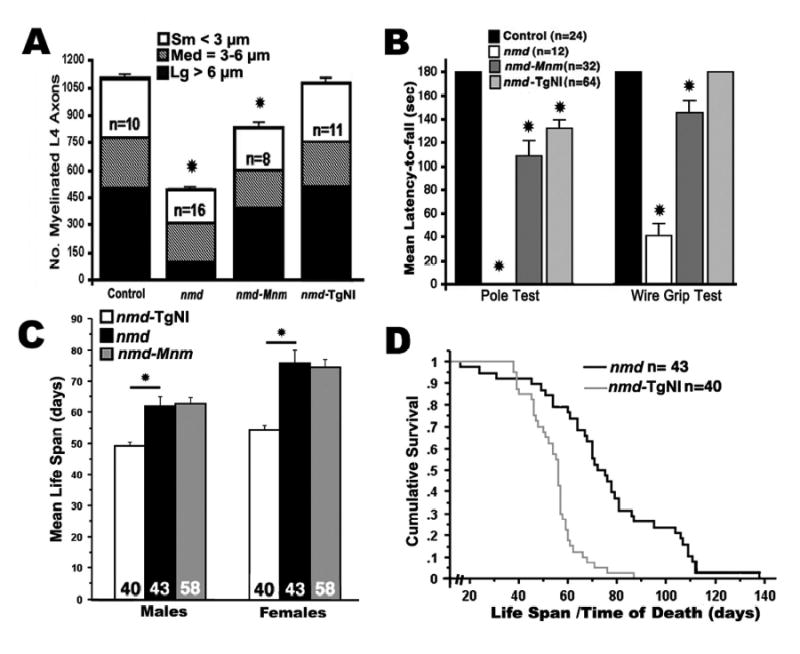
Histomorphometric assessment of L4 ventral nerve roots and muscle function tests in 30 to 50 day old mutant mice and age-matched B6 or sib controls. (A) Mean number of total myelinated axons from L4 ventral nerve roots was significantly reduced in 4–7 week old nmd mice versus age-matched B6 controls (477 ± 26 vs. 1089 ± 25, *p < 0.0001). In nmd-MnmC mice, their mean number of myelinated axons almost doubled, but is still significantly less than in the B6 control (806 ± 35, *p < 0.0001). No significant difference was observed between nmd-TgNI mice and B6 controls. The proportion of large (> 6 μm), medium (3–6 μm) and small (< 3 μm) caliber axons relative to the overall mean number of myelinated axons and the number (n) of L4 ventral roots sampled for each genotype is shown. (B) A significantly shorter mean latency to fall by all three genotypes of nmd mutant mice was observed in the pole test compared to controls (*p < 0.0001). Likewise, nmd and modified nmd-MnmC mice showed significant deficits in the wire grip test (*p = 0.003 and *p < 0.0001, respectively), while the grip strength of transgenic nmd-TgNI mice was not significantly different from littermate controls’. (C) Mean life span of nmd mice. Female nmd mice lived longer than male nmd mice (*p < 0.0001). Transgenic nmd-TgNI mice had the shortest life span compared to nmd and modified nmd-MnmC mice (p < 0.001). The mean life spans of nmd and modified nmd-MnmC mice were not significantly different. (D) Kaplan-Meier survival analysis of female nmd mice versus female transgenic nmd-TgNI mice reveals significant differences in cumulative survival rates between the two groups (log rank test, p < 0.0001).
The greatest contribution to muscle strength and power comes from large motor units composed of one alpha motor neuron with a large caliber axon innervating numerous myofibers (10). Nearly half (46%) of all motor axons in the L4 ventral nerve root are large caliber (> 6 μm) axons in B6 mice. A pole grip test was employed to asses the muscular deficit due to the greater than 75% loss of large motor axons in nmd mice (Fig. 3B). Similarly, the function of small motor units that control coordination and fine movement of the digits was quantified using a wire grip test. The mean latency to fall from a wire cage lid was significantly shorter in modified nmd-MnmC mice, with a 25% loss of small caliber axons, and showed an even greater reduction in nmd mice, with a 40% loss of small caliber axons. Although the total axonal density of L4 ventral root in modified nmd-MnmC mice was significantly reduced by approximately 25%, their overall performance in the pole test was generally comparable to that of transgenic nmd-TgNI mice. The presence of an underlying mild skeletal myopathy (Fig. 2K and L) and/or reduction in cardiac reserves in nmd-TgNI mice and nmd-MnmC mice may have adversely affected their overall performance.
Rescue of Neurogenic Atrophy Reveals Cardiomyopathy.
Despite the significant decrease in neurogenic atrophy observed in modified nmd-MnmC mice and the complete rescue of motor neuron loss in transgenic nmd-TgNI mice, these genetic interventions failed to increase the life span of nmd mice. In fact, as shown in Figure 3C, both male and female nmd-TgNI mice displayed significantly shorter mean life spans (49.2 ± 1.3 days, and 54.5 ± 1.7 days, p < 0.001) than gender matched nmd mice (61.8 ± 3.2 days, and 75.9 ± 4.0 days). In comparison, the mean life spans of nmd and modified nmd-MnmC mice were not significantly different. Additionally, the maximum life span (87 days for females) and cumulative survival rates of nmd-TgNI were significantly lower than those of either nmd or nmd-MnmC (138 days for female nmd) mice (Fig. 3C and D). Post-mortem examination of the cardiovascular system of all nmd mice (nmd, nmd-MnmC or nmd-TgNI) confirmed the presence of a grossly dilated heart containing one or more thrombi and consolidation of the lungs with concomitant pleural effusion suggesting that they died of congestive heart failure. Behavioral adaptations and changes in normal activity predicted the underlying heart disease in all nmd mice regardless of the degree of motor neuron rescue afforded by the MnmC modifier or TgNI transgene. Early clinical signs of heart disease included noticeable weight loss and a reduced breathing rate (bradypnea) accompanied by moderate facial or generalized edema. Except in cases of sudden death, mice that survived at least one week from the initial onset of clinical symptoms showed a distinctive presentation marked by an exaggerated hunched posture and labored breathing (dyspnea).
To correlate the clinical diagnostic findings with pathological changes, gross and histological examinations were performed in representative nmd mice and their control littermates shortly after heart rate (HR) and electrocardiography (ECG) data were obtained (described below). Clinically affected mice in end-stage heart failure have pale, flaccid and enlarged rounded hearts often containing one or more visible thrombi (Fig. 4A). Longitudinal sections revealed severe atrial and ventricular chamber dilation with marked thinning of the ventricular walls, which frequently contained mural thrombi (Fig. 4B and D). Detailed histopathologic evaluation of hearts from nmd mice with overt clinical signs revealed a full range of morphological changes including, but not exclusively, the presence of degenerating, apoptotic and necrotic cardiomyocytes with no signs of inflammation. With progressive cardiomyocyte dropout, potential secondary valvular insufficiency caused by the attenuation of the papillary muscles are likely to exacerbate atrial hypertrophy, systolic dysfunction and congestive heart failure (Fig. 4C and D). In addition, all nmd hearts develop moderate to severe interstitial fibrosis, indicated by the presence of collagen stained blue by Trichrome stain (Fig. 4E and F). Histochemical staining for succinate dehydrogenase (SDH) and cytochrome oxidase (COX) did not show a significant difference between nmd and control hearts suggesting that there is not a major defect in the functional integrity of the mitochondrial respiratory chain complexes II and IV (data not shown). Transmission electron micrograph (TEM) analysis of ventricular myocytes from 7 wk nmd hearts revealed an overabundance of mitochondria in various stages of degeneration, manifested as hydropic changes (mitochondrial swelling) accompanied by myofibrillar lysis and fragmentation. In addition, ventricular myocytes from nmd mice display attenuated intercalated disks with relatively few desmosomes and fasciae adherentes between gap junctions (data not shown). As shown in Figures 4G and H, nuclear morphological changes such as two or more chromatin condensations, chromatin margination and blebbing of the nuclear membrane, consistent with an apoptotic process, are frequently observed in nmd ventricular myocytes (23).
Figure 4.
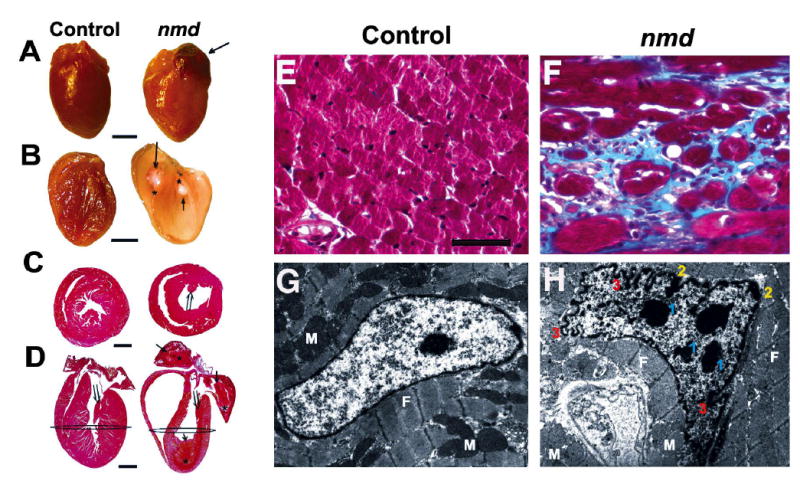
(A) Gross appearance of a cardiomyopathic nmd heart (right) and an unaffected littermate control (left) (Scale, bar = 2 mm). (B) The same hearts sectioned longitudinally, showing the nmd heart (right) with grossly attenuated walls and dilated chambers containing one or more thrombi (↑ and * mark the limits of each thrombus) compared to the control heart (left). (C) Cross sections of hearts taken at the level depicted in panel D showing longitudinally sectioned hearts stained with H& E (Scale bar = 2mm). The nmd heart has one or more organizing mural thrombi (⇒ and * mark the limits of each thrombus). Also, note the attenuated papillary muscles (⇑ in C and D) in the nmd versus control heart. Masson’s Trichrome stained histological samples of the ventricular myocardium (F) from an nmd mouse displayed severe interstitial fibrosis, indicated by the blue stained collagen, compared to the control in panel E (Scale, bar = 25 μm). Panel G shows an electron micrograph of a normal cardiomyocyte nucleus, while panel H shows an nmd ventricular cardiomyocyte nucleus. Morphological alterations include multiple nuclear condensations in addition to the nucleolus (1), chromatin margination (2), and marked ruffling of the nuclear membrane (3) suggestive of an apoptotic process. (M = mitochondria, F = myofibrils; Magnification = 6,700x).
Pre-clinical and early clinical non-invasive diagnoses of cardiomyopathy were evaluated by measuring total serum creatine kinase (CK) and its cardiac isoform (CK-MB), blood pressure, heart rate and electrocardiography (ECG). Apparently healthy 3–5wk old asymptomatic nmd, nmd-MnmC and nmd-TgNI mice had total plasma CK and cardiac CK-MB levels that were not significantly different from their unaffected littermates or B6 controls (pre-clinical nmd CK = 193.2 ± 27.5 U/L; CK-MB = 137.1 ± 16.0 U/L, n = 6, vs. control CK = 202.9 ± 18.8 U/L; CK-MB = 114.4 ± 18.8 U/L, n = 15, p > 0.05). However, approximately 3–7 days prior to clearly evident behavioral signs of heart disease, total plasma CK and cardiac-specific CK-MB levels in 5–9 wk old nmd, nmd-MnmC and nmd-TgNI mice become significantly elevated (clinical nmd CK = 1322.8 ± 144.7 U/L; CK-MB = 727.9 ± 102.7 U/L, n = 18, p < 0.001). This dramatic increase in total serum CK and CK-MB isoenzymes represents at least a 5 fold increase above pre-clinical nmd and control values. Hence, measurement of CK-MB can be used reliably to confirm the presence of cardiomyopathy and follow its progression in nmd mice. In fact, one modified nmd-MnmC mouse that was measured serially for CK-MB at 5 and 7 weeks of age captured the transition from a pre-clinical to clinical dilated cardiomyopathy phenotype that was confirmed by echocardiography (Table 1 and Fig. 6B and C). Interestingly, other markers of cardiomyopathy are not upregulated in nmd hearts such as ANP or BNP mRNA levels which showed no differences in northern blots between pre-clinical and clinical nmd mice and age-matched controls (data not shown).
Table 1.
Cardiac creatine kinase, heart rate and echocardiographic indices in nmd mice.
| M-Mode of the Left Ventricular Chamber | Diastole | Systole | Calculated | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | Clinical Presentation | CK-MB U/L | HR bpm | Wallmm | Vol.ml | Wallmm | Vol.ml | CO ml/min | EF % | FS % |
| Control | none | 163 | 359 | 0.599 | 42.6 | 1.227 | 10.5 | 8.025 | 75.4 | 43.0 |
| nmd-Mnm* | none | 131 | 360 | 0.678 | 36.2 | 0.889 | 16.2 | 4.163 | 53.8 | 26.7 |
| nmd-Mnm* | yes | 963 | 355 | 0.430 | 45.4 | 0.431 | 27.3 | 3.592 | 34.5 | 18.7 |
| nmd/nmd | yes | 1229 | 395 | 0.576 | 56.5 | 0.687 | 42.0 | 3.054 | 25.5 | 11.5 |
| nmd-TgNI | yes | 911 | 364 | 0.450 | 50.2 | 0.700 | 41.2 | 2.043 | 18.0 | 7.9 |
CK-MB, creatine kinase cardiac isoenzyme; HR, heart rate via doppler; Wall, ventricular free wall thickness via M-mode; CO, calculated cardiac output; EF, ejection fraction; FS, fractional shortening; measurements were obtained from 5–7-wk old mice.
The same mouse was tested serially at 5wk-of age and then again at 7-wk-of age.
Figure 6.
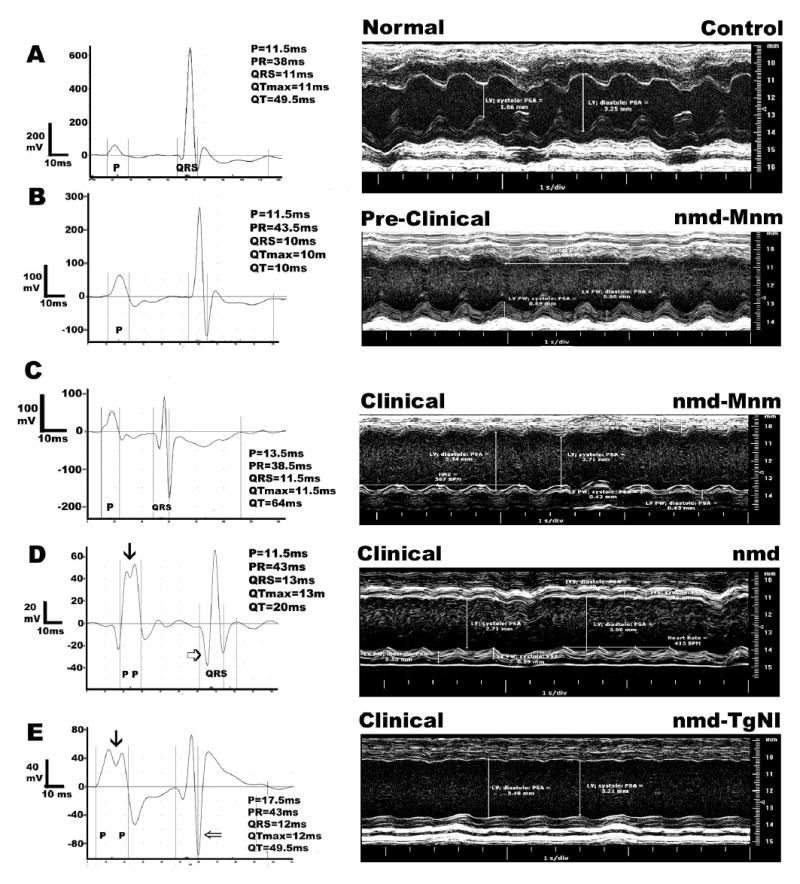
Representative electrocardiograms and M-mode echocardiographic images in 5–7 week old nmd mice with different clinical presentations based on normal/elevated levels of creatine kinase cardiac isoenzyme CK-MB. Panel A represents a normal B6 control. In panels B and C, the same modified nmd-MnmC mouse was tested before clinical symptoms were evident at 5 wks of age and again following a significant rise in its CK-MB values at 7 wks ( Table 1). Panels D and E were obtained from an nmd or transgenic nmd-TgNI mouse with overt clinical signs, respectively. The three mice with clinical heart failure showed similar changes in echocardiographic indices compared to the control mouse ( Table 1 and A, C, D and E). In contrast, the mouse in panel B, despite an apparently normal CK-MB level (Table 1), showed intermediate echocardiographic indices. The representative electrocardiograms further validate the presence of cardiomyopathy and its progression toward heart failure. Note also the various scales in each electrocardiogram (mV), which were necessary in order to show each individual tracing in greater detail.
Cardiovascular evaluation using other ancillary diagnostic methods, such as the measurement of end systolic blood pressure via tail cuff with minimal restraint showed that 5–9 week-old pre-clinical male and female nmd mice, with or without motor neuron rescue due to the MnmC modifier or transgene, were not significantly different from those of controls (94.0 ± 3.4 mmHg, n = 7, vs. 95.5 ± 2.6 mmHg, n = 8, p > 0.05). Simultaneous pulse rate (PR) measurements in this same group of mice showed equally comparable rates (nmd PR = 502 ± 28 bpm, n = 8; nmd-MnmC PR = 526 ± 14, n = 8, vs. B6 control PR = 539 ± 17, n = 8, p > 0.05). In contrast, all nmd mice with overt clinical signs showed significantly lower end systolic blood pressures (67.8 ± 4.2 mmHg, n = 13, p < 0.006). However, heart rate (HR) measurements in un-anesthetized and unrestrained 5–12 week-old nmd mice also showed modest, but significant reduction in HR (701 ± 20 bpm, n = 12, vs. 781 ± 7 bpm, n = 17, p < 0.01). As shown in Figure 5A, these differences became more pronounced in another group of nmd, nmd-MnmC and nmd-TgNI mice with overt clinical signs (460 ± 57 bpm, n = 12, p<0.0001). Simultaneous ECG recording using the same diagnostic modality likewise showed that even a small increase in the P-R interval in the pre-clinical nmd mice resulted in an observed decrease in heart rate (nmd-MnmC P–R duration = 23.14 ± 0.64 ms, n = 3, versus control P–R = 21.75 ± 0.53 ms, n = 9, p < 0.038). Although there was no apparent sinus arrhythmia, clearly there were increases in heart rate variability and/or electrocardiographic irregularities in atrial and ventricular conduction. Therefore in overtly clinical nmd mice with/without the transgene or MnmC modifier, prolongation of the P–R interval and/or the QRS complex was characterized by a concurrent reduction in heart rate. In addition, electrocardiograms of end-stage hearts often showed broadened (prolonged) and attenuated (reduced in amplitude) P and R waves. These electrocardiographic observations are consistent with the diagnosis of congestive heart failure due to systolic dysfunction.
Figure 5.
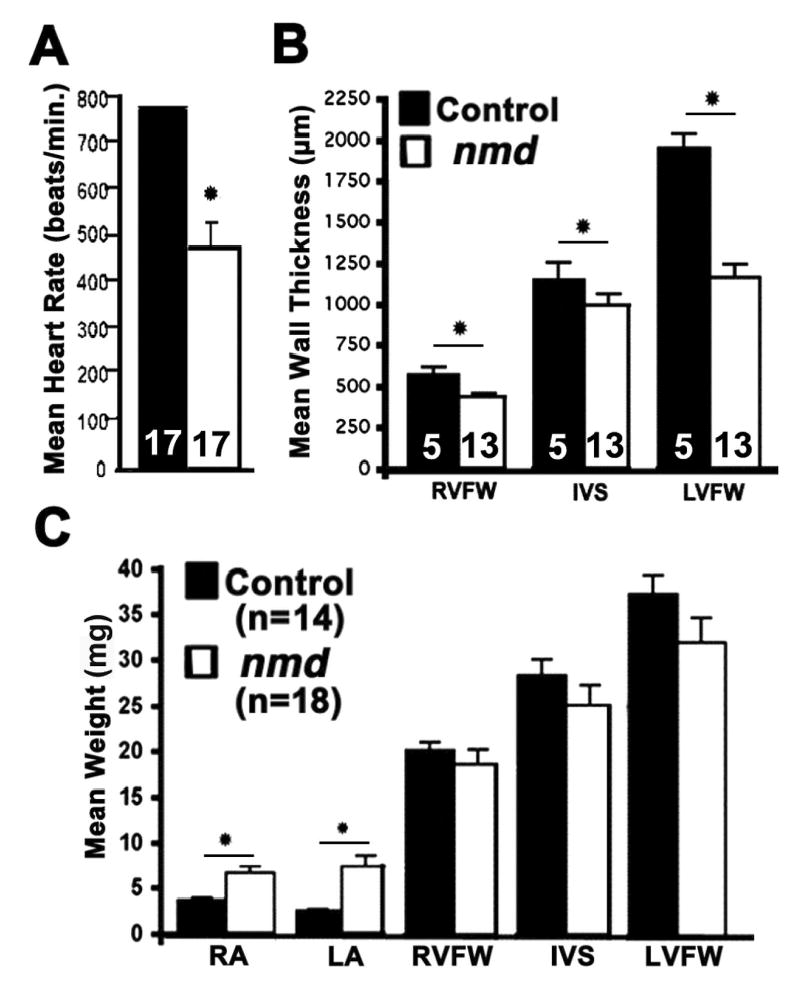
(A) In vivo non-invasive assessment of cardiac function in 4–10 week old mice. The mean resting heart rate of nmd mice (460 ± 57 beats/min) is significantly lower than that of sib controls (781 ± 7 beats/min, p < 0.0001). (B) Histomorphometric evaluation of post-mortem (in tetany) ventricular wall thickness in nmd hearts shows a significant reduction in the right ventricular free wall (RVFW, *p < 0.01), interventricular septum (IVS, * p < 0.05) and left ventricular free wall (LVFW, *p = 0.0007) thickness versus those of sib controls. (C) Mean ventricular weights of nmd mice are not significantly different from control values. However, a significant increase was observed in the mean weight of the nmd right (RA, * p = 0.008) and left atria (LA, * p = 0.005) versus control atrial weights
The histomorphometric changes accompanying these altered conduction values are summarized in Figures 5A and B. The post-mortem left ventricular free wall thickness in nmd mice with or without overt clinical signs was highly significantly reduced compared with age-matched controls. Similarly, the thickness of the right ventricular free wall and interventricular septum between all three nmd genotypes were significantly reduced relative to controls (Fig. 5B). A separate group of nmd mice with overt clinical signs and aged-matched control littermates were used for measurements of atrial and ventricular free wall and cardiac septal weights (Fig. 5C). The cardiac septum and ventricular free wall weights of nmd hearts were remarkably similar to control values, suggesting the absence of a compensatory ventricular hypertrophy. However, significant increases in atrial mass in nmd versus control hearts (6.7 ± 0.7 mg (n = 18) vs. 3.7 ± 0.27 mg (n = 14), p < 0.008) were unexpectedly observed.
To further validate the diagnosis of dilated cardiomyopathy with a sequela of congestive heart failure, we conducted definitive tests using echocardiography and ECG this time under generalized anesthesia. Echocardiography of nmd, nmd-MnmC and nmd-TgNI mice with overt clinical signs and elevated serum CK-MB values displayed several in vivo hallmarks of dilated cardiomyopathy including reduced ventricular wall thickness, increased ventricular chamber volume, reduced cardiac output, ejection fraction and fractional shortening (Fig. 6C–E, Table 1). Observed alterations in atrial and ventricular conduction recapitulated what was observed in unanesthetized mice, that is a paucity of arrhythmia and a marked increase in heart rate variability. These observations are exhibited as wide and/or split P wave (⇛), prolonged P–Q and/or QRS intervals and a generalized reduction or negation in the amplitude of the signals (⇒ and ⇨) that correlated with clinical presentation (Table 1, Fig. 6A–E). Previously measured post-mortem atrial enlargement in clinically affected nmd mice was confirmed by the atrial conduction in similarly affected nmd mice with the broadening of the P waves and the superimposition of the right and left atrial contractions shown as split P waves (Fig. 6C–E). Likewise, the observed post-mortem ventricular enlargement is corroborated by a prolonged QRS complex, increased negative deflection of the Q and S waves, and in the most severe cases, the presence of two superimposed R waves (data not shown). The combination of serial ancillary tests and echocardiography in the same mice provided an opportunity to correlate biochemical and functional parameters of the heart in order to assess with greater confidence the integrity of the cardiovascular system and the exact health status of the animal. Thus, a normal value for plasma CK-MB alone was not as informative as when combined with a normal electrocardiogram. However, a significantly high level of plasma CK-MB was predictive of a functionally compromised heart (Table 1, Fig. 6B and C).
Discussion
We have shown that neuronal expression of an Ighmbp2 transgene in nmd mice prevents motor neuron degeneration and restores normal axonal morphology and density. Interestingly, our tissue-specific rescue of motor neuron degeneration has uncovered a previously unrecognized requirement for IGHMBP2 in both cardiac and skeletal muscles. Regardless of the degree of motor neuron rescue, all nmd mice eventually develop dilated cardiomyopathy and congestive heart failure. In addition, the persistence of skeletal myocyte degeneration/regeneration suggests that normal levels of IGHMBP2 in skeletal muscles are essential for preventing an underlying myopathy (Fig. 2C–D). Autosomal recessive mutations in the human IGHMBP2 gene cause a phenotypically similar disease, SMARD1 (5, 6, 8, 9). Like the mutant nmd mouse, SMARD1 patients develop a progressive neurogenic limb muscular atrophy as infants. However, unlike in SMARD1 patients, the absence of diaphragmatic and intercostals muscle paralysis and normal axonal counts in the phrenic nerve of nmd mice suggest that this population of motor neurons is uniquely spared relative to neurons innervating limb skeletal muscles. Whether this is a species-specific manifestation or a consequence of residual Ighmbp2 expression (20–25% of normal) from the hypomorphic nmd splice-site mutation (1) remains to be determined. It is possible that a complete null mutation in the mouse Ighmbp2 gene would more faithfully reproduce this aspect of the human disease.
The Role of IGHMBP2 in Cardiac and Skeletal Muscle.
Gross morphological and histopathological examination of mutant nmd hearts shortly after birth revealed no discernable abnormalities. Thus, we hypothesize that IGHMBP2 is not critical for normal cardiac morphogenesis. From birth until 7 days postnatally, the major cardiomyocyte myosin heavy chain (MHC) isoform in the mouse is ß-MHC (manifested by slower heart rates). Thereafter, complete transition to the adult ∂-MHC isoform (manifested by faster heart rates) occurs (24). This transition appears to occur normally in nmd mice, since mutant mice weaned at three weeks of age are predominantly non-symptomatic, with plasma CK-MB, HRs, ECGs and echocardiogram measurements within normal range. However, normal levels of IGHMBP2 may be critical for cardiomyocyte maturation during the stage of rapid growth that occurs from 3 to 6 weeks of age. Appropriate compensatory responses and adequate remodeling of the heart to changing load and demand may be an essential function of IGHMBP2 in cardiac muscle (20, 25–28). Given IGHMBP2’s putative role in transcriptional activation or splicing, down-regulation of pro-survival genes and/or increased expression of pro-apoptotic genes could result in a cascade of signaling events culminating in apoptosis in motor neurons, as well as in cardiac and skeletal myocytes (20, 23, 27).
To date, descriptions of dilated cardiomyopathy in SMARD1 patients is lacking, but a single case of heart disease has been reported in an SMA patient and 5 SMARD1 infants were described with cardiac arrhythmia (8, 9, 29). Perhaps if SMARD1 patients were able to live longer they might manifest signs of heart disease as well. Alternatively, a significant ascertainment bias in the patient population may exist due to the mistaken limitation of molecular IGHMBP2 analyses only to children presenting with severe infantile neuropathy with diaphragmatic weakness. Based on our transgenic rescue results, we predict that treatment of neurodegeneration in SMARD1 will ameliorate the progressive motor neuron loss, while failing to prevent the development of progressive cardiomyopathy. The marked improvement in mobility and strength of our nmd-TgNI mice initially increased their probability of survival past weaning. Paradoxically, their overall increased activity may have hastened the manifestation of their underlying cardiac disease. It is interesting to note that the cardiomyocyte degeneration is much more severe than the myopathy observed in skeletal muscles of transgenic nmd mice. Skeletal muscle maintains a large pool of stem cells (myoblast satellite cells) capable of extensive regeneration evidenced by the presence of skeletal muscle fibers with centralized nuclei. Cardiomyocytes may have a higher innate requirement for IGHMBP2 levels than skeletal myofibers. Alternatively, a greater rate of cardiomyocyte cell death that exceeds the ability of cardiac stem or progenitor cells to regenerate (29) may account for the difference in severity between the two tissues.
MnmC is a neuron-specific genetic modifier.
Although the modified nmd-MnmC mice display a remarkable amelioration of the neurogenic atrophy phenotype, they still succumb to DCM. This suggests that the protective allele of the modifier gene is acting as a neuron-specific suppressor of motor neuron degeneration. In contrast to nmd-TgNI mice, the mean life span and cumulative survival of nmd-MnmC mice were comparable to those of nmd mice (Fig. 3C). Since nmd and nmd-MnmC mice do not have full control of their limbs, the consequent reduction in their activity due to deficits in muscle function may have lessened the demand on their cardiovascular systems.
An effective treatment for motor neuron diseases such as SMA or ALS is currently not available (30–32). Our successful transgenic rescue of the neuromuscular degeneration phenotype in nmd mice offers a promise for gene therapy but highlights potential difficulties in the search for a treatment strategy for SMARD1 patients. Our results also suggest that human IGHMBP2 mutation screens should be expanded to include familial DCM and atypical spinal muscular atrophy patients with cardiomyopathy. Complete functional rescue of the nmd mutation will likely require expression of IGHMBP2 not only in motor neurons, but also in cardiac and skeletal myocytes. Elucidating the role of Ighmbp2 in the manifestation of cardiomyopathy in nmd mice will highlight novel mechanisms in the pathogenesis of congestive heart failure.
Methods
Mice and Genotypic Selection.
Mice were bred and maintained under standard conditions in the Research Animal Facility at The Jackson Laboratory. Genotyping for the nmd mutation was performed essentially as described (1). Oligonucleotide primers F15 (5’ GCTGGAAACGATCACATACCG-3’) and R14 (5’-AGCTCCTGATGATCCAATGG-3’) were used to PCR amplify a 746 bp product from genomic DNA containing the nmd point mutation. A restriction fragment polymorphism detected using HindIII and DdeI enzymes was used to differentiate between wildtype-+/+, (396, 202 and 148 bp fragments), heterozygous-nmd/+, (396, 202, 148, 182 and 214 bp fragments) and homozygous-nmd/nmd (202, 148, 182 and 214 bp fragments) genotypes. Genotyping for the MnmC modifier locus was accomplished using two sets of flanking Chromosome 13 markers: HLAHF2 5’-GTTGTGAGACCCTCTCTTTG-3’ and HLAHR2 5’-GTTCCATGCTACAGATACCC-3’(CAST: 220 bp; C57BL/6J: 180 bp), and 9o13CA1F 5’-GCTGGGGAAACTTGGAGTTC-3’, and 9o13CA1R 5’-GCAAAATTGGCTTGTTCAAG-3’ (CAST: 152 bp; C57BL/6J: 140 bp). Transgenic mice were identified using Eno2i1F (5’-CTGAGTCTGCAGTCCTCGAG-3’) within the rat Eno2 intron 1 and Ighmbp2R17 (5’-GAGGTGAAGCTGTTGCTAGG-3’) within the Ighmbp2 cDNA to generate a 560 bp product.
Generation and Characterization of Transgenic Eno2-Ighmbp2 (TgNI) Mice.
A 3.1 kb mouse Ighmbp2 cDNA was amplified by RT-PCR from spinal cord RNA with primers F1 5’-CACGGTCCGACTGGAACTCG -3’ and R12 5’-GAAATGGCCAGTGTAGGACC-3’ essentially as described (33). The Ighmbp2 cDNA was cloned into the HindIII site of pNSE-Ex4 (kindly provided by Dr. J. Gregor Sutcliffe, Dept. of Molecular Biology, The Scripps Research Institute, La Jolla, CA) downstream of the 4 kb rat Eno2 promoter and upstream of an SV40 (simian virus) poly(A) sequence (34). Heterozygous transgenic C57BL/6J-TgN(Eno2-Ighmbp2) mice were generated from two independent founders (lines 17 and 90). Transgene expression was confirmed by RT-PCR (393 bp product) using primers within exon 1 of the rat Eno2 gene (Eno2e1F 5’-CTGAGTCTGCAGTCCTCGAG-3’) and within the Ighmbp2 cDNA (Ighmbp2-R17). Transgenic positive mice (+/+ for nmd) were bred to heterozygous B6.BKS - +/nmd mice and the F1 transgenic positive heterozygous (+/nmd) offspring were backcrossed to B6.BKS - +/nmd mice to generate transgenic positive mutant nmd/nmd mice (nmd-TgNI). Likewise, a full congenic of the CAST-derived nmd modifier Mnm on chromosome 13 was created in the background of C57BL/6J (1). Mice homozygous for the modifier (B6.CAST-Mnm) were bred to heterozygous nmd (+/nmd) mice and the F1 offspring intercrossed to generate homozygous modified mutant nmd/nmd mice (nmd-Mnm). All homozygous nmd mice with/without the presence of the transgene or the modifier were weighed daily with their littermate controls during their first week and were subsequently weighed weekly or bi-weekly until the manifestation of heart disease.
Functional Assessment of Muscle Strength and Endurance.
A wire grip test was used to assess muscle strength and dexterity in 4–7 week old mice. Mice were placed on top of an inclined (60 degrees from horizontal) wire-mesh cage cover (0.8 cm x 0.8 cm grids) for a maximum of 3 minutes. For the assessment of strength and endurance, we employed a pole test in which mice were vertically suspended for a maximum of 3 minutes on a coarsely threaded metal rod (1.27 cm diameter, 30 cm long) held by a ring stand 0.5 m above the cage bedding. Tests were terminated when a mouse fell. Latency to fall, in seconds, was recorded as a measure of limb muscle function.
L4 Ventral Root Axonal Count in Transgenic nmd Mice.
Mice between 28 and 49 days of age were cardiac perfused with 4% paraformaldehyde and 2.5% glutaraldehyde fixative in PBS. Intact dorsal and ventral lumbar segments L2 to L6 nerve roots were obtained by dorsal laminectomy. Isolated nerve roots were fixed for an additional two hours, then post-fixed with 2% osmium tetroxide and embedded in Embed-812 according to manufacturer’s protocol (Electron Microscopy Sciences, Fort Washington, PA) (35). One-μm thick axonal cross-sections were stained with p-Phenylenediamine (36). Total myelinated axons were counted for each nerve root and diameters of each axon were recorded.
Total Plasma Creatine Kinase (CK) and CK-MB.
One drop of ophthalmic anesthetic solution (0.5% Tetracaine Hydrochloride, Baush & Lomb, Pharmaceuticals Inc., Tampa) was administered in the eye and 150 μl of whole blood was obtained using a heparinized microcapillary tube via the retro-orbital sinus of 3 to 4-wk-old mice in the first cohort and 7 to 9-wk-old mice in the second cohort. Following centrifugation at 5,000x g, at least 75 μl of serum or plasma (using EDTA as anti-coagulant) was analyzed using the SYNCHRON CX Clinical System (Beckman Coulter, Inc. Fullerton CA) and reagents provided by the manufacturer.
Assessment of Cardiac Function and Histopathology.
Measurement of end-systolic blood pressure and pulse rate was obtained from un-anesthetized 5–10 week old mice with minimal restraint and recorded over a 5-day period following a one-week training period (Visitech BP-2000, Cary, NC). Non-invasive recording of heart rate (HR) and electrocardiogram (ECG) were accomplished in conscious 4–10 week old mice using the AnonyMouse™ ECG Screening System (Mouse Specifics, Inc., Boston, MA) (37). Standard electrocardiogram and echocardiogram was done in anesthetized mice (1–1.5% Isofluorane at 0.6 L/min.). Following hair removal (Nair Hair Remover) in the region of interest, a high-frequency Vevo 660 ultrasound system with built-in software for analysis was used (Visualsonics, Toronto, Ontario, Canada). For all echocardiographic images a 30 mHz RMV (real-time microvisualization) scan-head was used, along with a capture rate of 30 frames per second. In obtaining a 2-dimentional parasternal short axis (PSA) view of the left ventricle, the papillary muscles were used as the landmark. M-mode images were also produced from PSA views for the determination of heart rates and left ventricular wall (LV) thickness during diastole and systole. From these measurements the % fractional shortening (%FS), % ejection fraction (% EF) and diastolic/systolic volumes were calculated. In addition, 2-chamber apical views were imaged and then subsequently outlined to delineate the longitudinal area of the left ventricular chamber during systole and diastole. Obtained measurements from these images were thus used in calculating the fractional left ventricular cardiac output (LV CO). ECG recordings were taken over a six-minute period for each mouse. Heart rate, R-R interval, and respiratory rate were recorded simultaneously using lead I configuration (one positive (+) lead to left front limb and one negative (−) lead to the right front limb plus a ground lead to one of the hind foot)- while P, QRS and T wave intervals and amplitudes were measured using Chart 5.0.1 software (ADInstruments, Colorado Springs, CO, USA). Following ECG recording, hearts were isolated and fixed in Bouin’s fixative for 24 hr. Hematoxylin and eosin (H & E)-stained 6 μm sections were measured for cardiac chamber wall thickness at the mid-level of the left ventricular papillary muscle (Fig. 4D). Masson’s Trichrome stained sections were used to determine the degree of collagen deposition. For electron microscopic evaluation, samples from the atria, septum and ventricles were fixed overnight at 4°C in 3% glutaraldehyde in PBS and processed by standard methods. Hearts from an additional cohort of mice were used to determine atrial and ventricular wall weights.
Statistical Analysis .
Gender effects were not apparent except in the analysis of maximum lifespans; therefore, data measurements from males and females were combined and grouped according to age and genotype. Values were expressed as the mean ± SE and analyzed by paired Student’s t test or ANOVA using Statview 5.0.1 (SAS Institute Inc). A value of p < 0.05 was regarded as statistically significant.
Acknowledgments
We are grateful to Drs. Robert Burgess and Timothy O’Brien for critical review of the manuscript and Dr. Anthony Nicholson and Crystal Davis for physiology and in vivo imaging services. Special thanks go to Drs. Huykiang Kim and Alison Mann for their helpful suggestions. This work was supported by grants from NIH RO1NS38178 and RO1AR49043 to G.A.C.; T.P.M. was supported by fellowships from NIH T32 RR07068 and The American Heart Association.
References
- 1.Cox GA, Mahaffey CL, Frankel WN. Identification of the mouse neuromuscular degeneration gene and mapping of a second site suppressor allele. Neuron. 1998;21:1327–1337. doi: 10.1016/s0896-6273(00)80652-2. [DOI] [PubMed] [Google Scholar]
- 2.Molnar GM, Crozat A, Kraeft SK, Dou QP, Chen LB, Pardee AB. Association of the mammalian helicase MAH with the pre-mRNA splicing complex. Proc Natl Acad Sci U S A. 1997;94:7831–7836. doi: 10.1073/pnas.94.15.7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shieh SY, Stellrecht CM, Tsai MJ. Molecular characterization of the rat insulin enhancer-binding complex 3b2. Cloning of a binding factor with putative helicase motifs. J Biol Chem. 1995;270:21503–21508. doi: 10.1074/jbc.270.37.21503. [DOI] [PubMed] [Google Scholar]
- 4.Miao M, Chan SL, Fletcher GL, Hew CL. The rat ortholog of the presumptive flounder antifreeze enhancer-binding protein is a helicase domain-containing protein. Eur J Biochem. 2000;267:7237–7246. doi: 10.1046/j.1432-1327.2000.01836.x. [DOI] [PubMed] [Google Scholar]
- 5.Grohmann K, Schuelke M, Diers A, Hoffmann K, Lucke B, Adams C, Bertini E, Leonhardt-Horti H, Muntoni F, Ouvrier R, et al. Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nat Genet. 2001;29:75–77. doi: 10.1038/ng703. [DOI] [PubMed] [Google Scholar]
- 6.Grohmann K, Wienker TF, Saar K, Rudnik-Schoneborn S, Stoltenburg-Didinger G, Rossi R, Novelli G, Nurnberg G, Pfeufer A, Wirth B, et al. Diaphragmatic spinal muscular atrophy with respiratory distress is heterogeneous, and one form Is linked to chromosome 11q13–q21. Am J Hum Genet. 1999;65:1459–1462. doi: 10.1086/302636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Viollet L, Barois A, Rebeiz JG, Rifai Z, Burlet P, Zarhrate M, Vial E, Dessainte M, Estournet B, Kleinknecht B, et al. Mapping of autosomal recessive chronic distal spinal muscular atrophy to chromosome 11q13. Ann Neurol. 2002;51:585–592. doi: 10.1002/ana.10182. [DOI] [PubMed] [Google Scholar]
- 8.Pitt, M., Houlden, H., Jacobs, J., Mok, Q., Harding, B., Reilly, M. and Surtees, R. (2003) Severe infantile neuropathy with diaphragmatic weakness and its relationship to SMARD1. Brain [DOI] [PubMed]
- 9.Grohmann K, Varon R, Stolz P, Schuelke M, Janetzki C, Bertini E, Bushby K, Muntoni F, Ouvrier R, Van Maldergem L, et al. Infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1) Ann Neurol. 2003;54:719–724. doi: 10.1002/ana.10755. [DOI] [PubMed] [Google Scholar]
- 10.Nicholson SJ, Witherden AS, Hafezparast M, Martin JE, Fisher EM. Mice, the motor system, and human motor neuron pathology. Mamm Genome. 2000;11:1041–1052. doi: 10.1007/s003350010205. [DOI] [PubMed] [Google Scholar]
- 11.Franz WM, Muller OJ, Katus HA. Cardiomyopathies: from genetics to the prospect of treatment. Lancet. 2001;358:1627–1637. doi: 10.1016/S0140-6736(01)06657-0. [DOI] [PubMed] [Google Scholar]
- 12.Hoffman JR. Annals supplement on the American Heart Association proceedings. Ann Emerg Med. 2001;38:605. doi: 10.1067/mem.2001.119276. [DOI] [PubMed] [Google Scholar]
- 13.Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, Smoot L, Mullen MP, Woolf PK, Wigle ED, et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med. 2000;343:1688–1696. doi: 10.1056/NEJM200012073432304. [DOI] [PubMed] [Google Scholar]
- 14.Arber S, Hunter JJ, Ross J, Hongo M, Sansig G, Borg J, Perriard JC, Chien KR, Caroni P. MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell. 1997;88:393–403. doi: 10.1016/s0092-8674(00)81878-4. [DOI] [PubMed] [Google Scholar]
- 15.Nicol RL, Frey N, Olson EN. From the sarcomere to the nucleus: role of genetics and signaling in structural heart disease. Annu Rev Genomics Hum Genet. 2000;1:179–223. doi: 10.1146/annurev.genom.1.1.179. [DOI] [PubMed] [Google Scholar]
- 16.Fentzke RC, Korcarz CE, Lang RM, Lin H, Leiden JM. Dilated cardiomyopathy in transgenic mice expressing a dominant- negative CREB transcription factor in the heart. J Clin Invest. 1998;101:2415–2426. doi: 10.1172/JCI2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wallace DC. Mitochondrial defects in cardiomyopathy and neuromuscular disease. Am Heart J. 2000;139:S70–85. doi: 10.1067/mhj.2000.103934. [DOI] [PubMed] [Google Scholar]
- 18.Crone SA, Zhao YY, Fan L, Gu Y, Minamisawa S, Liu Y, Peterson KL, Chen J, Kahn R, Condorelli G, et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat Med. 2002;8:459–465. doi: 10.1038/nm0502-459. [DOI] [PubMed] [Google Scholar]
- 19.Schonberger J, Seidman CE. Many roads lead to a broken heart: the genetics of dilated cardiomyopathy. Am J Hum Genet. 2001;69:249–260. doi: 10.1086/321978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Towbin JA, Bowles NE. The failing heart. Nature. 2002;415:227–233. doi: 10.1038/415227a. [DOI] [PubMed] [Google Scholar]
- 21.Kang PM, Izumo S. Apoptosis and heart failure: A critical review of the literature. Circ Res. 2000;86:1107–1113. doi: 10.1161/01.res.86.11.1107. [DOI] [PubMed] [Google Scholar]
- 22.Ross J., Jr Dilated cardiomyopathy: concepts derived from gene deficient and transgenic animal models. Circ J. 2002;66:219–224. doi: 10.1253/circj.66.219. [DOI] [PubMed] [Google Scholar]
- 23.Elsasser A, Suzuki K, Schaper J. Unresolved issues regarding the role of apoptosis in the pathogenesis of ischemic injury and heart failure. J Mol Cell Cardiol. 2000;32:711–724. doi: 10.1006/jmcc.2000.1125. [DOI] [PubMed] [Google Scholar]
- 24.Dalloz F, Osinska H, Robbins J. Manipulating the contractile apparatus: genetically defined animal models of cardiovascular disease. J Mol Cell Cardiol. 2001;33:9–25. doi: 10.1006/jmcc.2000.1289. [DOI] [PubMed] [Google Scholar]
- 25.Chien KR. Genomic circuits and the integrative biology of cardiac diseases. Nature. 2000;407:227–232. doi: 10.1038/35025196. [DOI] [PubMed] [Google Scholar]
- 26.Davidoff AJ, Gwathmey JK. Pathophysiology of cardiomyopathies: Part I. Animal models and humans. Curr Opin Cardiol. 1994;9:357–368. [PubMed] [Google Scholar]
- 27.Schwartz K, Mercadier JJ. Molecular and cellular biology of heart failure. Curr Opin Cardiol. 1996;11:227–236. doi: 10.1097/00001573-199605000-00002. [DOI] [PubMed] [Google Scholar]
- 28.Wagoner LE, Walsh RA. The cellular pathophysiology of progression to heart failure. Curr Opin Cardiol. 1996;11:237–244. doi: 10.1097/00001573-199605000-00003. [DOI] [PubMed] [Google Scholar]
- 29.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–776. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 30.Wirth B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA) Hum Mutat. 2000;15:228–237. doi: 10.1002/(SICI)1098-1004(200003)15:3<228::AID-HUMU3>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 31.Strong MJ. The basic aspects of therapeutics in amyotrophic lateral sclerosis. Pharmacol Ther. 2003;98:379–414. doi: 10.1016/s0163-7258(03)00040-8. [DOI] [PubMed] [Google Scholar]
- 32.Hand CK, Rouleau GA. Familial amyotrophic lateral sclerosis. Muscle Nerve. 2002;25:135–159. doi: 10.1002/mus.10001. [DOI] [PubMed] [Google Scholar]
- 33.Cox GA, Phelps SF, Chapman VM, Chamberlain JS. New mdx mutation disrupts expression of muscle and nonmuscle isoforms of dystrophin. Nat Genet. 1993;4:87–93. doi: 10.1038/ng0593-87. [DOI] [PubMed] [Google Scholar]
- 34.Mucke L, Masliah E, Johnson WB, Ruppe MD, Alford M, Rockenstein EM, Forss-Petter S, Pietropaolo M, Mallory M, Abraham CR. Synaptotrophic effects of human amyloid beta protein precursors in the cortex of transgenic mice. Brain Res. 1994;666:151–167. doi: 10.1016/0006-8993(94)90767-6. [DOI] [PubMed] [Google Scholar]
- 35.Luft JH. Improvements in epoxy resin embedding methods. J Biophys Biochem Cytol. 1961;9:409–414. doi: 10.1083/jcb.9.2.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sadun AA. Vision: a multimodal sense. Bull Clin Neurosci. 1985;50:61–68. [PubMed] [Google Scholar]
- 37.Chu V, Otero JM, Lopez O, Morgan JP, Amende I, Hampton TG. Method for non-invasively recording electrocardiograms in conscious mice. BMC Physiol. 2001;1:6. doi: 10.1186/1472-6793-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]